")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 1
Expression of MMP-15 and MMP-24 in atherosclerotic and nonatherosclerotic coronary arteries
Authors Horozoglu C, Özdeş T, Erginel T, Ünaltuna N
Received 3 June 2014
Accepted for publication 20 August 2014
Published 15 October 2014 Volume 2014:1 Pages 15—20
DOI https://doi.org/10.2147/MNM.S68778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor William Parks
Cem Horozoglu,1 Taşkin Özdeş,2 Turgay Erginel,3 Nihan Erginel Ünaltuna1
1Department of Genetics, Institute for Experimental Medicine Research, Istanbul University, 2Department of Forensic Medicine, Ministry of Justice, 3Department of General Surgery, Istanbul Training and Research Hospital, Istanbul, Turkey
Abstract: Atherosclerosis is an inflammatory process involving the intima layer of elastic arteries as well as the coronary and popliteal arteries. Myocardial ischemia due to atherosclerosis is the most common cause of mortality worldwide, and occurs because of acute disruption of blood flow in the coronary arteries by atherosclerotic lesions, resulting in myocardial necrosis. Matrix metalloproteinases (MMPs) are members of a proteinase family related to several vascular disorders, in particular atherosclerosis. Thirteen members of the MMP family are thought to play a role in the atherosclerotic process. This study investigated the patterns of expression of the MMP-24 and MMP-15 genes, members of the membrane-type MMP subfamily, in vessels with atherosclerotic lesions in comparison with normal coronary arteries (n=30; 15 atherosclerotic coronary arteries and 15 lesion-free coronary arteries) obtained at autopsy. Gene expression of MMP-24 and MMP-15 was shown in atherosclerotic lesions using reverse transcription polymerase chain reaction methods. The MMP-24 expression pattern was not significantly different between diseased and normal coronary arteries (P=0.2603; U=73). Although MMP-15 expression was slightly higher in coronary arteries with atherosclerotic lesions, no statistically significant difference was observed (P=0.2301; U=46).
Keywords: atherosclerosis, matrix metalloproteinase-24, matrix metalloproteinase-15, gene expression
Introduction
Atherosclerosis is a subacute inflammatory process arising from lipid deposition in the vascular wall, defined by the infiltration of macrophages and T-cells interacting with each other and the arterial wall cells.1 The adaptive and innate immune systems play a role in development of vulnerability within the plaque. Atherosclerosis is diagnosed by directly measuring the degree of stenosis or by evaluating the effect of arterial stenosis on organ perfusion, generally during periods of progression of the disease.2–7
The genetic basis of atherosclerosis has long been known and is supported by candidate gene, linkage, and genome-wide association studies.8 Matrix metalloproteinases (MMPs) comprise a multi-gene family involving zinc-dependent and calcium-dependent endopeptidases that break down components of the extracellular matrix (ECM) to remodel it. A number of physiological processes, including tissue morphogenesis and tissue development and repair, as well as some pathological conditions, such as cardiovascular disorders, include remodeling of the ECM as an important step. ECM serves as a backbone for cells associated with blood vessels and contains a dynamic tissue pattern composed primarily of proteins (collagen, elastin, laminin, fibronectin, and proteoglycans) showing active interaction with cellular elements.9,10 One can therefore understand the importance of ECM remodeling, a primarily MMP-driven process, as one of the most crucial steps in atherosclerosis and its resulting complications.
MMPs were first reported in 1962. This family of proteases has grown in number with the passage of time, with over 60 members having been described so far; more than 20 are found in humans and 14 have been detected in healthy or diseased vessels.11 Based on their structure and substrate specifications, MMPs are classified into at least seven subgroups. MMPs have separate albeit partly overlapping substrates, and are capable of degrading all major ECM elements.12 Most are produced and released as inactive proenzymes or zymogens (pro-MMPs). They have enormous power for protein degradation, and their biological activity is strictly regulated. This control mechanism can be at the transcriptional or translational level during activation of pro-MMPs or at the level of their interaction with specific inhibitors of these peptidases.13 It has been reported that atherosclerotic human arteries contain increased amounts of MMP-1, MMP-2, MMP-3, MMP-7, MMP-9, MMP-10, MMP-11, and MMP-12 when compared with normal healthy arteries.14 Further, MMP-1, MMP-8, and MMP-12 expression levels have been linked to plaque rupture, as evidenced by morphological analyses or clinical parameters.14
MMP-9,15 MMP-10 and MMP-11,16,17 and MMP-1418 have been reported to be highly expressed in plaque shoulders and sites characterized by deposition of foam cells. MMP-7, on the other hand, is expressed at sites peripheral to lipid cores, which are rich in lipid-laden macrophages. The latter are a distinct group from those residing in the fibrous regions of atherosclerotic plaque and expressing MMP-1, MMP-3, and MMP-9.19 Some MMPs, including MMP-1, MMP-2, MMP-3, MMP-9, and MMP-14 (MT1-MMP), have been isolated from platelets. Among these, MMP-2 is related to platelet aggregation upon secretion whereas MMP-9 may oppose the proaggregatory effect of MMP-2.20,21
Atherosclerosis is a chronic progressive process characterized by accumulation and proliferation of intimal smooth muscle cells, infiltration of macrophages and T lymphocytes, a connective tissue matrix rich in collagen, elastin, fibronectin, and proteoglycans, and lipid deposition in the form of free cholesterol and cholesterol esters inside the cells or surrounding connective tissue, and is closely related to endothelial dysfunction.22–24 Therefore, examination of the components of the basal lamina, including fibronectin, laminin, and elastin, is of great importance in atherosclerosis. Our literature search revealed few studies investigating the role of MMP-15 and MMP-24, members of MMP family with an important role in the regulation of extracellular connective tissue in atherosclerosis.
We investigated the gene expression of MMP-15 (MT2-MMP) and MMP-24 (MT5-MMP) and investigated whether their expression differs between healthy coronary arteries and coronary arteries with atheromatous plaques. MMP-15 adheres to fibronectin, entactin, laminin, aggrecan, and perlecan as substrates. MMP-2, the atherogenic effect of which has been shown in many studies, is also among its substrates. MMP-24, on the other hand, uses fibronectin as a substrate.12 This study aimed to demonstrate the gene expression of MMP-15 and MMP-24 and to investigate its potential impact on the pathogenesis of atherosclerosis.
Materials and methods
Ethical approval
This study was approved by the local ethics committees of The Council of Forensic Medicine and Istanbul Medical Faculty in Turkey. The study complied with the code of ethics of the World Medical Association (Declaration of Helsinki).
Subjects
Thirty postmortem cases (all male, total n=30; 15 atherosclerotic coronary arteries and 15 lesion-free coronary arteries) were included in our study according to the following inclusion criteria: age 18–65 years, time from death to dissection of the coronary arteries within the time window specified for RNA procedures (the interval between time of death and autopsy was kept to a minimum of approximately 3–4 hours to minimize RNA degradation), a past medical history accessible by family members or experts, coronary artery disease as the cause of death or no cardiac disease as the cause of death, as indicated by morphological and pathological examinations at the forensic medicine institute. Female cases were not included in the study because of differences between males and females with respect to genetic susceptibility to myocardial infarction. Both groups were evaluated in terms of hypoxia and serological, toxicological, and microbiological findings. Tissues from cases of noncardiac death were used as controls, and conditions that may contribute to atherosclerosis or myocardial infarction were excluded. Controls were also chosen based on not having had a cardiac history, metabolic syndrome, or dyslipidemia, and not showing ischemic disease at autopsy.
Given that the left anterior descending artery is the most commonly involved culprit artery in atherosclerosis, and individual coronary arteries may have different gene expression levels, we sampled only the left anterior descending artery. The dissected coronary vessels were evaluated in terms of atheroma load, and atheromatous and nonatheromatous tissue samples were harvested from suitable sites. The samples were immediately frozen in liquid nitrogen during autopsy, and were stored at −80°C until molecular analysis.
Molecular analysis
All analyses were performed using RNAase and DNAase materials. No chemicals that could alter RNA expression were used.
The coronary artery tissues were homogenized in TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) using a homogenizator, followed by extraction of RNA with TRIzol reagent. The quality and amount of RNA was assessed using an RNA 6000 Nano Kit® (Agilent Technologies, Santa Barbara, CA, USA) and a 2100 Bioanalyzer® (Agilent Technologies). At the end of analysis, cases with higher RNA integrity number levels were included. Next, synthesis of complementary DNA from RNA samples was carried out using a RevertAid First Strand cDNA Fermentas® synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA). Primers used for real-time (RT) polymerase chain reaction (PCR) were designed to be compatible from sequence exon boundaries for real-time PCR gene amplification in RT-PCR was detected using Fast-Plus EvaGreen® Master Mix (Bio-Rad, Hercules, CA, USA). A separate PCR reaction was set for each target and housekeeping gene, and the study was designed so as to have triplet repeats. As a result of both our pilot studies, we used glyceraldehyde 3-phosphate dehydrogenase as the housekeeping gene. The PCR protocol and primers are described in Table 1.
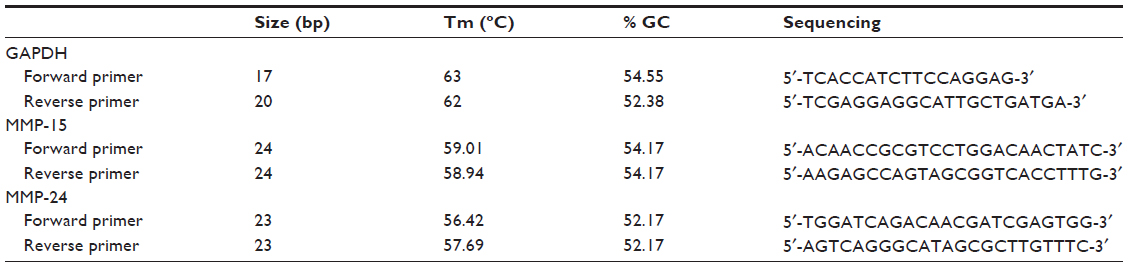 | Table 1 MMP-24, MMP-15, and GAPDH primers for RT-PCR (real-time polymerase chain reaction) |
Statistics
The study data were analyzed using Statistical Package for the Social Sciences and GraphPad analysis software packages, and relative expression of the genes was determined using the ΔΔCt method. Analysis of the means and intragroup distributions for the control and lesion groups revealed a non-normal distribution. Our sample size was small, so we used the Mann–Whitney U test, which is nonparametric.
Results
The gene expression levels of the target genes were determined using the ΔΔCt method. A calibrator was formed to normalize the resulting ΔΔCt level. Separate calibrators were used for the atherosclerotic coronary artery group and the nonatherosclerotic coronary artery group due to a heterogeneous intragroup distribution. After excluding those showing extreme gene expression, selection of the calibrator was based on consideration of data such as geometric and arithmetic means, and the lowest Ct level. The value of the calibrator sample was fixed to 1. The value selected as the calibrator was only used for normalization and excluded from the statistical analysis. Relative quantitation levels were obtained from the normalized Ct values, and were evaluated separately and comparatively in atherosclerotic and nonatherosclerotic coronary arteries.
Our analysis showed that MMP-24 and MMP-15 were expressed in coronary arteries with and without atherosclerotic plaque. To assess its possible contribution to the pathogenesis of atherosclerosis, we compared levels of MMP-24 in atherosclerotic and nonatherosclerotic coronary arteries and found that MMP-24 levels were similar in both groups (Figure 1) (P=0.2603, U=73). Gene expression of MMP-15, on the other hand, showed a slight elevation in atherosclerotic coronary arteries when compared with nonatherosclerotic coronary arteries, but this finding was not statistically significant (Figure 2) (P=0.2301, U=46).
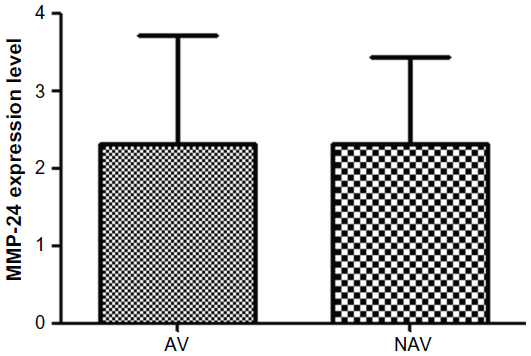 | Figure 1 MMP-24 expression in atherosclerotic vessels versus nonatherosclerotic vessels. |
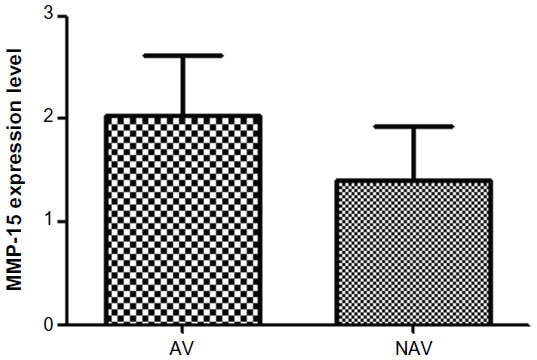 | Figure 2 MMP-15 expression in atherosclerotic vessels versus nonatherosclerotic vessels. |
Discussion
Secretion of MMP occurs in connective tissue and proinflammatory cells, including fibroblasts, osteoblasts, endothelial cells, macrophages, neutrophils, and lymphocytes.25,26 MMPs have an important role in the remodeling process, both physiologically and pathologically, including atherosclerotic plaque formation and rupture, development of arterial aneurysms, and critical limb ischemia.10,27 It has been reported that atherosclerotic human arteries contain increased amounts of MMP-1, MMP-2, MMP-3, MMP-7, MMP-9, MMP-10, MMP-11, and MMP-12 when compared with normal healthy arteries.14 Further, MMP-1, MMP-8, and MMP-12 expression levels have been linked to plaque rupture, as evidenced by morphological analyses and clinical parameters.14 MMP-9, MMP-10 and MMP-11, and MMP-14 have been reported to be highly expressed in plaque shoulders and sites characterized by deposition of foam cells.15–17 Reduction of fibrillar collagen by activated MMPs is believed to be the primary mechanism involved in fibrous cap thinning. Some MMPs (1, 2, 3, 8, 9, 11, 12, 13, 14, and 16) are found at sites rich in macrophages.28 Nevertheless, regions characterized by cleaved collagen contain MMP-1, MMP-3, and MMP-8, suggesting that these particular MMPs may be responsible for fibrous cap thinning.28
A number of genetic association studies have examined the role of specific MMP types, including MMP-1, MMP-3, MMP-8, and MMP-9, in the etiopathogenesis of atherosclerosis.29–32 These studies have provided some important data, such as the MMP-3 6A6A genotype being linked with atherosclerosis and the 5A allele being linked with plaque rupture. MMP-9 polymorphisms were found to be unrelated to atherosclerosis in coronary artery disease31 but were correlated with cardiovascular risk. Peeters et al demonstrated that increased levels of MMP-8 and MMP-9 in carotid plaques are indicators of unstable plaque morphology.32
We showed gene expression of MMP-24 and MMP-15 in atherosclerotic and nonatherosclerotic coronary arteries. Comparison of atherosclerotic and nonatherosclerotic coronary arteries revealed that gene expression of MMP-24 was similar in both groups (Figure 1) (P=0.2603, U=73), while gene expression of MMP-15 showed a slight but statistically insignificant increase in atherosclerotic coronary arteries (Figure 2) (P=0.2301, U=46).
In vitro and in vivo studies have shown that different types of MMP contribute to the pathogenesis of atherosclerosis in different ways. Our literature search regarding MMP-15 and MMP-24, the membrane-type MMPs investigated in our study, yielded insufficient data with respect to the pathogenesis of atherosclerosis. Thus, it is important to determine differences in gene expression between MMP-15 and MMP-24 in the coronary arteries in order to elucidate the effects of MMPs on the pathogenesis of atherosclerosis. Studies of MMP-14, an MMP similar to MMP-15 and MMP-24, have shown that MMP-14 plays a role in the pathogenesis of atherosclerosis. This information helped us to create our hypothesis.33 In addition, the knowledge that MMP-2 is one of the substrates of MMP-15 also supports our hypothesis.12
MMPs are downregulated by tissue specific inhibitors, collectively referred as TIMP (tissue inhibitor of metalloproteinases). This interaction is defined as the MMP/TIMP balance. Perturbation of this balance leads to transformation of physiological processes to pathological ones. It has been suggested that the limited information available on the biological properties and pathological role of MMPs and their inhibitors may have led to unsatisfactory results in clinical trials.34 In a study of cardiomyocyte dysfunction, the role of microRNA in MMP-9 gene expression was demonstrated.35 It has been reported that several internal and external factors, including estrogen levels,36 heroin use,37 and antidiabetic drugs such as atorvastatin,38 impair the MMP/TIMP balance. Although our cases had undergone biochemical, microbiological, and toxicological screening for major parameters before inclusion in the current study, it was not practically impossible to determine the above-mentioned external and internal factors, any of which may have caused the difference in gene expression levels between atherosclerotic and nonatherosclerotic vessels. The reason why it is not practically possible is that it is fairly more difficult to obtain clinical data in judicial cases than hospital cases. In judicial cases patient anamnesis could be obtained from the patient’s testators or successors. It is, however, practically impossible to be certain about the accuracy of patient data due to having died. Blood samples obtained from the judicial cases to determine the eligibility for study entry, have a lower quality and amount compared to those obtained from patients. Biochemical, microbiological, serological, and toxicological examinations could be performed to a limited extent in these blood samples unfavorable for analysis. It should not be forgotten that exact information can only be obtained from postmortem examination. However, both supply and laboratory processing of postmortem tissue are fairly difficult. These are limitations of our study.
The next steps in our research will include increasing the sample size, investigating the cell types involved in the gene expression shown in coronary arteries using in situ hybridization, and confirmation of the results with protein-based methods.
Disclosure
The authors report no conflicts of interest in this work.
References
Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. | |
Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50:S364–S369. | |
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Cytokine network and T cell immunity in atherosclerosis. Semin Immunopathol. 2009;31:23–33. | |
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. | |
Mangge H, Almer G, Truschnig-Wilders M, Schmidt A, Gasser R, Fuchs D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr Med Chem. 2010;17:4511–4520. | |
Salagianni M, Galani IE, Lundberg AM, et al. Toll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation. 2012;126:952–962. | |
Sanz J, Fayad ZA. Imaging of atherosclerotic cardiovascular disease. Nature. 2008;451:953–957. | |
Stylianou IM, Bauer RC, Reilly MP, Rader DJ. Genetic basis of atherosclerosis: insights from mice and humans. Circ Res. 2012;110:337–355. | |
Woessner JF. Introduction to serial reviews: the extracellular matrix. FASEB J. 1993;7:735–736. | |
Hobeika MJ, Thompson RW, Muhs BE, Brooks PC, Gagne PJ. Matrix metalloproteinases in peripheral vascular disease. J Vasc Surg. 2007;45:849–857. | |
Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. | |
Busti C, Falcinelli E, Momi S, Gresele P. Matrix metalloproteinases and peripheral arterial disease. Intern Emerg Med. 2010;5:13–25. | |
Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A. 1990;87:5578–5582. | |
Bäck M, Ketelhuth DF, Agewall S. Matrix metalloproteinases in atherothrombosis. Prog Cardiovasc Dis. 2010;52:410–428. | |
Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. | |
MonteroI I, Orbe J, Varo N, et al. C-reactive protein induces matrix metalloproteinase-1 and -10 in human endothelial cells: implications for clinical and subclinical atherosclerosis. J Am Coll Cardiol. 2006;47:1369–1378. | |
Schonbeck U, Mach F, Sukhova GK, et al. Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J Exp Med. 1999;189:843–853. | |
Johnson JL, Sala-Newby GB, Ismail Y, Aguilera CM, Newby AC. Low tissue inhibitor of metalloproteinases 3 and high matrix metalloproteinase 14 levels defines a subpopulation of highly invasive foam-cell macrophages. Arterioscler Thromb Vasc Biol. 2008;28:1647–1653. | |
Halpert I, Sires UI, Roby JD, et al. Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc Natl Acad Sci U S A. 1996;93:9748–9753. | |
Santos-Martínez MJ, Medina C, Jurasz P, Radomski MW. Role of metalloproteinases in platelet function. Thromb Res. 2008;121:535–542. | |
Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase A during platelet activation mediates aggregation. Nature. 1997;386:616–619. | |
Drouet L. Atherothrombosis as a systemic disease. Cerebrovasc Dis. 2008;13 Suppl 1:S1–S6. | |
Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis. American Heart Association. Circulation. 1994;89:2462–2478. | |
Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. | |
De B, Natchus MG, Cheng M, et al. The next generation of MMP inhibitors. Design and synthesis. Ann N Y Acad Sci. 1999;878:40–60. | |
Kontogiorgis CA, Papaioannou P, Hadjipavlou-Litina DJ. Matrix metalloproteinase inhibitors: a review on pharmacophore mapping and (Q)SARs results. Curr Med Chem. 2005;12:339–355. | |
Amalinei C, Caruntu ID, Giusca SE, Balan RA. Matrix metalloproteinases involvement in pathologic conditions. Rom J Morphol Embryol. 2010;51(2):215–228. | |
Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114. | |
Zhang B, Ye S, Herrmann SM, et al. Functional polymorphism in the regulatory region of gelatinase B gene in relation to severity of coronary atherosclerosis. Circulation. 1999;99:1788–1794. | |
Pearce E, Tregouet DA, Samnegård A, et al. Haplotype effect of the matrix metalloproteinase-1 gene on risk of myocardial infarction. Circ Res. 2005;97:1070–1076. | |
Abilleira S, Bevan S, Markus HS. A role of genetic variants of matrix metalloproteinases in coronary and carotid atherosclerosis. J Med Genet. 2006;43:897–901. | |
Peeters W, Moll FL, Vink A, et al. Collagenase matrix metalloproteinase-8 expressed in atherosclerotic carotid plaques is associated with systemic cardiovascular outcome. Eur Heart J. 2011;32:2314–2325. | |
Ray BK, Shakya A, Turk JR, Apte SS, Ray A. Induction of the MMP-14 gene in macrophages of the atherosclerotic plaque: role of SAF-1 in the induction process. Circ Res. 2005;95:1082–0890. | |
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–673. | |
Mishra PK, Metreveli N, Tyagi SC. MMP-9 gene ablation and TIMP-4 mitigate PAR-1-mediated cardiomyocyte dysfunction: a plausible role of dicer and miRNA. Cell Biochem Biophys. 2010;57:67–76. | |
Voloshenyuk TG, Gardner JD. Estrogen improves TIMP-MMP balance and collagen distribution in volume-overloaded hearts of ovariectomized females. Am J Physiol Regul Integr Comp Physiol. 2010;299:683–893. | |
Kovatsi L, Batzios S, Nikolaou K, et al. Alterations in serum MMP and TIMP concentrations following chronic heroin abuse. Toxicol Mech Methods. 2013;23:377–781. | |
McGinn S, Poronnik P, Gallery ED, Pollock CA. The effects of high glucose and atorvastatin on endothelial cell matrix production. Diabet Med. 2004;21:1102–1107. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.