")
Back to Journals » The Application of Clinical Genetics » Volume 17
Familial LCAT Deficiency and Low HDL-C Levels: In silico Characterization of Two Rare LCAT Missense Mutations
Authors Ciro Acosta S, Díaz-Ordóñez L , Gutierrez-Medina JD, Silva-Cuero YK, Arango-Vélez LG, García-Trujillo AO, Pachajoa H
Received 1 September 2023
Accepted for publication 16 December 2023
Published 20 February 2024 Volume 2024:17 Pages 23—32
DOI https://doi.org/10.2147/TACG.S438135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Sebastian Ciro Acosta,1 Lorena Díaz-Ordóñez,1,2 Juan David Gutierrez-Medina,1,3 Yisther Katherine Silva-Cuero,1,2 Luis Guillermo Arango-Vélez,4,5 Andrés Octavio García-Trujillo,4,5 Harry Pachajoa1,2,6
1Centro de Investigaciones en Anomalias Congenitas y Enfermedades Raras (CIACER), Universidad Icesi, Cali, Colombia; 2Departamento de Ciencias Basicas Medicas, Facultad de Salud, Universidad Icesi, Cali, Colombia; 3Centro de Investigaciones Clinicas, Fundacion Valle del Lili, Cali, Colombia; 4Servicio de Endocrinologia, Fundacion Valle del Lili, Cali, Colombia; 5Departamento de Medicina interna, Seccion de Endocrinologia, Universidad Icesi, Cali, Colombia; 6Genetic Division, Fundacion Valle del Lili, Cali, Colombia
Correspondence: Lorena Díaz-Ordóñez, Centro de Investigaciones en Anomalias Congenitas y Enfermedades Raras (CIACER), Universidad Icesi, Street 18 Number 122-135, Cali, Valle del Cauca, 760031, Colombia, Tel +57 602 5552334, Email [email protected]
Abstract: Mutations in the lecithin-cholesterol acyltransferase (LCAT) gene, which catalyzes the esterification of cholesterol, result in two types of autosomal recessive disorders: Familial LCAT deficiency (FLD) and Fish Eye Disease (FED). While both phenotypes are characterized by corneal opacities and different forms of dyslipidemia, such as low levels of high-density lipoprotein-cholesterol (HDL-C), FLD exhibits more severe clinical manifestations like splenomegaly, anemia, and renal failure. We describe the first clinically and genetically confirmed case of FLD in Colombia which corresponds to a 46-year-old woman with corneal opacity, hypothyroidism, and dyslipidemia, who does not have any manifestations of renal failure, with two pathogenic heterozygous missense variants in the LCAT gene: LCAT (NM_000229.2):c.803G>A (p.Arg268His) and LCAT (NM_000229.2):c.368G>C (p.Arg123Pro). In silico analysis of the mutations predicted the physicochemical properties of the mutated protein, causing instability and potentially decreased LCAT function. These compound mutations highlight the clinical heterogeneity of the phenotypes associated with LCAT gene mutations.
Keywords: eye, LCAT, cholesterol/trafficking, genomics, VLDL, lecithin cholesterol acyltransferase deficiency, LCAT deficiency, alpha-LCAT deficiency, fish eye disease
Introduction
Familial LCAT deficiency (FLD; MIM 245900) and Fish Eye Disease (FED; MIM 136120) are two autosomal recessive disorders caused by mutations in the lecithin-cholesterol acyltransferase (LCAT) gene which is located in the q22.1 region of chromosome 16 and is made up of 6 exons that code for a 440 amino acid residue glycoprotein. LCAT is expressed mainly in the liver, although it is also found in smaller amounts in the brain, testicles and plasma.1
LCAT catalyzes the esterification of unesterified cholesterol (UC) in plasma, the maturation of high-density lipoproteins (HDL) and is essential for the reverse cholesterol transport from peripheral tissues to the liver.2 The enzyme reversibly binds to lipoproteins and is responsible for transferring the acyl chain from the second position of lecithin to the hydroxyl group of UC housed within plasma lipoproteins, thus generating esterified cholesterol (EC) and lysolecithin.3 Since EC is significantly more hydrophobic than UC in plasma, the molecule moves into the core of lipoproteins allowing their maturation.1
The prevalence of mutations in the LCAT gene is estimated to be below 1/1,000,000 and of the population with extremely low HDL levels, approximately 2–9% is related to some level of LCAT deficiency.4,5 Decreased LCAT enzymatic activity is characterized clinically by bilateral corneal opacity due to the accumulation of cholesterol deposits in the corneal stroma and decreased levels of cholesterol housed in HDL in an esterified form known as high-density lipoprotein-cholesterol (HDL-C), which is the result of the decrease in the maturation of HDL and the low capacity of LCAT to esterify UC inside lipoproteins.6
LCAT enzymatic activity is classified into two groups: alpha LCAT and beta LCAT enzymatic activity. Alpha LCAT activity esterifies the UC in HDL, whereas beta LCAT activity catalyzes the reaction in low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL).2,4 Patients that retain only de beta LCAT enzymatic activity will present low levels of HDL-C and milder clinical manifestations because beta LCAT is still active on VLDL and LDL: the characteristic bilateral corneal opacity, low HDL-C levels, high LDL-C and TG levels, and normal to elevated plasma EC levels.6 However, if both groups of enzymatic activity are affected, cholesterol esterification will be almost null in lipoproteins and will lead to familial LCAT deficiency (FLD, MIM: 25900) which is a severe pathology with clinical manifestations such as low HDL-C levels, corneal opacities, elevated plasma TG levels, hemolytic anemia, splenomegaly, proteinuria and progressive renal failure lead by the accumulation of UC in tissues like the glomeruli and erythrocytes’ membrane.3
Here we describe the first clinically and genetically confirmed case of FLD in Colombia, which corresponds to a 46-year-old woman with corneal opacity, hypothyroidism, dyslipidemia, and episodes of anemia, with no splenomegaly and no manifestations of renal failure, with two compound heterozygous variants in the LCAT gene: LCAT (NM_000229.2):c.803G>A (p.Arg268His) and LCAT (NM_000229.2):c.368G>C (p.Arg123Pro). This research contributes to enriching the spectrum of variants of this rare disease, as well as highlighting the clinical heterogeneity of this phenotype.
Experimental Procedures
Sample Processing
Blood samples were collected in 4mL EDTA tubes and genomic DNA extraction was performed using the QIAamp DNA Mini Kit (QIAGEN, Germany) following the manufacturer’s protocol. The concentration and purity (260/280 and 260/230 ratios) of the nucleic acids were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Long PCR was performed using the Promega GoTaq® Long PCR Master Mix with the primers 5´-GGTTGCCCGTTGATTCTGTTG-3´ and 3´-ACTGAACTAACTCGGGTCCT-5´, generating a 6336 bp amplicon covering all the exons of the LCAT gene. The PCR conditions were as follows: an initial denaturation at 94°C for 2 minutes, followed by 40 cycles of denaturation at 93°C for 30 seconds, annealing at 60.5°C for 45 seconds, and extension at 72°C for 6 minutes, with a final extension step at 72°C for 5 minutes. To ensure proper amplification, PCR products were separated by gel electrophoresis with 1% agarose at 100V for 40 minutes, stained with ethidium bromide, and visualized using UV light. Since non-specific amplifications were obtained, it was necessary to perform amplicon purification by band excision using the E.Z.N.A.® Cycle Pure Kit (Omega bio-tek, USA).
Purified amplicons were used for single-stranded Sanger sequencing of all the exons of the LCAT gene using the BigDye Terminator v.3.1 (Applied Biosystems, Foster City, USA) following the manufacturer’s instructions. The primers used for each exon sequencing are described in Table 1. The Sanger sequencing products were purified with the BigDye® Xterminator™ Purification Kit (Applied Biosystems, Thermo Fisher Scientific, USA) and loaded in the 3500 Genetic Analyzer (Thermo Fisher Scientific, USA). Sequence data were analyzed using MEGA X software7 and the GenBank reference sequence of the LCAT gene (NG_009778.1).
 |
Table 1 Sequencing Primers |
Mutation Analysis
In order to predict the potential impact of a gene variant in protein structure or function, in silico bioinformatic tools that analyze the stability and functionality of the mutated protein were used: SIFT predictor (https://sift.bii.a-star.edu.sg/) which uses sequence homology to predict whether an amino acid substitution will affect protein function;8 PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) which uses annotated UniProt entries to predict whether the protein variation occurs within an important structural or functional site of the protein based mainly on a well annotated crystal structure o modeled protein;9 I-mutant 2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html) which from an experimental thermodynamic database predicts changes in protein stability based on changes in free energy10 and CADD (https://cadd.gs.washington.edu/) which analyzes multiple parameters built through other predictors and databases and integrates them into a single global score.11 Finally, to review the impact that the change of amino acids has from the physical-chemical point of view on the interactions and functionality of the protein, the HOPE platform (www.cmbi.umcn.nl/hope) was used, which based on the properties of the wild-type amino acid contrasted with the mutant, predicts the changes and alterations that could occur.12
Case Presentation
A 46-year-old Colombian woman, daughter of non-consanguineous parents, was referred to the genetic area due to a differential diagnosis between LCAT deficiency phenotypes and Tangier disease because of corneal opacity without visual acuity alteration, but with progressive deterioration of night vision, since the age of 10 years. The patient appeared phenotypically healthy, except for the corneal opacity (Figure 1). At the time of consultation, the patient did not present hepatomegaly or splenomegaly, proteinuria, anemia or other clinical characteristics. However, the patient had presented anemia episodes in 2018 and 2019. Electrolytes information revealed hypercalciuria and a significant hypercalcemia, 31.86 mg/dL and 10.30 mg/dL, respectively. The patient had no symptoms of coronary atherosclerosis. The results of the tests of liver and renal function were normal. Hemogram without evidence of leukocytosis, leukopenia, or any white cellular abnormality. No evidence of relevant alteration in hemoglobin levels. Platelets in adequate ranges without evidence of thrombocytosis or thrombocytopenia. Vitamin D, serum and urine creatinine, ferritin and phosphorus levels in serum are within the standard parameters.
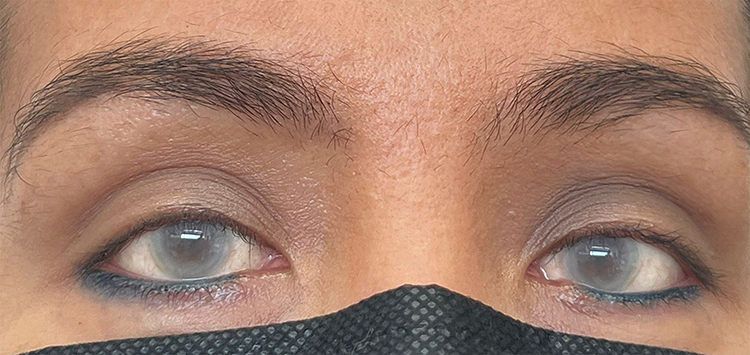 |
Figure 1 Patient´s corneal opacity. |
The patient was diagnosed with primary hypothyroidism at the age of 30 years (TSH 9.09mUI/mL). Throughout her life she presented with chronic dyslipidemia characterized by persistently low HDL-C levels stood out, even down to 2.8 mg/dL (Figure 2), high levels of triglycerides (216 mg/dL) and low levels of Apo-A1 (34.6 mg/dL). Total cholesterol of 95.0 mg/dL and non-HDL cholesterol of 92.2 mg/dL are within the standard population. VLDL levels have been above the reference value (30 mg/dL) and LDL-C levels have been continuously low, 43 mg/dL and 38.80mg/dL, respectively.
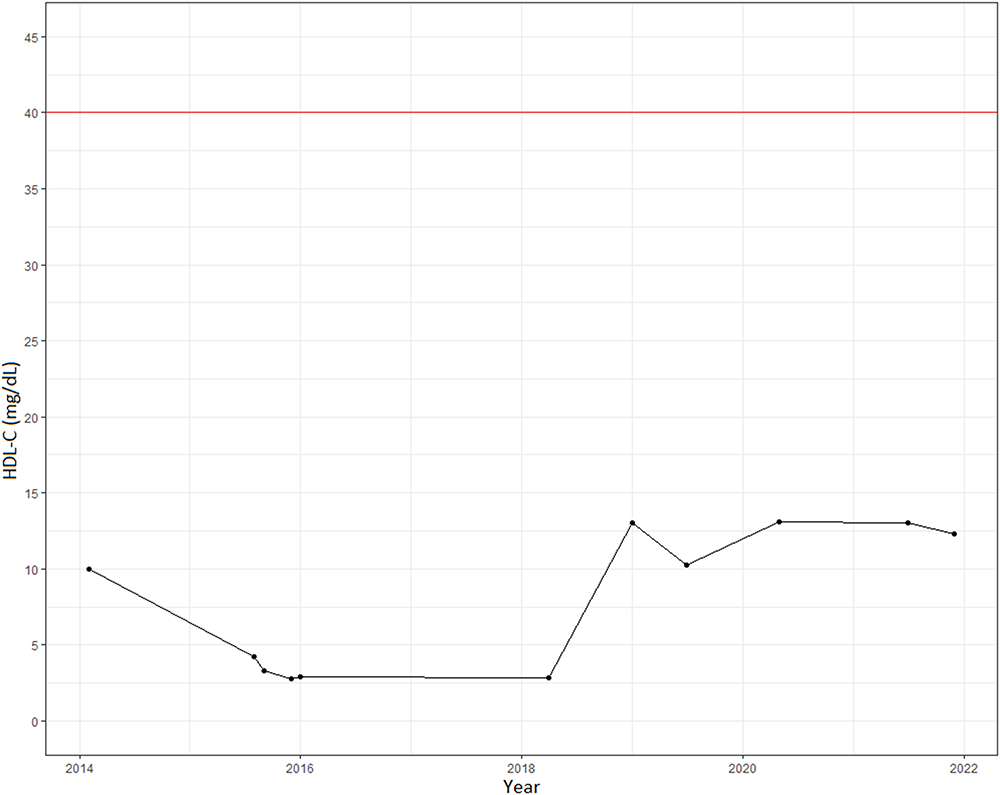 |
Figure 2 Temporal record of the patient’s HDL-C levels. Red line corresponds to the minimum value of the clinical reference. |
At the age of 39 years, lipoprotein electrophoresis revealed hypoalphalipoproteinemia (14.4% of total band signal) and a consequent increase of beta fraction (78.6% of total band signal), indicating that, even though the LDL-C values are low but among the reference levels (0–150 mg/dL), most of the circulating cholesterol was in beta migrating particles (LDL). The esterified cholesterol levels were very low: 6% of total cholesterol (reference value 60–80%). In addition, two of her three brothers have had recurrently low levels of HDL-C throughout their lives (down to 15–16 mg/dL).
The 6 exons in the LCAT gene of the proband were analyzed using the Sanger method to identify the mutations responsible for the pathology. Two missense variants were found: c.368G>C (p.Arg123Pro) classified as pathogenic (criteria: PM2, PM5, PP3, PP2, PP5 according to the American College of Medical Genetics and Genomics, ACMG)13 and c.803G>A (p.Arg268His) classified as pathogenic (criteria: PS3, PP3, PM2, PM5, PP5, according to the ACMG) located in exons 3 and 6 of the LCAT gene, respectively (Figure 3). Once the mutations were confirmed, carrier analysis was performed on the parents and three siblings, which determined that the proband inherited the variant Arg123Pro from her mother and the mutation Arg268His from her father. In addition to the patient, two of her male siblings are carriers of the variant located in exon 6 (Arg268His) (Figure 3).
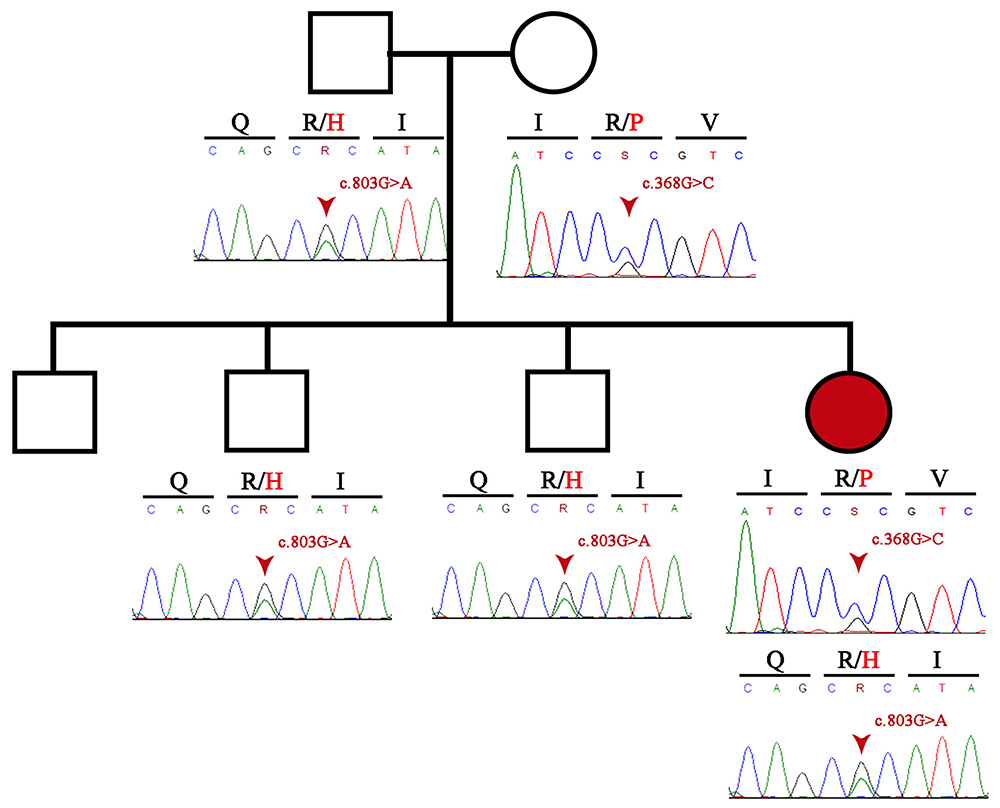 |
Figure 3 Pedigree information of the patient and Sanger sequencing electropherogram of both patient and her family. |
Analysis of c.368G>C (p.Arg123Pro)
This mutation was classified as pathogenic according to the criteria of the ACMG. This variant has been described in the literature in a homozygous 44-year-old Spanish woman,14 although there is no functional information on its impact on the function of the protein. This variant is classified in this category because of other known mutations in the same codon and its Genome Aggregation Database (gnomAD) allelic frequency of 0.00000398. Variant rs199717050 (Arg123His), the mutation in the same codon reported in our patient, was associated with decreased (β = –0.72) HDL-C levels (P discovery = 5.9×10−10, P conditional = 2.5×10−12) in the Finnish population.15
Using in silico prediction tools it was possible to determine that the region of the mutation corresponds to an LCAT conserved region (Figure 4) and SIFT predicts that the residue change has a deleterious effect on the function of the protein (0.018). This variant has a score of 1.0 (maximum score) on the PolyPhen-2 predictor and according to I-mutant 2.0 this change decreases, in silico, the stability of the new mutated protein (ΔG= −1.60). The mutation also obtained a high score in the CADD predictor (26.5). Finally, according to HOPE, the amino acid change corresponds to a smaller one with a neutral charge, possibly altering the physicochemical characteristics of the protein.
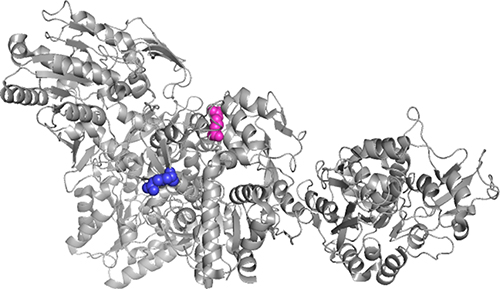 |
Figure 4 Visualization through PyMOL of the crystal structure of the human Lecithin-Cholesterol Acyltransferase (4X96 Protein Data Bank) reported by Glukhova et al 2015.16 Protein Data Bank accession number 4X96. Arginine residue 123 is visualized in blue and the purple residue corresponds to Arginine 268. |
Analysis of c.803G>A (p.Arg268His)
The effects of this variant are quite similar. The mutation was analyzed with the PolyPhen-2 and SIFT predictors that gave a score of 1.0 and 0.0 respectively, being the maximum values for predictions of deleterious effects of a mutation. I-mutant 2.0 classified it as a variant that decreases protein stability in silico (ΔG= −1.05) and it obtained a CADD score of 28.5. HOPE predicts physicochemical changes in the protein and loss of internal and external interactions, since the amino acid change differs in size and charge with the new one (Figure 4). The allelic frequency of this variant in gnomAD is 0.000036 and according to the ACMG classification the mutation corresponds to a pathogenic variant with known cases of this mutation related to LCAT deficiency.17–19
Discussion
This is the first clinically and genetically diagnosed case of FLD in Colombia and the first report of a compound heterozygous patient with the LCAT variants Arg123Pro and Arg268His. Mutations in codon 123 of the LCAT gene have been described previously. Blanco-vaca et al described a Spanish woman homozygous for the Arg123Cys variant who was diagnosed with FED based on the appearance of corneal opacities at the age of approximately 54 years and vanishingly low plasma concentrations of cholesteryl esters.20 However, our patient´s corneal opacity and progressive deterioration of night vision started since the age of 10 years, a rather soon manifestation when compared to the initiation of ocular symptoms in most of FED cases. In addition, Bérard et al described a compound heterozygous case with the same mutation as the Spanish patient.21 This compound heterozygous case, harboring one of our patient’s mutations, sheds some light on the possibility of our patient being a FLD case. The patient’s plasma LCAT concentrations and alpha LCAT enzymatic activity were significantly reduced, indicating a virtual absence of LCAT. Furthermore, Arg123 is a conserved residue in the human, rabbit, rodent, C. albicans and yeast LCAT genes, suggesting a role in LCAT function or stability.22 Although there is no information about the specific alpha or beta LCAT enzymatic activity of mutation Arg123Pro, LCAT enzymatic activity was 15.2 nmol/mL/hour (reference range, 81±12 mL/min/hour) in a homozygous patient.20
Splenomegaly, anemia, and renal failure are some differential conditions that most FLD patients share, in contrast to most FED cases. Although important contrasting symptoms, the appearance and progression of renal failure are variable among FLD cases, and it is likely related to the biochemical phenotype rather than to the inherited mutation.23 Clinical and biochemical heterogeneity is a challenging characteristic regarding the diagnosis of LCAT phenotypes. However, on the basis of the clinical and biochemical features of our case, the anemia episodes, and that both of our patient’s mutations and variants in the same codons have been described in FLD patients,14,19–22 we diagnosed our compound heterozygous patient as an FLD case who had not developed renal failure, proteinuria, or splenomegaly, pointing to the possibility that additional genetic or environmental factors may have contributed to the apparently benign course of the patient’s disease.
Likely due to the complexity of the LCAT biochemical reaction and despite the availability of a 3D model enzyme, it is impossible to predict the phenotype associated with the mutations.24 The dyslipidemia profile is indistinguishable between subjects classified as FLD or FED. The differential diagnosis between these two phenotypes is limited to alternatives that are not available in clinical laboratories in Colombia, such as the measurement of the ability of individual plasma to esterify cholesterol in endogenous lipoproteins (alpha LCAT with beta LCAT enzymatic activity) and in a standardized exogenous HDL (alpha LCAT activity only), both of which are null in FLD cases but low or normal in FED.3,24 Other useful alternatives to distinguish between these conditions are the CE/TC ratio in plasma, which is always reduced in FLD but not in FED; and through the expression of LCAT mutants in cultured cells, and subsequent measurement of LCAT concentration and activities in cell media.3 Nevertheless, we were unable to calculate this ratio as well as performing these experiments.
An association with increased risk of cardiovascular disease has been described in LCAT phenotypes by preserving the esterification of cholesterol in atherogenic lipoproteins such as LDL. Nevertheless, at the time of the patient’s approach, no calcified atherosclerotic plaques were documented by coronary computed tomography (calcium score 0 Agatston units). Cardiovascular disease in LCAT diseases has been described with a median age at presentation of 56 years, meaning that the patient is still at risk of developing such condition.3,25
Despite FLD being a recessive disorder, the two siblings of the proband are carriers of the Arg268His mutation who have HDL-C levels that are persistently below normal. Authors have described the behavior of lipoproteins in carriers of LCAT gene mutations who don´t have clinical manifestations as severe as a homozygous or compound heterozygous patient, but whose HDL-C levels are persistently below the values of non-carrier subjects, meaning they express an intermediate phenotype.4,26
Of the compound heterozygous mutations in the patient, Arg123Pro was found in a 44-year-old woman in Spain.14 The patient was homozygous for this LCAT gene variant and had corneal dystrophy, anemia and an altered lipid profile resembling our patient: HDL-C levels down to 6.6 mg/dL, triglycerides up to 173 mg/dL, LDL-C 131 mg/dL, VLDL 45 mg/dL and Apo-A1 62 mg/dL. However, unlike our patient, the Spanish LCAT patient had proteinuria and the histology study from the renal biopsy confirmed segmental hyaline lesions, irregular mesangial enlargement, and parietal thickening of the glomerular capillary walls. Despite having the same mutation, our patient didn´t have any sign of renal compromise or proteinuria.
According to the in silico predictors, this variant is classified as a destabilizing and deleterious mutation, and according to the ACMG criteria it is classified as pathogenic. The Arg123Pro mutation is predicted to generate changes in the stability and functionality of the protein because of the difference in size and charge from the original residue as it is smaller and has a neutral charge, as well as being more hydrophobic than arginine.
The mutated residue is found in the membrane binding region, a region enriched with hydrophobic residues responsible for anchoring HDL to membranes to initiate the cholesterol esterification process (Figure 4).27 Variations in the physicochemical properties of the new residue affect the formation of chemical bonds with other residues and could cause a loss of external interactions, which interferes in the function of the protein.12
The Arg268His variant was previously reported as a cause of LCAT enzyme activity deficiency, specifically of FLD in compound heterozygous patients.12 This mutation is found in the Cap domain of the protein, specifically a part of the lid region (residues 257–271), which opens and closes the access through a hydrophobic tunnel that leads to the catalytic site of the enzyme. This tunnel is made up of hydrophobic residues whose function is also to protect these residues from interaction with water.16,28 In addition, this residue forms salt bridges with Asp 359 and Glu 265, which keep the lid in a closed conformation; but when they are broken the lid changes to an open conformation.29 This variant corresponds to a substitution of a polar amino acid for a smaller one with a neutral charge, unlike arginine, which has a positive charge that allows it to form salt and hydrogen bonds with two leucines at positions 247 and 309, interactions that will be affected by this mutation.12
It is believed that the Cap domain interacts with Apo-AI (major HDL apolipoprotein) and is essential for the activation of the LCAT enzyme, the interaction involves a conformational change in the lid to an open state for a better binding with the substrate.30,31 In this case, the Arg268His mutation could interfere with the interaction of HDL with Apo-AI and the activation and the conformational change of the lid, causing poor cholesterol esterification specifically in HDL. In addition, Holleboom et al concluded that this residue is important for the expression and function of the enzyme based on the severe reduction of its expression in an in vivo study in the case of a mutation in the same codon (Arg268Cys).32
Our study has some limitations. We were unable to obtain specific data of the siblings’ corneal photographs, renal function, and specific blood data. In addition, limited to our research and clinical resources, we could not perform any of the assays that would clearly differentiate FLD from FED, such as the measurement of alpha and beta LCAT enzymatic activity. Also, we were not able to contact the patient for further ophthalmologic information such as intraocular pressure. Finally, we were not able to perform a molecular characterization of the variants, which would have required expression of the variants in cell systems and evaluation of their ability to promote alpha and beta LCAT enzymatic activity.
Conclusion
This is the first clinically and genetically confirmed report of FLD in Colombia. Moreover, this is the first case of a compound heterozygous patient with the LCAT (NM_000229.2):c.803G>A (p.Arg268His) and LCAT (NM_000229.2):c.368G>C (p.Arg123Pro) mutations. The in silico analysis of the mutations determined the affect of the physicochemical properties of the protein, mainly by altering the interaction of residues in their own domains or external interactions with other proteins, possibly causing instability and decreased function of the LCAT enzyme. This case highlights the clinical heterogeneity caused by LCAT mutations, demonstrating the possibility of other factors that may contribute to these phenotypes. The high risk of developing accelerated atherosclerotic disease as previously described may be an indicator for pharmacological (statin) and non-pharmacological interventions for its prevention in this patient. The description of these two variants will allow a better characterization of FLD and LCAT phenotype patients and support the identification of other individuals in heterozygosity and their appropriate clinical approach, as well as highlight the necessity to further evaluate the prevalence and clinical presentation of LCAT deficiency syndromes in Latin American countries.
Abbreviations
FED, Fish eye disease; LCAT, lecithin-cholesterol acyltransferase; UC, unesterified cholesterol; HDL, high-density lipoproteins; EC, esterified cholesterol; FLD, familial LCAT deficiency; LDL, low-density lipoproteins; VLDL, very low-density lipoproteins; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; TG, triglycerides; ACMG, American College of Medical Genetics; gnomAD, Genome Aggregation Database; RFLP, restriction fragment length polymorphism.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
All research was conducted according to the Declaration of Helsinki and the research protocol was registered with the number 1504 upon approval from the IRB Biomedical Research Ethics Committee of the Fundacion Valle del Lili. The patient and patient’s family provided their written informed consent to participate in this study and the publication of the case details and accompanying images.
Consent for Publication
The patient and patient’s family provided their written informed consent authorizing to perform genetic tests, use case details, pictures and publish the case along with the accompanying images.
Acknowledgments
The authors would like to thank the patient for agreeing to the publication of this report. We also thank the people who have contributed to the development and execution of this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Yang K, Wang J, Xiang H, Ding P, Wu T, Ji G. LCAT- targeted therapies: progress, failures and future. Biomed Pharmacother. 2022;147:112677. doi:10.1016/J.BIOPHA.2022.112677
2. Goñi Ros N, González-Tarancón R, Sienes Bailo P, Salvador-Ruperez E, Puzo Bayod M, Puzo Foncillas J. A novel pathogenic variant in LCAT causing FLD. A case report. Acta clinica Belgica. 2022;77(6):970–975. doi:10.1080/17843286.2021.2007598
3. Kuroda M, Bujo H, Yokote K, et al. Current status of familial LCAT deficiency in Japan. Journal of Atherosclerosis and Thrombosis. 2021;28(7):679–691. doi:10.5551/jat.RV17051
4. Mehta R, Elías-López D, Martagón AJ, et al. LCAT deficiency: a systematic review with the clinical and genetic description of Mexican kindred. Lipids Health Dis. 2021;20(1):70. doi:10.1186/s12944-021-01498-6
5. Savel J, Lafitte M, Pucheu Y, Pradeau V, Tabarin A, Couffinhal T. Very low levels of HDL cholesterol and atherosclerosis, a variable relationship--A review of LCAT deficiency. Vasc Health Risk Manag. 2012;8:357–361. doi:10.2147/VHRM.S29985
6. Kanai M, Koh S, Masuda D, Koseki M, Nishida K. Clinical features and visual function in a patient with fish-eye disease: quantitative measurements and optical coherence tomography. Am J Ophthalmol Case Rep. 2018;10:137–141. doi:10.1016/J.AJOC.2018.02.016
7. Kumar S, Stecher G, Li M, Knyaz C, Tamura K, Battistuzzi FU. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi:10.1093/molbev/msy096
8. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812. doi:10.1093/NAR/GKG509
9. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;76:7–20.
10. Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucl Acids Res. 2005;33:W306–W310. doi:10.1093/nar/gki375
11. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13(1). doi:10.1186/S13073-021-00835-9
12. Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf. 2010;11(1):548. doi:10.1186/1471-2105-11-548
13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
14. Morales E, Alonso M, Sarmiento B, Morales M. LCAT deficiency as a cause of proteinuria and corneal opacification. BMJ Case Rep. 2018;bcr2017224129. doi:10.1136/bcr-2017-224129
15. Davis JP, Huyghe JR, Locke AE, et al. Common, low-frequency, and rare genetic variants associated with lipoprotein subclasses and triglyceride measures in Finnish men from the METSIM study. PLoS genetics. 2017;13(10):e1007079. doi:10.1371/journal.pgen.1007079
16. Glukhova A, Hinkovska-Galcheva V, Kelly R, Abe A, Shayman JA, Tesmer JJG. Structure and function of lysosomal phospholipase A2 and lecithin: cholesterol acyltransferase. Nat Commun. 2015;6(1):1–12. doi:10.1038/ncomms7250
17. Calabresi L, Pisciotta L, Costantin A, et al. The molecular basis of lecithin: cholesterol acyltransferase deficiency syndromes. Arteriosclerosis Thrombosis Vasc Biol. 2005;25(9):1972–1978.
18. Strøm EH, Sund S, Reier-Nilsen M, Dørje C, Leren TP. Lecithin: Cholesterol Acyltransferase (LCAT) Deficiency: renal lesions with early graft recurrence. Ultrastruct Pathol. 2011;35(3):139–145. doi:10.3109/01913123.2010.551578
19. Pisciotta L, Calabresi L, Lupattelli G, et al. Combined monogenic hypercholesterolemia and hypoalphalipoproteinemia caused by mutations in LDL-R and LCAT genes. Atherosclerosis. 2005;182(1):153–159. doi:10.1016/j.atherosclerosis.2005.01.048
20. Blanco-Vaca F, Qu SJ, Fiol C, et al. Molecular basis of fish-eye disease in a patient from Spain. Characterization of a novel mutation in the LCAT gene and lipid analysis of the cornea. Arteriosclerosis Thrombosis Vasc Biol. 1997;17(7):1382–1391. doi:10.1161/01.atv.17.7.1382
21. Bérard AM, Clerc M, Brewer B
22. Peelman F, Verschelde JL, Vanloo B, et al. Effects of natural mutations in lecithin: cholesterol acyltransferase on the enzyme structure and activity. J Lipid Res. 1999;40(1):59–69. doi:10.1016/S0022-2275(20)33339-3
23. Lamiquiz-Moneo I, Civeira F, Gómez-Coronado D, et al. Lipid profile rather than the LCAT mutation explains renal disease in familial LCAT deficiency. J Clin Med. 2019;8(11):1860. doi:10.3390/jcm8111860
24. Pavanello C, Calabresi L. Genetic, biochemical, and clinical features of LCAT deficiency: update for 2020. Curr Opin Lipidol. 2020;31(4):232–237. doi:10.1097/MOL.0000000000000697
25. Schaefer EJ, Anthanont P, Diffenderfer MR, Polosecki E, Asztalos BF. Diagnosis and treatment of high-density lipoprotein deficiency. Prog Cardiovasc Diseases. 2016;59(2):97–106. doi:10.1016/j.pcad.2016.08.006
26. Vitali C, Bajaj A, Nguyen C. A systematic review of the natural history and biomarkers of primary lecithin: cholesterol acyltransferase deficiency. J Lipid Res. 2022;63(3):100169. doi:10.1016/j.jlr.2022.100169
27. Oldoni F, Baldassarre D, Castelnuovo S. Complete and partial lecithin: cholesterol acyltransferase deficiency is differentially associated with atherosclerosis. Circulation. 2018;138(10):1000–1007. doi:10.1161/CIRCULATIONAHA.118.034706
28. Casteleijn MG, Parkkila P, Viitala T, Koivuniemi A. Interaction of lecithin: cholesterol acyltransferase with lipid surfaces and apolipoprotein A-I-derived peptides. J Lipid Res. 2018;59(4):670–683. doi:10.1194/JLR.M082685
29. Charlton-Menys V, Pisciotta L, Durrington PN, et al. Molecular characterization of two patients with severe LCAT deficiency. Nephrol Dial Transplant. 2007;22(8):2379–2382. doi:10.1093/ndt/gfm311
30. Manthei KA, Yang SM, Baljinnyam B, et al. Molecular basis for activation of lecithin: cholesterol acyltransferase by a compound that increases HDL cholesterol. Elife. 2018;7. doi:10.7554/ELIFE.41604
31. Giorgi L, Niemelä A, Kumpula EP, et al. Mechanistic insights into the activation of lecithin−cholesterol acyltransferase in therapeutic nanodiscs composed of apolipoprotein A-I mimetic peptides and phospholipids. Mol Pharm. 2022;19:4135–4148. doi:10.1021/acs.molpharmaceut.2c00540
32. Holleboom AG, Kuivenhoven JA, Peelman F, et al. High prevalence of mutations in LCAT in patients with low HDL cholesterol levels in The Netherlands: identification and characterization of eight novel mutations. Human Mutation. 2011;32(11):1290–1298. doi:10.1002/humu.21578
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.