Back to Journals » Cancer Management and Research » Volume 12
Familial Pancreatic Cancer: Current Perspectives
Authors Llach J, Carballal S, Moreira L
Received 18 July 2019
Accepted for publication 15 January 2020
Published 31 January 2020 Volume 2020:12 Pages 743—758
DOI https://doi.org/10.2147/CMAR.S172421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Joan Llach, Sabela Carballal, Leticia Moreira
Departmento de Gastroenterología, Hospital Clínic de Barcelona, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Institut d’ Investigacions Biomediques August Pi i Sunyer (IDIBAPS), Universidad de Barcelona, Barcelona, Spain
Correspondence: Leticia Moreira Villarroel 170, Barcelona 08036, Spain
Tel +34 932275400
Fax +34 932279381
Email [email protected]
Abstract: Pancreatic cancer (PC) is a highly lethal disease, mostly incurable when detected. Thus, despite advances in PC treatments, only around 7% of patients survive 5-years after diagnosis. This morbid outcome is secondary to multifactorial reasons, such as late-stage diagnosis, rapid progression and minimal response to chemotherapy. Based on these factors, it is of special relevance to identify PC high-risk individuals in order to establish preventive and early detection measures. Although most PC are sporadic, approximately 10% cases have a familial basis. No main causative gene of PC has been identified but several known germline pathogenic mutations are related with an increased risk of this tumor. These inherited cancer syndromes represent 3% of all PC. On the other hand, in 7% of cases of PC, there is a strong family history without a causative germline mutation, a situation known as familial pancreatic cancer (FPC). In recent years, there is increasing evidence supporting the benefit of genetic germline analysis in PC patients, and periodic pancreatic screening in PC high-risk patients (mainly those with a lifetime risk greater than 5%), although there is no general agreement in the group of patients and individuals to study and screen. In the present review, we expose an update in the field of hereditary and FPC, with the aim of describing the current strategies and implications in genetic counseling, surveillance and therapeutic interventions.
Keywords: pancreatic cancer, hereditary, familial, mutation, screening, personalized medicine
Introduction
Pancreatic cancer (PC) (referred to pancreatic ductal adenocarcinoma) is one of the most aggressive cancer, a rapidly progressive disease and mostly incurable when detected. Thus, despite advances in PC treatments, only around 7% of patients survive 5-year after diagnosis.1 In fact, it is expected to become the second leading cause of cancer death worldwide by 2030.2,3
PC development has been commonly related with environmental factors, such as smoking and alcohol consumption. But, while most PC are sporadic, approximately 10% of the cases have a familial basis.4 No main causative gene of PC has been identified but several known germline pathogenic mutations are related with an increased risk of this tumor. These inherited cancer syndromes represent 3% of all PC. On the other hand, in 7% of cases of PC, there is a strong family history without a causative germline mutation, situation known as familial pancreatic cancer (FPC).1
Due to the low incidence of PC, with a lifetime risk of 1.5% in general population (seer.cancer.gov/statfacts/html/pancreas.html), screening is not feasible, but it should be considered in high-risk individuals (those with five to tenfold increased risk for PC). This scenario includes hereditary syndromes associated with increased risk of PC and members of FPC. The purpose of screening is the detection of precursor lesions or early cancer. Identification of PC in an early stage could be essential to improve survival, as demonstrated by the small portion of people with localized cancers who reach a 5-year survival rate of 31.5%.5 Moreover, recent data suggest that some specific germline mutations (mainly related to homologous repair) could be therapeutically targetable and guide personalized therapy.5–7 Thus, identifying patients with predisposing genetic factors or FPC seems an attractive strategy for improving clinical outcomes.
In the present review, we expose an update in the field of hereditary and FPC, with the aim of describing the current strategies and implications in genetic counseling, surveillance and therapeutic interventions.
Hereditary Syndromes with Higher Risk of Pancreatic Ductal Adenocarcinoma
We define “hereditary pancreatic cancer” when this tumor occurs as a consequence of a known germline pathogenic mutation. These inherited cancer syndromes represent 3% of all PC.
Hereditary pancreatic cancer (HPC) can present in the context of several hereditary syndromes. Diagnosis is usually based upon clinical criteria of the different syndromes associated, followed by a confirmation with a genetic test.
The most frequent genetic alterations are BRCA2, PALB2, ATM, and CDKN2A/p16, and, less frequently, BRCA1, APC, MLH1, MSH2, MSH6, PMS2, PRSS1, and STK11. Hereditary breast and ovarian cancer syndrome (HBOC) due to BRCA2 mutations is the most common form of HPC, and those genes with the highest risk of developing this neoplasm are STK11, PRSS1 CDKN2A/p16, corresponding to Peutz-Jeghers syndrome (PJS), hereditary pancreatitis (HP) and Familial atypical multiple mole melanoma (FAMMM), respectively. Except in hereditary pancreatitis, the rest of hereditary syndromes predispose to other tumors, and the pancreas is not the main organ affected. The increased risk of PC is calculated based on previous prospective and retrospective series of individuals with some of the above-mentioned germ cell mutations that were reported to have a significantly elevated incidence of PC. In those groups in which PC has been seen to be higher than 5% lifetime or 10 times more often than in general population, screening should be recommended. Hereditary syndromes associated with an increased risk of PC and specific germline mutations are summarized in Table 1.
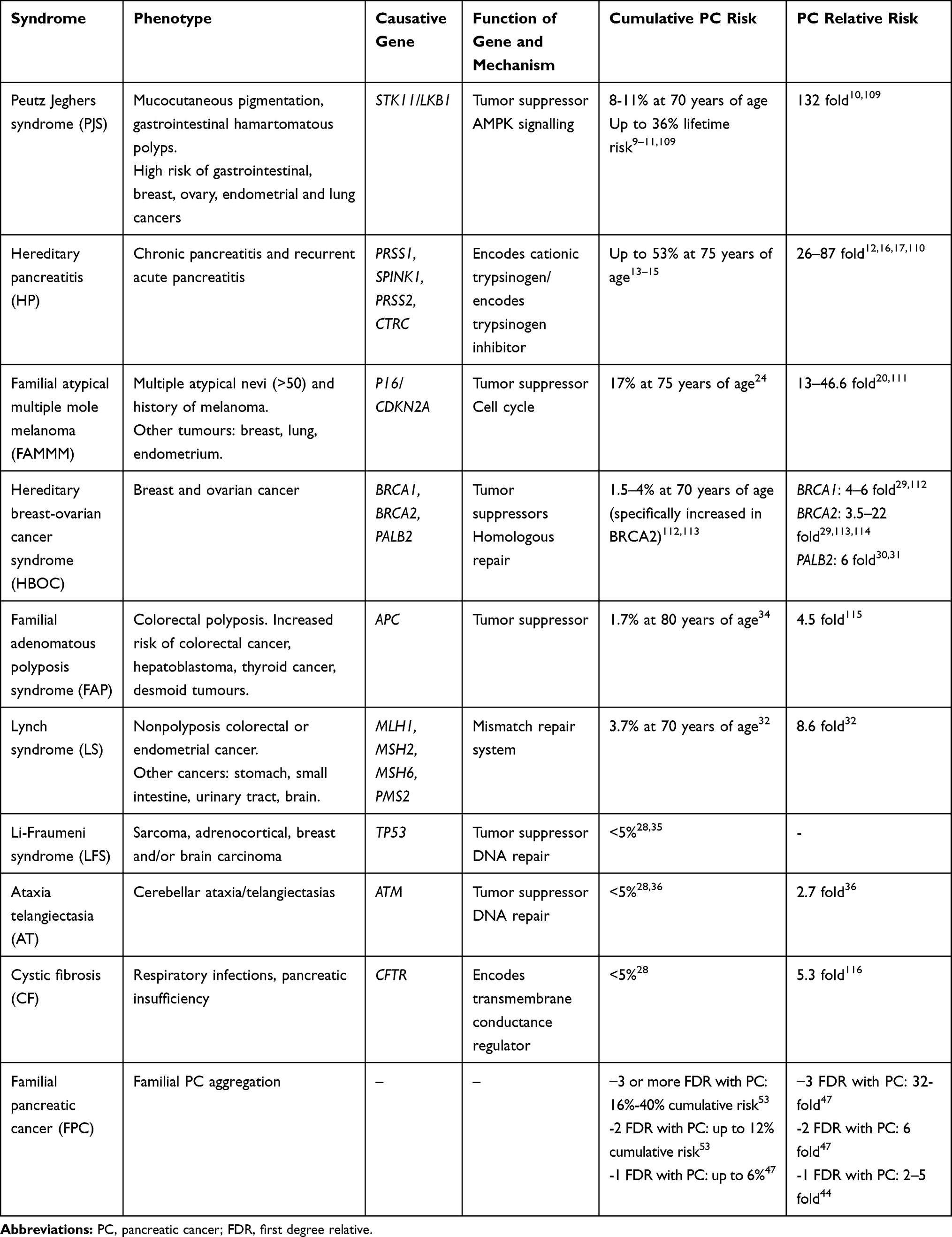 |
Table 1 Hereditary/Familial Syndromes Associated with Increased Risk of Pancreatic Cancer |
Peutz-Jeghers syndrome (PJS)
PJS is an autosomal dominant inherited syndrome characterized by the presence of hamartomatous polyps along the gastrointestinal tract and mucocutaneous hyperpigmentation (both seen in more than 95% and 88% of cases, respectively).8 Most common neoplasms are breast and colon cancer, followed by pancreatic, stomach and ovarian cancer. The cumulative risk of developing any cancer and specifically PC at 70 years of age is 85% and 11%, respectively (132-fold increased PC risk).9,10 In another analysis of 240 PJS patients harboring SKT11 mutation, cumulative PC risk was 8% at 60 years of age.11
A causal germline mutation in the STK11 gene (also known as LKB1), a tumor suppressor gene related with the development of hamartomas and located on chromosome 19p13.3, has been identified in 70.9% of cases.9 The predominant tumor histology is adenocarcinoma and clinical diagnosis of PJS requires the presence of at least two of the following: a) mucocutaneous pigmentation; b) two or more Peutz-Jeghers-type gastrointestinal hamartomas; c) family history of PJS.
Hereditary Pancreatitis (HP)
HP is a rare disease with an autosomal-dominant inheritance pattern commonly associated with a mutation in the PRSS1 gene. This gene is located on 7q35 chromosome and PRSS1 mutations have an estimated penetrance of 80%. In some other cases, HP is associated with the SPINK gene, located on 5q32 chromosome. HP is a hereditary form of chronic pancreatitis in which symptoms begin between the first and second decades of life. The increased risk for the development of pancreatic cancer in these individuals is estimated to be 26-fold to as high as 87-fold,12–15 and cumulative risk of PC varies between 7.2% and 53.5%.16,17 Diagnosis is based on the medical history supported by complementary imaging tests and an autosomal-dominant pattern of inheritance.
Familial Atypical Multiple Mole Melanoma (FAMMM)
FAMMM is an autosomal dominant inherited syndrome with incomplete penetrance, characterized by the presence of multiple atypical nevi progressing to melanoma.18 Patients with FAMMM have a 13-46-fold increased risk of pancreatic cancer compared to the general population, and increased incidence of other cancers such as breast, lung or endometrium is also known.19,20 FAMMM is associated with germline mutations in the p16/CDKN2A gene located on chromosome 9p21. The estimated prevalence of CDKN2A mutations among the general population is 0.01%.21 Although germline mutations in CDKN2A are the main hereditary cause of familial melanoma, there are other genes, including CDK4 and BAP13.22 The increased risk of pancreatic cancer has been especially associated with the pathogenic variant of p16 (known as p16-Leiden).20 In terms of mutations in this gene, a study in a Spanish population published in 2014 found a higher prevalence of pancreatic cancer (prevalence rate 2.97, p=0.006) in patients with multiple melanoma who were CDKN2A carriers compared to patients with multiple melanoma with no identified mutation.23 Cumulative PC risk in FAMMM families harboring CDKN2A mutation is 17% at 75 years of age.24 The diagnostic clinical criteria for FAMMM are a high number of common and atypical nevi (>50) and history of melanoma in one or more first or second-degree relatives.25 The incidence of mutations in CDKN2A is in fact greater in individuals with three or more melanomas and/or in families with at least one member with melanoma and two or more relatives of the first or second degree diagnosed with adenocarcinoma of the pancreas. According to the family melanoma database, the Melanoma Genetics Consortium (GenoMEL; https://genomel.org/), the presence of pancreatic cancer is a strong positive predictive indicator of pathogenic mutation in CDKN2A. Thus, of 466 families with melanoma, 185 (40%) were mutation carriers in CDKN2A. Within families with mutation, 49 (28%) had a family history of pancreatic cancer. And of the 66 families with melanoma and pancreatic cancer, 49 (74%) were carriers of mutation in CDKN2A. The frequency of mutation in families with only melanoma was significantly lower, only 33% (compared to 74% when there were both neoplasms in the family).
Hereditary Breast and Ovarian Cancer Syndrome (HBOC)
HBOC is an autosomal dominant inherited syndrome that is genetically caused by inactivating mutations in BRCA1 and BRCA2 genes that are involved in the homologous recombination repair pathway. This disorder is associated with an increased risk of breast cancer, ovarian cancer and other types of cancer, including prostate, male breast, melanoma, pancreatic and gastric cancer. BRCA2 is widely accepted to be a PC precursor, while there is not so evidence in BRCA1, although there is evidence of a 2-fold increased risk of PC in BRCA1 carriers.26–28 According to a recent study including 613 BRCA1 and 459 BRCA2 mutation carriers, Mersch et al29 reported that BRCA2 mutation carriers had 21.7-fold increased risk of PC (95% confidence interval [CI]: 13.1–34.0), whereas the presence of a BRCA1 mutation did not significantly increase the risk of pancreatic adenocarcinoma.
PALB2 is another gene that encodes a protein that may function as tumor suppressor. This protein binds to and colocalizes with the breast cancer 2 early onset protein (BRCA2) in nuclear foci and likely permits the stable intranuclear localization and accumulation of BRCA2. Increasing evidence is progressively available supporting PALB2 as a susceptibility gene for pancreatic cancer.30 Mutations on this gene seem to be less frequent and, although robust evidence is lacking, individuals presenting these mutations could have a 6-fold increased PC risk.31
Lynch Syndrome (LS) or Hereditary Nonpolyposis Colorectal Cancer
LS is an autosomal dominant inherited syndrome where tumors develop from germline mutations in mismatch repair genes (MLH1, MSH2, MSH6 and PMS2). Mismatch repair dysfunction results in loss of protein expression and microsatellite instability (MSI) in tumors. LS is associated mainly with an increased risk of colorectal cancer, although it also carries a risk of other tumors such as endometrial, ovarian, stomach, bile duct, small bowel, pancreatic, ureter and renal pelvis cancer, as well as skin cancer (sebaceous tumors, in the variant known as Muir-Torre syndrome) and central nervous system tumors (glioblastomas and astrocytomas, in the variant known as Turcot syndrome). With regard to PC, Kastrinos et al described that this syndrome increases PC risk by 8.6-fold and a cumulative PC risk of 3.7% at 70 years of age.32 There is recent evidence relating this increased risk of PC in LS mainly with MLH1 mutation carriers.33
Familial Adenomatous Polyposis (FAP)
FAP is an autosomal dominant inherited syndrome associated with germline mutations in the APC gene (chromosome 5q21–q22). Diagnostic suspicion is based on 2 main phenotypes: classic form of FAP, characterized by hundreds of synchronous colorectal adenomas that inevitably progress into malignancies if prophylactic colectomy is not carried out, and the attenuated form, which presents between 10 and 99 adenomas. Moreover, FAP is associated with a wide spectrum of extracolonic tumors, including hepatoblastoma, duodenal, pancreatic, thyroid, bile duct and brain adenocarcinoma. Cumulative PC risk in FAP patients is 1.7% at 80 years of age.34
Other Hereditary Syndromes
Li-Fraumeni syndrome (LFS; TP53 gene-), Ataxia telangiectasia (AT; ATM gene) or Cystic fibrosis (CF; CFTR gene) are other syndromes associated with PC with lower cumulative risk (<5%).35,36 Among these entities, evidence relating germline mutations in ATM with and a high risk of PC is increasing rapidly; although AT is an autosomal recessive disease, the risk of PC is associated with a monoallelic mutation in ATM gene, with a heterogeneous phenotype due to incomplete penetrance. The increased risk is at least 2-fold in comparison with the general population.27,28,37 Finally, some other genes have been identified as potential candidates of susceptibility to PC, such as CHEK2,38 FANNC, FANCG,39,40 CPA1, BUB1B28 and PALLD,41 although more studies are needed to confirm these associations.
Familial Pancreatic Cancer
Definition and Epidemiology
“Familiar pancreatic cancer” (FPC) has been defined by consensus opinion as: “families with two or more first-degree relatives (FDRs) with PC who don´t meet criteria for a known PC-associated hereditary syndrome”.8,42 Therefore, definition of FPC excludes the situations in which a causative germline mutation are identified.43 This scenario accounts for approximately 80% of PC clustering, suggesting that additional genetic, epigenetic, or environmental factors may contribute to PC development.
Phenotype and Risk
First reports demonstrating that PC risk is increased in individuals with a family history were published in 1970s. Then, several case–control and cohort studies have been published quantifying this risk according to the number of family members affected, describing a 2- to 5-fold excess of PC in FDRs of patients with PC.44 These observations led to the creation of family registries, both in USA and Europe, aiming to study the familial aggregation of PC; such as the National Familial Pancreas Tumour Registry (NFPTR, Johns Hopkins, USA), the European Registry of Familial Pancreatic Cancer and Hereditary Pancreatitis (EUROPAC) and the German National Case Collection for Pancreatic Cancer (FaPaCa).13,45,46
The NFPTR demonstrated a nine-fold greater risk of developing PC among individuals with an FDR with PC in the setting of FPC, compared to a 1.8-fold greater risk for those with an FDR with sporadic PC.47 Additionally, among FPC kindreds, having two or three FDRs with PC was associated with a 6.4-fold and 32-fold greater risk of developing PC, respectively.47 Therefore, the risk for developing PC in FPC kindreds is dependent on the number of FDRs affected (Table 1).
As occurs in sporadic cases, smoking is the strongest exogenic risk factor in the setting of FPC, particularly in people younger than 50 years. It increases the risk of PC by 2 to 3.7 times over the inherited predisposition and decreases the age of onset in 10 years.47,48
Similar to other familial cancers, FPC shows a trend toward a younger onset than sporadic cases (FPC 58–68 years vs 61−74)44 and an ethnic deviation.49 The lifetime risk of PC also increases with decreasing age of onset of PC in family members.50
In addition, European registries have observed an anticipation phenomenon in FPC scenario. Thus, a large European study investigated 106 FPC families through three generations and found that from one generation to the next, the age of death from PC was younger with each generation. Subsequent studies performed by EUROPAC and FaPaCa confirmed this event, showing an earlier development of PC by 10 years in 59% to 80% of FPC families.45,51
Finally, one study, using complex segregation analysis has shown evidence for a yet-unidentified autosomal dominant, high-risk allele influencing the onset age of PC present in 7/1000 individuals. This study observed a 32% lifetime risk for PC development at age 85 years.52
The complexity in cancer risk assessment has led to the development of a risk prediction model (PancPRO) to provide more detailed risk estimates for individuals from FPC kindreds that take into account the ages at diagnosis, family size, and the relationship between family members.53
Indication of Germline Analysis
Traditionally germline testing to rule out a hereditary syndrome in the context of a PC is indicated only if the patient or family meets the criteria of one of the known syndromes described above. Recently this approach has been changing for less restricted criteria. Families that meet CPF criteria and do not meet the criteria of any other hereditary syndrome represent up to 80% of families with PC aggregation. Some recent studies27,28 have reported germline mutations in the most frequent genes (BRCA1, BRCA2, PALB2, CDKN2A, ATM) related to hereditary pancreatic syndromes, in up to 10% of these families even without other extra-pancreatic manifestations. Moreover, there is recent evidence37,54,55 revealing that 5–8% of the patients with PC without cancer family history are carriers of a germline mutation in a gene that predisposes to this neoplasia.
Based upon this, there is a trend to support the use of multigenic panels in any patient with PC (regardless age of diagnosis or cancer family history) or at least perform the germline genetic test in families with CPF criteria or patients with early onset PC.56 The analysis should include the following pancreatic cancer predisposition genes: BRCA1, BRCA2, PALB2, CDKN2A, ATM, TP53, MLH1, MSH2, MSH6, PMS2, STK11, as well as PRSS1/SPINK1 and CFTR if the clinical is suggestive of hereditary pancreatitis or cystic fibrosis, respectively. If a hereditary syndrome is diagnosed (with an identified germline mutation), the presymptomatic genetic study it recommended on the relatives to establish who are carriers of the germline mutation, and therefore require surveillance.
In case of ruling out a hereditary syndrome, the diagnosis of familial pancreatic cancer (if it meets the criteria for family aggregation) or sporadic pancreatic cancer will be established. In case of familial pancreatic cancer, the appropriate screening recommendations should be established (Figure 1).
 |
Figure 1 Diagnosis flow-chart of hereditary/familial pancreatic cancer. Conventional approach to rule out a hereditary PC (left part of the figure) is based on the fulfillment of clinical criteria for known hereditary conditions. New proposal suggests performing germline genetic testing in all PC cases or, at least, in early onset PC or if family meets criteria of FPC.1 PSJ, HP, FAMMM, HBOC, FAP, LS, LFS, AT, CF.2BRCA1, BRCA2, PALB2, CDKN2A, ATM, TP53, MLH1, MSH2, MSH6, PMS2, STK11, as well as PRSS1/SPINK1 and CFTR if the clinical is suggestive of hereditary pancreatitis or cystic fibrosis, respectively.3 Clinical criteria of familiar pancreatic cancer (FPC): ≥2 first-degree relatives (FDRs). Abbreviations: PC, pancreatic cancer; PSJ, Peutz Jeghers syndrome; HP, hereditary pancreatitis; FAMMM, familial atypical multiple mole melanoma; HBOC, hereditary breast-ovarian cancer syndrome; FAP, familial polyposis syndrome; LS, Lynch syndrome; LFS, Li-Fraumeni syndrome; AT, Ataxia telangiectasia; CF, cystic fibrosis; FPC, familial pancreatic cancer. |
Surveillance Strategy
Screening of PC is recommended when pancreatic cancer risk is significantly increased (>10-fold increased risk or lifetime PC risk >5%). Since the best methods of surveillance have not been well established, at-risk patients should be referred to centers with ongoing research programs in pancreatic cancer surveillance. Most scientific societies and expert opinion consensus recommend screening in members of FPC kindreds with a PC-affected FDR.8,42
Although large studies confirming mortality benefit of pancreatic screening are lacking, emerging data suggest screening in individuals at high risk is associated with downstaging of incident cancers.57,58 Thus, these screening programs are progressively extended.8,58–60
WHO
There is not a general consensus about individual that should be under PC screening programs. The most accepted indications are summarized in Figure 2.
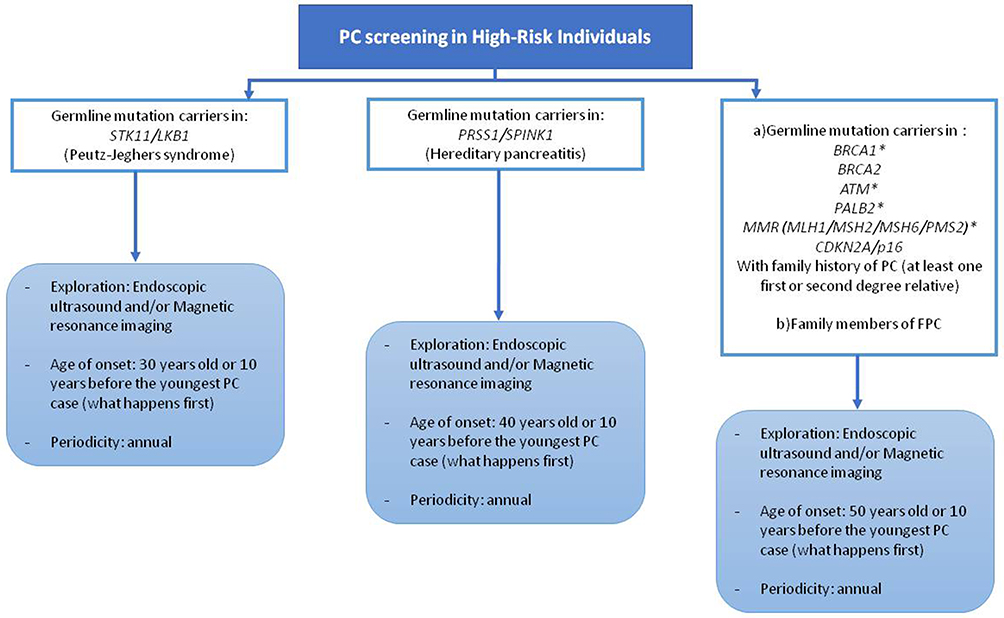 |
Figure 2 Candidates for pancreatic cancer screening. *Evidence and consensus for screening in these mutation carriers is not solid. PC, pancreatic cancer; FPC, familial pancreatic cancer. |
According to the International Symposium of Inherited Diseases of the Pancreas in 2007 (ISIOP)4 potential subjects of surveillance are:
- Individuals diagnosed with PJS or HP
- CDKN2A, BRCA1/BRCA2, mutation carriers with at least one affected first or second degree-relative
- Individuals with three or more affected first-degree, second-degree or third-degree relative
- Individuals with two affected relatives including one FDR
However, the International Cancer of the Pancreas Screening (CAPS) recommendations (2013) differ slightly from those described above:59
- Individuals diagnosed with PJS
- CDKN2A, BRCA2 or MMR gene mutation carriers with at least one affected first degree-relative
- Individuals with at least two FDR affected
A recent prospective study published in 2016 with a long-term follow-up showed that surveillance of CDNK2A mutation carriers is relatively successful, detecting most PCs at a resectable stage, while the benefit of surveillance in families with FPC is less evident.58
Although there is general agreement in screening patients in context of PJS, FAMMM, PH, and HBOC (due to BRCA2 mutation), FPC, there is heterogeneity of indications between centers regarding other syndromes and mutations. This discordance reflects the lack of solid evidence supporting the convenience of screening in some specific groups, such as MMR, BRCA1, or ATM mutations. In this sense, there is an ongoing prospective clinical trial (CAPS5, The Cancer of the Pancreas Screening-5) (https://clinicaltrials.gov/ct2/show/NCT02000089) that classifies individuals in different risk groups based upon their relative risk of PC, establishing different screening recommendations in each group. Figure 3 describes the 5 groups that are candidates for screening according to this study. More studies and solid evidence are needed to define the appropriate indications for PC screening.
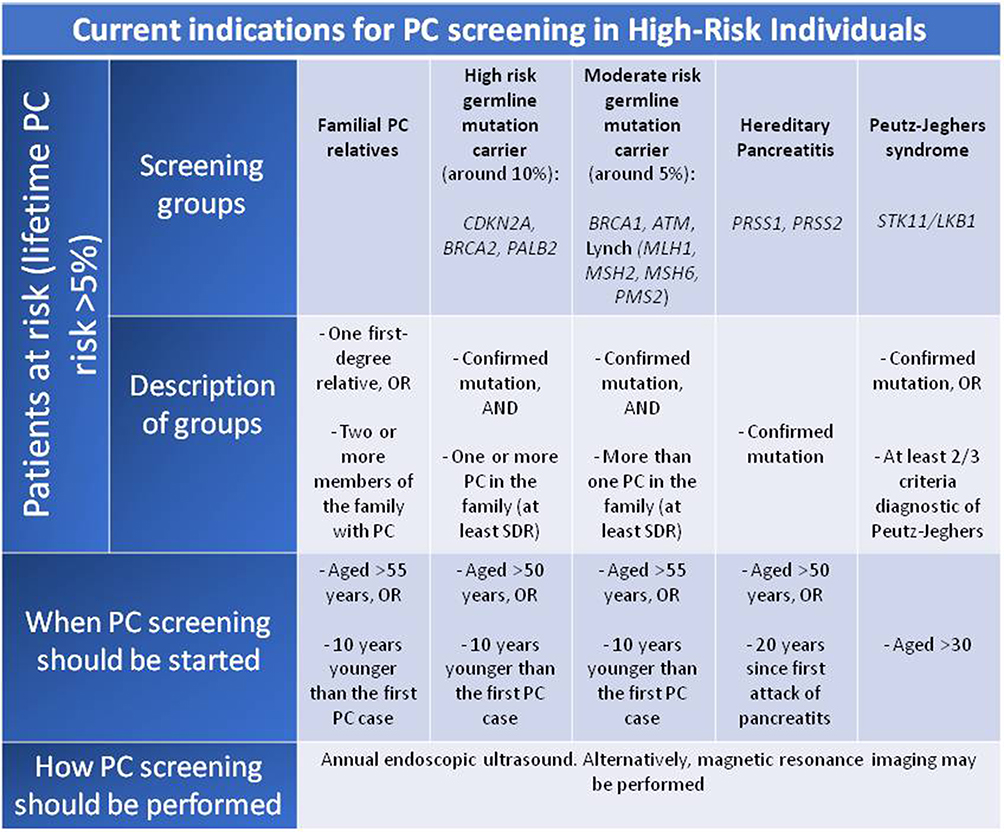 |
Figure 3 Classification of pancreatic cancer high-risk individuals and screening recommendation based upon CAPS5 study. The Cancer of the Pancreas Screening-5. Abbreviations: PC, pancreatic cancer; SDR, second degree relative. |
WHEN
The mean age of PC diagnosis in patients with a PC high-risk syndrome is 68.18, but it is observed an anticipation effect; thus, successive generations usually develop the tumor younger.50 The CAPS Consortium recommends screening in patients with family history of PC at the age of 50, while it is also accepted start 10 years before the youngest PC case. However, in PJS screening should be offered at the age of 30 years (or 10 years before the youngest case) and in HP at the age of 40 years (or 10 years before the youngest case). A recent study published in 2018 that included 86 individuals who underwent PC screening (median of 29.8 months follow-up) concluded that PC screening in patients with known genetic mutations under the age of 50 has low yield.61 For now, no consensus for the age to initiate PC screening exists and it must be individualized, but in general terms, the recommended age of onset of screening is 50 years (or 10 years before the age of the younger affectionate relative), up to 75–80 years, except in PJS and HP that entail an increased risk of early cancer and therefore the screening strategy is different as described above.62 Regarding the periodicity, the most accepted recommendation is to perform the exploration annually.
HOW
Whereas computed tomography is the imaging test of choice when evaluating PC resectability, it has a low sensitivity for small pancreatic neoplasms.63,64 Magnetic resonance imaging (MRI) and endoscopic ultrasonography (EUS) are the most used in the detection of small asymptomatic pancreatic lesions.65 Harinck et al observed that MRI and EUS results are complementary since MRI seems to show a greater sensitivity in the identification of cystic lesions and EUS in the detection of solid lesions.66 However, given its ability to take ultrasound-guided fine needle aspiration and to detect lesions measuring less than 10 mm, EUS should be if possible the first option.57,60,67
WHY
A systematic review published in 2015 analyzed the benefits and harms of pancreatic cancer screening in familial high-risk individuals. Sixteen studies on pancreatic cancer screening were included. Pancreatic cancer screening resulted in a higher curative resection rate (60% vs 25%, P = 0.011) and longer median survival time (14.5 vs 4 months, P < 0.001) compared with the control group.68 Recently, Canto et al observed in a long-term (16-year) follow-up study of individuals at high-risk for PC (354 individuals at high risk were included) that most PCs detected during surveillance (9/10) were resectable, and 85% of these patients survived for 3 years.57 Compared to natural progression of PC, it supposes an important increase of survival in these patients. Nevertheless, pancreatic resection is an aggressive surgery with potential risks, and while PC screening appears to be promising, more data are needed to conclude about its long-term benefits.
When possible, pancreatic surveillance should be performed at centers with the appropriate expertise to manage individuals at increased risk for PC. In asymptomatic patients, prophylactic total pancreatectomy is not indicated. The aim of screening is to detect PC precursor lesions or early-stage cancer (such as intraductal papillary mucinous tumor, intraepithelial pancreatic neoplasm grade 3, mucinous cystic neoplasms and early cancers – T1N0M0-) with the ultimate goal of decrease morbid-mortality in these individuals.60 If a pancreatic lesion is observed, there is no consensus on the type of pancreatic resection that should be classified, whether partial or total pancreatectomy. The type of surgical intervention must be individualized, discussing with the patient the risks including the potential of overtreatment and unnecessary resections.
Biomarkers for Screening and Early Detection of PC in High-Risk Individuals
Based upon the aggressiveness of PC and its high mortality, there is no doubt of the imperative need for the development of novel and robust strategies for the detection of precursor lesions and early-stage PC, and the subset of individuals at high-risk for PC represents the group with the greatest benefit of these biomarkers to facilitate the screening of this neoplasia.
An ideal biomarker should be non-invasive, cost-effective and with a high sensitivity and specificity to detect early tumors or precursor lesions (PanIN and cystic lesions with high-grade dysplasia) in order to improve the overall survival.
There are several studies with promising biomarkers for the early detection of PC, mainly in blood, but also in urine,69 and stool DNA70 and more interestingly in pancreatic juice; as well as biomarkers in cystic fluid to distinguish between malignant or benign lesions. However, there are very limited data on the usefulness of biomarkers in the screening of asymptomatic individuals at high-risk for PC.
Blood-Based Markers
Carbohydrate antigen 19–9 (CA 19–9) and carcinoembryonic antigen, as well as other glycoproteins such as AACT, THBS1 and HPT,71 have been evaluated for the diagnostic of PC, but none have proven their usefulness in a screening scenario.
However, there are promising results of studies evaluating several potential markers for early detection of this tumor, and therefore attractive for a screening program, such as IGFBP2,72 TSP-1,73 panels of proteins74 and microRNAs.75 For example, Liu et al76 reported that combination of plasmatic miR-16, miR-196a and serum Ca 19–9 distinguish PC from non-PC with a sensitivity of 92.0% and specificity of 95.6% (including stage I tumors). Furthermore, recently an elegant study showed that Trefoil factors (TFF1, TFF2, and TFF3) gene expression in precursor lesions and early stages of PC, especially in combination with CA 19.9, discriminate significantly between early stage of PC from controls (area under curve = 0.93) and from chronic pancreatitis (area under curve = 0.93).77
Another interesting approach is the measurement of circulating exosomes. Exosomes are small membrane vesicles (30–100 nm) released from diverse cell types under both normal and pathological conditions. These secreted vesicles carry intracellular cargo, including mRNA, miRNA, lipids, metabolites, and/or proteins and can be used as diagnostic markers, i.e. one study78 observed high levels of glypican-1 in patients with pancreatic cancer in comparison with healthy controls.
Also, there is a special focus in circulating tumor DNA (ct-DNA) for early detection and, specifically in PC, mutated KRAS is the main ct-DNA marker, detected in up to 50% of patients with localized pancreatic cancer and 85% of patients with advanced disease,79 although there is still an important heterogeneity in the results and detection methods to generalized their use in a screening set.
Finally, to increase the diagnostic accuracy of circulating markers, especially in a surveillance scenario, recently some researchers are evaluating combination of biomarkers. For example, Bartsch et al80 developed a panel of 3 biomarkers (miR-196b/LCN2/TIMP1) that can discriminate between precursor lesions (PanINs and IPMNs with advanced dysplasia) and stage I PC from healthy controls (p=0.02) within a cohort of FPC individuals.
Cystic Fluid Markers
Benign pancreatic cystic lesions are frequently difficult to distinguish from the malignant cysts, and the main goal of the biomarkers in this setting is to obtain an accurate diagnosis. In this sense, identification of genetic mutations in cystic fluid is a helpful tool, i.e. GNAS mutation is associated to IPMN, KRAS mutation is present in up to 90% of mucinous cystic lesions, and VHL mutations are related to serous cystic lesions.81
Pancreatic Juice Markers
In the last years, there is an increased interest in pancreatic juice as a source of different kind of diagnostic biomarkers, such as proteins,82 genetic mutations (such as KRAS, TP53 and SMAD4 in PC samples)80,83 and methylated DNA markers.84
Specifically in the screening context of high-risk patients, analysis of pancreatic juice biomarkers added to regular endoscopic ultrasound could be an interesting and feasible screening approach, and there are some research groups that had evaluated some options. For example, in a study85 with 272 individuals under a PC screening program, KRAS mutations in duodenal juice samples were detected in 73% of PC patients, 50% of screening individuals with and without pancreatic abnormalities and 19% of controls (p<0.005). Other study86 using digital next-generation sequencing of a 12-gene panel in pancreatic juice samples from high-risk individuals under pancreatic surveillance, patients with PC or precursor lesions and controls observed a significantly higher proportion of mutations in samples from pancreatic cancer or high-grade dysplasia in comparison with all other subjects (46.6 ± 69.7 vs 6.2 ± 11.6, p=0.02) representing a 72.2% sensitivity and 89.4% specificity (area under the curve= 0.872) and especially mutations in TP53/SMAD4 could distinguish patients with pancreatic cancer or high-grade dysplasia from all other subjects with 61.1% sensitivity and 95.7% specificity (area under the curve= 0.819).
Thus, there have been great advances in the study of biomarkers for the diagnosis of pancreatic cancer, but there is still a long way to go to generalize their use in routine clinical practice. Further studies are needed to evaluate their potential diagnostic utility, especially in individuals undergoing pancreatic screening and surveillance.
Treatment Implication
Due to the aggressiveness and lack of response to chemotherapy, PC is one of the most lethal cancers worldwide and constant research is focused on the achievement of better therapeutic strategies. In this sense, during the last years, there is increasing evidence of the benefit of tailored treatment strategies based on the presence of specific germline mutations, as is already done in other tumors (such as ovary and breast). The main promising targetable genetic alterations in PC are related to DNA damage associated agents, especially related to homologous recombination. Among the germline genetic mutations involved in this pathway, there are BRCA1, BRCA2, PALB2, ATM, CHEK2, and some studies address the potential targeted therapy in these hereditary syndromes, suggesting the better response to platinum-based chemotherapy and to poly (adenosine diphosphate) ribose polymerase (PARP) inhibitors. Table 2
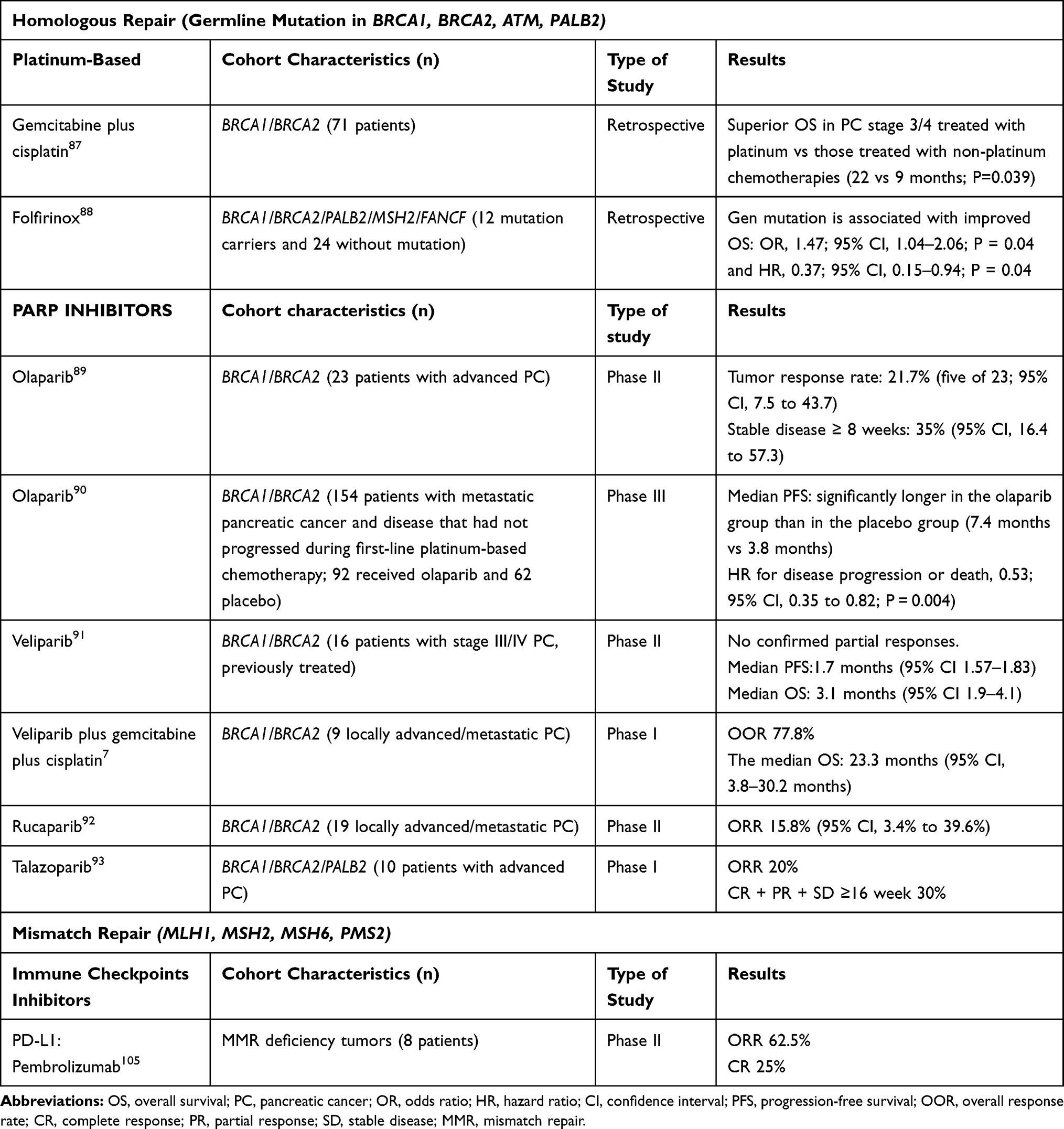 |
Table 2 Potential Personalized Therapy Based Upon Germline Mutations |
Platinum-based agents cause DNA double-strand breaks and homologous recombination deficiency leads to lack in DNA repair process, favoring the tumor response. Thus, retrospective studies have observed a better overall survival in germline BRCA1/BRCA2 mutation carriers. Interestingly, Golan87 reported in a cohort of 71 BRCA1/BRCA2 mutation carriers a superior overall survival in patients with locally advanced and metastatic PC treated with platinum in comparison with those treated with non-platinum chemotherapies (22 vs 9 months; p=0.039). Another small retrospective study88 including 12 patients with germline mutations (7 BRCA1, 5 BRCA2, 3 PALB2, 1 MSH2, 1 FANCF) treated with FOLFIRINOX (folinic acid, fluorouracil, irinotecan, and oxaliplatin) observed a trend is a superior overall survival in patients with germline mutations in comparison with patients without mutation, although this difference was not statistically significant (14 vs 5 months; Hazard ratio [HR], 0.58; 95% confidence interval, 0.29–1.14; log-rank p=0.08). However, multivariate logistic (Odds ratio [OR], 1.47; 95% CI, 1.04–2.06; p= 0.04) and Cox regression (HR, 0.37; 95% CI, 0.15–0.94; p= 0.04) associated germline mutations with better overall survival.
Regarding, PARP inhibitors, as platinum agents, cause DNA double-strand breaks and hinder DNA damage and repair. Recent data support the potential benefit of these drugs as second-line therapy in advanced PC and in stable disease in BRCA germline mutation carriers. Among these agents, olaparib is the best known and studied. One Phase II study89 included 23 BRCA1/BRCA2 mutation carriers with advanced PC treated with olaparib after progression during gemcitabine treatment (or felt to be unsuitable for gemcitabine). Tumor response rate was 21.7% (five of 23; 95% CI, 7.5 to 43.7) and stable disease during ≥8 weeks was observed in 35% (95% CI, 16.4 to 57.3). Recently, Golan et al90 published a very elegant randomized, double-blind, placebo-controlled, Phase III trial to evaluate the efficacy of olaparib as maintenance therapy in patients who had a germline BRCA1/BRCA2 mutation and metastatic pancreatic cancer and disease that had not progressed during first-line platinum-based chemotherapy. Within a total of 154 patients, 92 received olaparib and 62 placebo. The median progression-free survival was significantly longer in the olaparib group than in the placebo group (7.4 months vs 3.8 months; HR for disease progression or death, 0.53; 95% CI, 0.35 to 0.82; P = 0.004). Remarkably, no significant differences in health-related quality of life between groups were observed, and grade 3 or higher adverse effects were present in 40% of the olaparib group and 23% of the placebo group (between-group difference, 16 percentage points; 95% CI, −0.02 to 31).
Veliparib is another promising agent that has been studied in this setting as monotherapy91 and in combination with gemcitabine and cisplatin7 in locally advanced and metastatic PC. Specifically, the study that combined the 3 agents included 9 patients BRCA positive, with an objective response rate of 77.8% and a median overall survival of 23.3 months (95% CI, 3.8–30.2 months) in comparison with the median overall survival in BRCA negative patients was 11 months (95% CI, 1.5–12.1 months).
Other PARP inhibitors (rucaparib92 and talazoparib93) have been studied in small cohorts of germline BRCA carriers, revealing a modest effect (Table 2).
The results of the mentioned studies have given rise to further trials in order to confirm the effectiveness of these drugs in a homologous recombination setting. For example, a randomized Phase 2 trial is currently ongoing (NCT01585805) evaluating cisplatin and gemcitabine with and without veliparib for BRCA+ PC. Also, there are trials evaluating PARP inhibitors as a first-line therapy instead of second-third line treatment.
Finally, immunotherapy in cancer is gaining weight in several types of tumors, although the response in pancreatic cancer is limited, probably due to the tumor microenvironment, restricted T-cell infiltration, and tumor immunogenicity.94 There are several studies with immune checkpoint inhibitors95,96 [Cytotoxic T-Lymphocyte-Associated Antigen-4 (CTLA-4), PD-1, or PD-L1], engineered T-cells97 (Chimeric Antigen Receptors, CAR-T), agonistic immunotherapy (CD40),98 myeloid immunotherapy99,100 (CCR2, CSF-1R, CXCR2), as well as therapeutic vaccines101,102 (GVAX, hTERT), although the results are heterogeneous but some of them promising. Based upon disappointed results to a single-agent therapy, there is a continuous effort to modulate the immune response in PC with a multi-target strategy, such as immunomodulators in combination with vaccines to induce anti-tumor T cells.
Based on the presence of specific germline mutations, there are favorable results observed in mismatch repair (MMR) deficiency tumors (associated with Lynch syndrome) treated with immune checkpoint blockade103 and some studies evaluating the role of these therapies in combination with PARP inhibitors in patients with BRCA and PALB2 mutations, as well as describing specific immune profiles in correlation with specific mutational signatures in PC.104 Thus, in 2017 a study105 evaluated the response to pembrolizumab (anti-PD-L1 antibody) in 86 patients with MMR-deficiency tumors, including 8 PC, and an overall response rate of 62.5% and complete response of 25% in the PC subgroups. Although these results are encouraging, the proportion of MMR-deficiency tumors in the context of hereditary syndromes (Lynch syndrome) is very low, and most of them are secondary to somatic mutations.106
Finally, another attractive approach of immunotherapy, especially in individuals at high-risk for PC, is the use of vaccines to prevent the progression of premalignant or early-stage lesions,107,108 although the preclinical data is still limited.
Conclusions
The underlying genetic cause has not been identified in a high percentage of families with PC aggregation, although recent evidence suggests that germline mutations are more common than previously thought because many cases do not meet classical criteria for genetic testing. Due to the increasing use of next-generation sequencing and multigene panels associated with the less restrictive indications of genetic testing and therapeutic implications, the approach of genetic counseling is rapidly changing, and a multidisciplinary group within specialized centers is mandatory to achieve the best management strategies in terms of screening, surveillance, and treatment.
Although in the last years significant progress has been made in the identification and surveillance of individuals with an increased risk of pancreatic cancer, there is a long way to go in order to improve the global management of these individuals. For example, non-invasive biomarkers are promising as screening methods and diagnosis of preneoplasic lesions, such as epigenetic modifications, exosomal markers, and circulating cell-free DNA in blood and pancreatic juice. These biomarkers could be used as complementary with the imaging tools, in order to avoid under and overdiagnosis lesions in PC-high risk populations.
Nowadays diagnosis of a hereditary syndrome in context of PC has not only a genetic consoling purpose but also potential targeted therapies based upon specific germline mutation, opening a window into the era of precision medicine.
Further larger and longer prospective, randomized studies are needed to evaluate the appropriateness of germline genetic testing, screening programs (who, how, when, including noninvasive biomarker panels) and tailored therapies in the context of hereditary/familial pancreatic cancer.
Disclosure
The authors declare that they have no conflict of interests in this work.
References
1. Ohmoto A, Yachida S, Morizane C. Genomic features and clinical management of patients with hereditary pancreatic cancer syndromes and familial pancreatic cancer. Int J Mol Sci. 2019;20.
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
3. Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi:10.1158/0008-5472.CAN-14-0155
4. Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56:1460–1469. doi:10.1136/gut.2006.108456
5. Yurgelun MB, Chittenden AB, Morales-Oyarvide V, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med. 2019;21:213–223. doi:10.1038/s41436-018-0009-5
6. Golan T, Sella T, O’Reilly EM, et al. Overall survival and clinical characteristics of BRCA mutation carriers with stage I/II pancreatic cancer. Br J Cancer. 2017;116:697–702. doi:10.1038/bjc.2017.19
7. O’Reilly EM, Lee JW, Lowery MA, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer. 2018;124:1374–1382. doi:10.1002/cncr.31218
8. Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:
9. Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi:10.1158/1078-0432.CCR-06-0083
10. van Lier MG, Wagner A, Mathus-Vliegen EM, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:
11. Lim W, Olschwang S, Keller JJ, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology. 2004;126:1788–1794. doi:10.1053/j.gastro.2004.03.014
12. Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi:10.1056/NEJM199305203282001
13. Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261. doi:10.1016/S1542-3565(04)00013-8
14. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–446. doi:10.1093/jnci/89.6.442
15. Rebours V, Levy P, Ruszniewski P. An overview of hereditary pancreatitis. Dig Liver Dis. 2012;44:8–15. doi:10.1016/j.dld.2011.08.003
16. Shelton CA, Umapathy C, Stello K, et al. Hereditary pancreatitis in the United States: survival and rates of pancreatic cancer. Am J Gastroenterol. 2018;113:1376. doi:10.1038/s41395-018-0194-5
17. Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103:111–119. doi:10.1111/j.1572-0241.2007.01597.x
18. Hruban RH, Canto MI, Goggins M, et al. Update on familial pancreatic cancer. Adv Surg. 2010;44:293–311. doi:10.1016/j.yasu.2010.05.011
19. Lynch HT, Fusaro RM, Lynch JF, et al. Pancreatic cancer and the FAMMM syndrome. Fam Cancer. 2008;7:103–112. doi:10.1007/s10689-007-9166-4
20. de Snoo FA, Bishop DT, Bergman W, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res. 2008;14:7151–7157. doi:10.1158/1078-0432.CCR-08-0403
21. Bishop DT, Demenais F, Goldstein AM, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94:894–903. doi:10.1093/jnci/94.12.894
22. Soura E, Eliades PJ, Shannon K, et al. Hereditary melanoma: update on syndromes and management: genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74:
23. Potrony M, Puig-Butille JA, Aguilera P, et al. Increased prevalence of lung, breast, and pancreatic cancers in addition to melanoma risk in families bearing the cyclin-dependent kinase inhibitor 2A mutation: implications for genetic counseling. J Am Acad Dermatol. 2014;71:888–895. doi:10.1016/j.jaad.2014.06.036
24. Vasen HF, Gruis NA, Frants RR, et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87:809–811. doi:10.1002/1097-0215(20000915)87:6<809::AID-IJC8>3.0.CO;2-U
25. Eckerle Mize D, Bishop M, Resse E, et al. Familial atypical multiple mole melanoma syndrome. In: Riegert-Johnson DL, Boardman LA, Hefferon T, Roberts M, editors. Cancer Syndromes. Bethesda (MD): National Center for Biotechnology Information (US); 2009. Available from: http://www.ncbi.nlm.nih.gov/books/NBK7030.
26. Thompson D, Easton DF, Breast Cancer Linkage C. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–1365. doi:10.1093/jnci/94.18.1358
27. Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17:569–577. doi:10.1038/gim.2014.153
28. Roberts NJ, Norris AL, Petersen GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6:166–175. doi:10.1158/2159-8290.CD-15-0402
29. Mersch J, Jackson MA, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–275. doi:10.1002/cncr.29041
30. Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi:10.1126/science.1171202
31. Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–2229. doi:10.1158/0008-5472.CAN-10-3958
32. Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–1795. doi:10.1001/jama.2009.1529
33. Moller P, Seppala TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut. 2018;67:1306–1316. doi:10.1136/gutjnl-2017-314057
34. Giardiello FM, Offerhaus GJ, Lee DH, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993;34:1394–1396. doi:10.1136/gut.34.10.1394
35. Leoz ML, Sanchez A, Carballal S, et al. Hereditary gastric and pancreatic cancer predisposition syndromes. Gastroenterol Hepatol. 2016;39:481–493. doi:10.1016/j.gastrohep.2015.11.009
36. Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. doi:10.1158/2159-8290.CD-11-0194
37. Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319:2401–2409. doi:10.1001/jama.2018.6228
38. Bartsch DK, Krysewski K, Sina-Frey M, et al. Low frequency of CHEK2 mutations in familial pancreatic cancer. Fam Cancer. 2006;5:305–308. doi:10.1007/s10689-006-7850-4
39. van der Heijden MS, Yeo CJ, Hruban RH, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63:2585–2588.
40. Couch FJ, Johnson MR, Rabe K, et al. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005;65:383–386.
41. Pogue-Geile KL, Chen R, Bronner MP, et al. Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Med. 2006;3:e516. doi:10.1371/journal.pmed.0030516
42. Stoffel EM, McKernin SE, Khorana AA. Evaluating susceptibility to pancreatic cancer: ASCO clinical practice provisional clinical opinion summary. J Oncol Pract. 2019;15:108–111. doi:10.1200/JOP.18.00629
43. Steinberg WM, Barkin JS, Bradley EL
44. Matsubayashi H, Takaori K, Morizane C, et al. Familial pancreatic cancer and surveillance of high-risk individuals. Gut Liver. 2019;13:498–505. doi:10.5009/gnl18449
45. Schneider R, Slater EP, Sina M, et al. German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer. 2011;10:323–330. doi:10.1007/s10689-010-9414-x
46. Wang L, Brune KA, Visvanathan K, et al. Elevated cancer mortality in the relatives of patients with pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:2829–2834. doi:10.1158/1055-9965.EPI-09-0557
47. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–2638. doi:10.1158/0008-5472.CAN-03-3823
48. Rulyak SJ, Lowenfels AB, Maisonneuve P, et al. Risk factors for the development of pancreatic cancer in familial pancreatic cancer kindreds. Gastroenterology. 2003;124:1292–1299. doi:10.1016/S0016-5085(03)00272-5
49. Lynch HT, Rubinstein WS, Locker GY. Cancer in Jews: introduction and overview. Fam Cancer. 2004;3:177–192. doi:10.1007/s10689-004-9538-y
50. Brune KA, Lau B, Palmisano E, et al. Importance of age of onset in pancreatic cancer kindreds. J Natl Cancer Inst. 2010;102:119–126. doi:10.1093/jnci/djp466
51. McFaul CD, Greenhalf W, Earl J, et al. Anticipation in familial pancreatic cancer. Gut. 2006;55:252–258. doi:10.1136/gut.2005.065045
52. Klein AP, Beaty TH, Bailey-Wilson JE, et al. Evidence for a major gene influencing risk of pancreatic cancer. Genet Epidemiol. 2002;23:133–149. doi:10.1002/gepi.1102
53. Wang W, Chen S, Brune KA, et al. PancPRO: risk assessment for individuals with a family history of pancreatic cancer. J Clin Oncol. 2007;25:1417–1422. doi:10.1200/JCO.2006.09.2452
54. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35:3382–3390. doi:10.1200/JCO.2017.72.3502
55. Young EL, Thompson BA, Neklason DW, et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer. 2018;18:697. doi:10.1186/s12885-018-4573-5
56. Stoffel EM, McKernin SE, Brand R, et al. Evaluating susceptibility to pancreatic cancer: ASCO provisional clinical opinion. J Clin Oncol. 2019;37:153–164. doi:10.1200/JCO.18.01489
57. Canto MI, Almario JA, Schulick RD, et al. Risk of neoplastic progression in individuals at high risk for pancreatic cancer undergoing long-term surveillance. Gastroenterology. 2018;155:740–751 e2. doi:10.1053/j.gastro.2018.05.035
58. Vasen H, Ibrahim I, Ponce CG, et al. Benefit of surveillance for pancreatic cancer in high-risk individuals: outcome of long-term prospective follow-up studies from three European expert centers. J Clin Oncol. 2016;34:2010–2019. doi:10.1200/JCO.2015.64.0730
59. Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62:339–347. doi:10.1136/gutjnl-2012-303108
60. Canto MI, Hruban RH, Fishman EK, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142:
61. DaVee T, Coronel E, Papafragkakis C, et al. Pancreatic cancer screening in high-risk individuals with germline genetic mutations. Gastrointest Endosc. 2018;87:1443–1450. doi:10.1016/j.gie.2017.12.019
62. Vangala DB, Cauchin E, Balmana J, et al. Screening and surveillance in hereditary gastrointestinal cancers: recommendations from the European Society of Digestive Oncology (ESDO) expert discussion at the 20th European Society for Medical Oncology (ESMO)/World Congress on Gastrointestinal Cancer, Barcelona, June 2018. Eur J Cancer. 2018;104:91–103. doi:10.1016/j.ejca.2018.09.004
63. Canto MI, Goggins M, Yeo CJ, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol. 2004;2:606–621. doi:10.1016/S1542-3565(04)00244-7
64. Canto MI, Goggins M, Hruban RH, et al. Screening for early pancreatic neoplasia in high-risk individuals: a prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:
65. Signoretti M, Bruno MJ, Zerboni G, et al. Results of surveillance in individuals at high-risk of pancreatic cancer: a systematic review and meta-analysis. United European Gastroenterol J. 2018;6:489–499. doi:10.1177/2050640617752182
66. Harinck F, Konings IC, Kluijt I, et al. A multicentre comparative prospective blinded analysis of EUS and MRI for screening of pancreatic cancer in high-risk individuals. Gut. 2016;65:1505–1513. doi:10.1136/gutjnl-2014-308008
67. Stoita A, Penman ID, Williams DB. Review of screening for pancreatic cancer in high risk individuals. World J Gastroenterol. 2011;17:2365–2371. doi:10.3748/wjg.v17.i19.2365
68. Lu C, Xu CF, Wan XY, et al. Screening for pancreatic cancer in familial high-risk individuals: a systematic review. World J Gastroenterol. 2015;21:8678–8686. doi:10.3748/wjg.v21.i28.8678
69. Radon TP, Massat NJ, Jones R, et al. Identification of a three-biomarker panel in urine for early detection of pancreatic adenocarcinoma. Clin Cancer Res. 2015;21:3512–3521. doi:10.1158/1078-0432.CCR-14-2467
70. Kisiel JB, Yab TC, Taylor WR, et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer. 2012;118:2623–2631. doi:10.1002/cncr.26558
71. Nie S, Lo A, Wu J, et al. Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J Proteome Res. 2014;13:1873–1884. doi:10.1021/pr400967x
72. Yoneyama T, Ohtsuki S, Honda K, et al. Identification of IGFBP2 and IGFBP3 as compensatory biomarkers for CA19-9 in early-stage pancreatic cancer using a combination of antibody-based and LC-MS/MS-based proteomics. PLoS One. 2016;11:e0161009. doi:10.1371/journal.pone.0161009
73. Jenkinson C, Elliott VL, Evans A, et al. Decreased serum thrombospondin-1 levels in pancreatic cancer patients up to 24 months prior to clinical diagnosis: association with diabetes mellitus. Clin Cancer Res. 2016;22:1734–1743. doi:10.1158/1078-0432.CCR-15-0879
74. Gerdtsson AS, Wingren C, Persson H, et al. Plasma protein profiling in a stage defined pancreatic cancer cohort – implications for early diagnosis. Mol Oncol. 2016;10:1305–1316. doi:10.1016/j.molonc.2016.07.001
75. Hernandez YG, Lucas AL. MicroRNA in pancreatic ductal adenocarcinoma and its precursor lesions. World J Gastrointest Oncol. 2016;8:18–29. doi:10.4251/wjgo.v8.i1.18
76. Liu J, Gao J, Du Y, et al. Combination of plasma microRNAs with serum CA19-9 for early detection of pancreatic cancer. Int J Cancer. 2012;131:683–691. doi:10.1002/ijc.v131.3
77. Jahan R, Ganguly K, Smith LM, et al. Trefoil factor(s) and CA19.9: a promising panel for early detection of pancreatic cancer. EBioMedicine. 2019;42:375–385. doi:10.1016/j.ebiom.2019.03.056
78. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi:10.1083/jcb.201211138
79. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi:10.1126/scitranslmed.3007094
80. Bartsch DK, Gercke N, Strauch K, et al. The combination of MiRNA-196b, LCN2, and TIMP1 is a potential set of circulating biomarkers for screening individuals at risk for familial pancreatic cancer. J Clin Med. 2018;7.
81. Springer S, Wang Y, Dal Molin M, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology. 2015;149:1501–1510. doi:10.1053/j.gastro.2015.07.041
82. Chen R, Pan S, Yi EC, et al. Quantitative proteomic profiling of pancreatic cancer juice. Proteomics. 2006;6:3871–3879. doi:10.1002/(ISSN)1615-9861
83. Yu J, Sadakari Y, Shindo K, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut. 2017;66:1677–1687. doi:10.1136/gutjnl-2015-311166
84. Kisiel JB, Raimondo M, Taylor WR, et al. New DNA methylation markers for pancreatic cancer: discovery, tissue validation, and pilot testing in pancreatic juice. Clin Cancer Res. 2015;21:4473–4481. doi:10.1158/1078-0432.CCR-14-2469
85. Eshleman JR, Norris AL, Sadakari Y, et al. KRAS and guanine nucleotide-binding protein mutations in pancreatic juice collected from the duodenum of patients at high risk for neoplasia undergoing endoscopic ultrasound. Clin Gastroenterol Hepatol. 2015;13:963–9 e4. doi:10.1016/j.cgh.2014.11.028
86. Suenaga M, Yu J, Shindo K, et al. Pancreatic juice mutation concentrations can help predict the grade of dysplasia in patients undergoing pancreatic surveillance. Clin Cancer Res. 2018;24:2963–2974. doi:10.1158/1078-0432.CCR-17-2463
87. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111:1132–1138. doi:10.1038/bjc.2014.418
88. Sehdev A, Gbolahan O, Hancock BA, et al. Germline and somatic DNA damage repair gene mutations and overall survival in metastatic pancreatic adenocarcinoma patients treated with FOLFIRINOX. Clin Cancer Res. 2018;24:6204–6211. doi:10.1158/1078-0432.CCR-18-1472
89. Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi:10.1200/JCO.2014.56.2728
90. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi:10.1056/NEJMoa1903387
91. Lowery MA, Kelsen DP, Capanu M, et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2018;89:19–26. doi:10.1016/j.ejca.2017.11.004
92. Shroff RT, Hendifar A, McWilliams RR, et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol. 2018;2018.
93. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7:620–629. doi:10.1158/2159-8290.CD-16-1250
94. Johnson BA
95. Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi:10.1097/CJI.0b013e3181eec14c
96. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi:10.1056/NEJMoa1200694
97. Posey AD
98. Beatty GL, Torigian DA, Chiorean EG, et al. A Phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–6295. doi:10.1158/1078-0432.CCR-13-1320
99. Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi:10.1158/0008-5472.CAN-13-3723
100. Byrne KT, Leisenring NH, Bajor DL, et al. CSF-1R-dependent lethal hepatotoxicity when agonistic CD40 antibody is given before but not after chemotherapy. J Immunol. 2016;197:179–187. doi:10.4049/jimmunol.1600146
101. Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. doi:10.1158/2326-6066.CIR-14-0027
102. Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33:1325–1333. doi:10.1200/JCO.2014.57.4244
103. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi:10.1056/NEJMoa1500596
104. Connor AA, Denroche RE, Jang GH, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol. 2017;3:774–783. doi:10.1001/jamaoncol.2016.3916
105. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi:10.1126/science.aan6733
106. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology. 2017;152:68–74 e2. doi:10.1053/j.gastro.2016.09.060
107. Cappello P, Rolla S, Chiarle R, et al. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology. 2013;144:1098–1106. doi:10.1053/j.gastro.2013.01.020
108. Keenan BP, Saenger Y, Kafrouni MI, et al. A LISTERIA vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 2014;146:1784–94 e6. doi:10.1053/j.gastro.2014.02.055
109. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi:10.1053/gast.2000.20228
110. Raimondi S, Lowenfels AB, Morselli-Labate AM, et al. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi:10.1016/j.bpg.2010.02.007
111. Lynch HT. Familial cancer. Foreword. Fam Cancer. 2008;7:1–2. doi:10.1007/s10689-007-9168-2
112. Brose MS, Rebbeck TR, Calzone KA, et al. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94:1365–1372. doi:10.1093/jnci/94.18.1365
113. van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005;42:711–719. doi:10.1136/jmg.2004.028829
114. Breast Cancer Linkage C. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–1316. doi:10.1093/jnci/91.15.1310
115. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–398. doi:10.1111/ajg.2006.101.issue-2
116. Maisonneuve P, Marshall BC, Lowenfels AB. Risk of pancreatic cancer in patients with cystic fibrosis. Gut. 2007;56:1327–1328. doi:10.1136/gut.2007.125278
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.