")
Back to Journals » International Journal of Nanomedicine » Volume 11
Exposure to nickel oxide nanoparticles induces pulmonary inflammation through NLRP3 inflammasome activation in rats
Authors Cao Z, Fang Y, Lu Y, Qian F, Ma Q, He M, Pi H, Yu Z, Zhou Z
Received 22 February 2016
Accepted for publication 10 May 2016
Published 22 July 2016 Volume 2016:11 Pages 3331—3346
DOI https://doi.org/10.2147/IJN.S106912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Zhengwang Cao,1 Yiliang Fang,1 Yonghui Lu,1 Fenghua Qian,2 Qinglong Ma,1 Mingdi He,1 Huifeng Pi,1 Zhengping Yu,1 Zhou Zhou1
1Department of Occupational Health, 2Department of Haematology, Southwest Hospital, Third Military Medical University, Chongqing, People’s Republic of China
Abstract: With recent advances in the manufacture and application of nickel oxide nanoparticles (NiONPs), concerns about their adverse effects on the respiratory system are increasing. However, the underlying cellular and molecular mechanisms of NiONP-induced pulmonary toxicity remain unclear. In this study, we focused on the impacts of NiONPs on pulmonary inflammation and investigated whether the NLRP3 inflammasome is involved in NiONP-induced pulmonary inflammation and injury. NiONP suspensions were administered by single intratracheal instillation to rats, and inflammatory responses were evaluated at 3 days, 7 days, or 28 days after treatment. NiONP exposure resulted in sustained pulmonary inflammation accompanied by inflammatory cell infiltration, alveolar proteinosis, and cytokine secretion. Expression of Nlrp3 was markedly upregulated by the NiONPs, which was accompanied by overexpression of the active form of caspase-1 (p20) and interleukin (IL)-1β secretion in vivo. NiONP-induced IL-1β secretion was partially prevented by co-treatment with a caspase-1 inhibitor in macrophages. Moreover, siRNA-mediated Nlrp3 knockdown completely attenuated NiONP-induced cytokine release and caspase-1 activity in macrophages in vitro. In addition, NiONP-induced NLRP3 inflammasome activation requires particle uptake and reactive oxygen species production. Collectively, our findings suggest that the NLRP3 inflammasome participates in NiONP-induced pulmonary inflammation and offer new strategies to combat the pulmonary toxicity induced by NiONPs.
Keywords: NLRP3 inflammasome, nickel oxide nanoparticles, inflammation, cytokines
Introduction
Nanoparticles (NPs) are widely used in industrial applications, ceramics, cosmetics, and biological medicine due to their specific optical, mechanical, and electrical properties. With the development of nanotechnology, public concerns about the biological effects of NPs on the environment and human health have been raised.1 Several studies have demonstrated that, on an equal mass basis, NPs are more toxic than larger sized particles.2–4 Nickel oxide NPs (NiONPs), a representative type of metal oxide NP, are manufactured for various applications, such as ceramics, sensors, paint formulations, catalysts, and storage batteries.5,6 The increasing production and application of NiONPs has led to an increase in the environmental burden and a serious hazard to human health. However, the effects of NiONP exposure on human health remain largely unknown. Therefore, it is vital to assess their harmful effects.
Occupational exposure to NiONPs occurs in refining, electroplating, alloy production, and welding.7 Inhalation is the major route of NiONP exposure.8 Inhaled NiONPs are encountered by alveolar macrophages (AMs) and epithelial cells in the lungs. AMs phagocytize and clear particles through the mucociliary escalator and/or lymphatic systems. Inhalation of NiONPs has been associated with pulmonary inflammation, granulomas, and interstitial fibrosis.3,9 Accumulating evidence indicates that NiONP exposure can lead to neutrophil, eosinophil, and macrophage accumulation, followed by activation of cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6.10 Inflammatory cells and cytokines play vital roles during the early inflammatory response and in lung injury induced by NiONPs. Consistent with these results, Capasso et al11 have shown that NiONPs are able to elicit the pro-inflammatory response through activation of nuclear factor (NF)-κB in lung epithelial cells. Although the pro-inflammatory response to NiONPs has been studied, the underlying molecular mechanism remains unknown.
Recent studies have revealed the key roles of inflammasomes in inflammation, innate immunity, and tissue remodeling.12 Among the several types of inflammasomes, the NLRP3 (nucleotide oligomerization domain-like receptor family, pyrin domain containing 3) inflammasome is involved in sensing endogenous danger signals and orchestrating inflammatory responses and cell stress, and it is also associated with various inflammatory diseases, such as acute lung injury, silicosis, diabetes, and obesity.13,14 Recently, numerous studies have indicated that the NLRP3 inflammasome is triggered by environmental fibers and NPs, including silica, carbon nanotubes (CNTs), titanium oxide NPs, cerium oxide NPs, and silver NPs.15,16 In addition, recent studies have shown that NPs with various physicochemical properties activate the NLRP3 inflammasome, suggesting the important role of this inflammasome in mediating the host defense system against NP exposure. Activation of the NLRP3 inflammasome in macrophages, in addition to IL-1β signaling, has been shown to be necessary for CNT-induced inflammation.17,18 Moreover, Nlrp3-deficient mice have been shown to exhibit reduced pulmonary inflammation and inflammatory cytokine production following exposure to asbestos compared with wild-type mice.19 Ni2+ contamination of multi-walled CNTs is related to particle bioactivity and is correlated with NLRP3 inflammasome activation.20 However, whether NLRP3 inflammasome activation participates in NiONP-induced IL-1β release and pulmonary inflammation is unclear. Furthermore, the mechanisms of the NP-mediated activation of the NLRP3 inflammasome remain elusive.
In the present study, we aimed to investigate whether NLRP3 inflammasome activation is involved in NiONP-induced pulmonary inflammation. We evaluated pulmonary inflammation at different time points after NiONP exposure in rats and assessed cytotoxicity and NLRP3 inflammasome activation following exposure to different concentrations of NiONPs in RAW264.7 cells. We found that NiONP exposure resulted in sustained pulmonary inflammation and NLRP3 inflammasome activation in vivo and in vitro. Our results provide insights into NiONP-mediated pulmonary toxicity and offer new strategies to combat pulmonary toxicity induced by NiONPs.
Materials and methods
NiONP suspensions
Nickel oxide nanopowder (particle size: <50 nm, 99.8% purity) was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). NiONPs were sterilized by heating at 200°C for 2 hours before suspension. The sterilized NiONPs were diluted in Dulbecco’s Modified Eagle’s Medium (DMEM) or saline and sonicated without any dispersant using a probe sonicator (130 W; Sonics & Materials Inc., Newton, MA, USA) on ice for 1 hour. The suspension concentration was adjusted to 2.0 mg/mL, and the suspension was stored at 4°C.
Dispersion and characterization
The methods used have been described in our earlier study.21 Briefly, the primary particle sizes of the NPs were measured by high-resolution transmission electron microscopy (Carl Zeiss Meditec AG, Jena, Germany) using an accelerating voltage of 200 kV. The hydrodynamic diameter and zeta potential were measured in three different media. The stock suspensions of NiONPs were resonicated for 10 minutes and diluted with DMEM to 19 μg/mL (equal to 4 μg/cm2 in six-well plates). The hydrodynamic diameter and polydispersity index were analyzed using a Zeta PALS + BI-90 Plus (Brookhaven Instruments Corp., New York, NY, USA), and zeta potential was measured using a Zetasizer Nano-ZS 900 (Malvern Instruments, Malvern, UK).
In vitro experiments
Cell culture and treatments
A murine macrophage cell line, RAW264.7, was purchased from the cell bank of the Institute of Biochemistry and Cell Biology (Shanghai, People’s Republic of China). The cells were grown in DMEM (SH30022.01B; HyClone Laboratories, Inc., Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone) and 1% v/v penicillin/streptomycin (Sigma-Aldrich Co.). The cells were cultured in a 5% CO2 humidified incubator at 37°C and seeded at a density of 1.0×106 cells/well in six-well plates. When the cells reached 80% confluence, they were washed with pre-warmed phosphate-buffered serum (PBS), the medium was changed to serum-free Opti-MEM (Thermo Fisher Scientific, Waltham, MA, USA), and they were primed with 200 ng/mL ultrapure lipopolysaccharides (LPS; Sigma-Aldrich Co.) for 3 hours. Next, the cells were treated with NiONP suspensions at different concentrations (0 μg/cm2, 1 μg/cm2, 2 μg/cm2, and 4 μg/cm2) for 12 hours. The stock suspensions were resonicated for 10 minutes and diluted to appropriate concentrations before the cell treatments. The cells were preincubated with a caspase-1 inhibitor (z-yvad-fmk, 20 μM; BioVision, San Francisco, CA, USA), phagocytosis inhibitor (cytochalasin D, 5 μM; Sigma-Aldrich Co.), and reactive oxygen species (ROS) scavenger (N-acetylcysteine [NAC], 5 mM; Sigma-Aldrich Co.) for 1 hour before NiONP stimulation.
Detection of intracellular ionic Ni
The detection of intracellular mobilized ionic Ni was performed as described in our earlier study.21 Briefly, the treated cells were washed with 1 mM ethylenediaminetetraacetic acid/Hank’s balanced salt solution (HBSS) to chelate the extracellular ionic Ni, and they were then incubated with 7 μM Newport Green 2,7-dichlorofluorescein diacetate (DCF-DA; Thermo Fisher Scientific)/Pluronic F127 (Thermo Fisher Scientific) in 5% FBS/HBSS at 37°C for 30 minutes. After recovery in 5% FBS/HBSS for 30 minutes, Newport Green fluorescence was visualized under a Leica confocal laser scanning microscope (Leica TCS SP2, Wetzlar, Germany).
Cytotoxicity assay
Cell viability was assessed using a Cell Counting Kit-8 (Dojindo Molecular Technologies, Kunamoto, Japan) according to the manufacturer’s instructions. After the treatments, 90 μL fresh medium and 10 μL Cell Counting Kit-8 reagent were added to each well and incubated at 37°C for 2 hours. After centrifugation, 80 μL cell medium was transferred to a new 96-well plate and absorbance was measured at 450 nm using an Infinite™ M200 Microplate Reader (Tecan, Mannedorf, Switzerland) to minimize the interference caused by the NiONPs. The results are expressed as percentages of the control value.
NLRP3 siRNA transfection
RAW264.7 cells were cultured to 50%–60% confluence in DMEM without penicillin/streptomycin. The cells were then transfected with 100 nmol/L Nlrp3 siRNA (sc-45470; Santa Cruz Biotechnology Inc., Dallas, TX, USA) or control nonspecific siRNA (sc-37007; Santa Cruz Biotechnology Inc.) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After transfection for 24 hours, the cells were exposed to 2 μg/cm2 for 12 hours.
Intracellular ROS detection
The intracellular ROS were detected by measuring the oxidation of DCF-DA (Beyotime Company, Haimen, People’s Republic of China). After treatment, the cells were incubated with DCF-DA at 37°C for 30 minutes. Subsequently, the fluorescence was measured at 488 nm for excitation and 525 nm for emission using an Infinite™ M200 Microplate reader.
In vivo experiments
Animals
Adult male Sprague Dawley (SD) rats (8 weeks old) were obtained from the Experimental Animal Centre of the Third Military Medical University (Chongqing, People’s Republic of China). The rats were housed under standard conditions (temperature, 22°C±2°C; relative humidity, 60%–65%) and a 12-hour light/dark cycle (lights on at 7 am), and they were allowed ad libitum access to rodent chow and water during the experiment. All procedures and animal handling methods were approved by the Local Animal Use Committee and were performed according to the Guidelines on the Handling and Training of Laboratory Animals of the Third Military Medical University.
Intratracheal instillation
The NiONP suspension was sonicated for 30 minutes on ice to ensure for particle dispersion and mixed with 0.4 mL saline, and 800 μg (3.3 mg/kg) nickel oxide was intratracheally instilled once to the male SD rats. The NiONP dose was selected according to an earlier study and was equivalent to ~0.2 μg/cm2 epithelium, considering a total alveolar surface area of the rat lung of 4,000 cm2.9,22 The negative control groups received 0.4 mL saline.
Bronchoalveolar lavage fluid and cell counts
The animals were sacrificed under anesthetization by intraperitoneal injection of pentobarbital at 3 days, 7 days, and 28 days after receiving the NiONP treatment. Bronchoalveolar lavage fluid (BALF) was collected by introducing ice-cold physiological saline (15 mL for each rat) through a cannula into the right lung. After centrifuging the BALF (600× g, 5 minutes), the cells were re-suspended in PBS, and the supernatant was frozen at −80°C until further analysis.
The total cell counts in the BALF were determined using a TC10 Automated Cell Counter (Bio-Rad Laboratories Inc., Hercules, CA, USA). For differential cell counts, the samples were prepared by cytospin (Shandon Southern Products Ltd., Cheshire, UK) at 50× g for 5 minutes and stained with Diff-Quik (Nanjing Jiancheng, Nanjing, People’s Republic of China). A total of 300 cells were counted to determine the numbers of macrophages, eosinophils, and neutrophils according to their morphological characteristics.
BALF analysis
The amounts of alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) released in the BALF were determined by Alkaline Phosphatase Assay Kit (Nanjing Jiancheng) and LDH Cytotoxicity Assay Kit (Hoffman-La Roche Ltd., Basel, Switzerland) according to the manufacturers’ instructions.
AM isolation and culture
AMs were isolated by differential adherence. After lavage, the obtained BALF cells were collected by centrifugation at 600× g for 5 minutes at 4°C and washed once with PBS. The washed cells were suspended at a concentration of 3–5×105 cells/mL in RPMI-1640 medium supplemented with 10% FBS and 1% v/v penicillin/streptomycin. After incubation for 2 hours, nonadherent cells were washed off with PBS, and then attached cells were stained with Wright’s staining, revealing that >95% were AMs. AMs were prepared for RNA extraction.
Myeloperoxidase activity
The lung tissue was homogenized on ice in five volumes of PBS and centrifuged at 15,000× g at 4°C for 10 minutes. Myeloperoxidase (MPO) activity in the lung tissues was measured using a MPO Assay Kit (Nanjing Jiancheng) following the manufacturer’s instructions. The MPO activity results are expressed as unit per gram of lung tissue weight (U/g).
Caspase-1 activity assay
Caspase-1 activity was measured using a commercially available kit (Applygen Technologies Inc., Beijing, People’s Republic of China) following the manufacturer’s instructions.
Cytokine assays
The release of cytokines into the cell culture supernatants and BALF were detected by enzyme-linked immunosorbent assay (ELISA). The IL-1β, TNF-α, and IL-18 levels were detected using a mouse IL-1β ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA), rat IL-1β ELISA Kit (Boster, Wuhan, People’s Republic of China), rat IL-18 ELISA Kit (Boster), and mouse TNF-α ELISA Kit (BioLegend, San Diego, CA, USA), following the manufacturer’s instructions.
RNA extraction and quantification
Total RNA was extracted from the cultured cells and lung tissues after NiONP exposure using Trizol reagent (TaKaRa, Shiga, Japan), according to the manufacturer’s instructions. cDNA was then obtained using a Reverse Transcription Kit (TaKaRa). Real-time polymerase chain reaction (PCR) was performed using a CFX96 Real-Time System (Bio-Rad Laboratories Inc.) with SYBR Green PCR Master Mix (TaKaRa). The specific primers used are as follows: mouse Nlrp3: forward, 5′-GACACGAGTCCTGGTGACTTT-3′ and reverse, 5′-GGGCTTAGGTCCACACAGAA-3′; mouse IL-1β: forward, 5′-TGGTGTGTGACGTTCCCATTA-3′ and reverse, 5′-CAGCACGAGGCTTTTTTGTTG-3′; mouse TNF-α: forward, 5′-GGCAGGTCTACTTTGGAGTCATTG-3′ and reverse, 5′-ACATTCGAGGCTCCAGTGAATTCGG-3′; mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH): forward, 5′-TGGCCTCCAAGGAGTAAGAAAC-3′ and reverse, 5′-AGTTGGGATAGGGCCTCTCTTG-3′; rat IL-1β: forward, 5′-GCACAGTTCCCCAACTGGTA-3′ and reverse, 5′-TGTCCCGACCATTGCTGTTT-3′; rat IL-18: forward, 5′-ACCGCAGTAATACGGAGCAT-3′ and reverse, 5′-TCTGGGATTCGTTGGCTGTT-3′; and rat GAPDH: forward, 5′-TGCCACTCAGAAGACTGTGGATG-3′ and reverse, 5′-GCCTGCTTCACCACCTTCTTGAT-3′. Relative mRNA expression was calculated using the 2−ΔΔCT method and normalized to endogenous GAPDH.
Western blot analysis
RAW264.7 cells and lung tissues were homogenized in lysis buffer (Beyotime Company) containing a cocktail of protease inhibitors (Hoffman-La Roche Ltd.) and centrifuged at 15,000× g for 25 minutes. Supernatants were collected, and protein concentrations were quantified using a bicinchoninic acid (BCA) Protein Assay Kit (Beyotime Company). Cell culture supernatants were concentrated with Amicon Ultra Centrifugal Filters (EMD Millipore, Billerica, MA, USA). Proteins (100 μg/sample) were mixed with sample buffer and boiled for 5 minutes at 97°C, separated by 8%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto a polyvinylidene fluoride membrane (EMD Millipore), and blocked in 5% nonfat powdered milk in Tris-buffered saline with Tween 20 (TBST) for 1 hour. The membranes were then incubated with Nlrp3 (1:200; Santa Cruz Biotechnology Inc.), IL-1β (1:1,000; Abcam, Cambridge, UK), caspase-1 (1:200; BioVision), MPO (1:100; Santa Cruz Biotechnology Inc.), or β-actin (1:1,000; Sigma-Aldrich Co.) overnight at 4°C. After the membranes were subjected to three 5-minute washes in Tris-buffered saline containing 0.1% Tween-20, they were incubated with the appropriate secondary antibodies conjugated with horseradish peroxidase (1:2,000; Abcam) for 1 hour at room temperature. Protein bands were visualized and analyzed using a Western blotting detection system according to the manufacturer's instructions (Bio-Rad Laboratories Inc.).
Histopathology and immunohistochemistry
The left lungs were fixed by in situ perfusion of the airway with 4% paraformaldehyde (Sigma-Aldrich Co.) at 25 cm H2O pressure. The fixed lung tissues were embedded in paraffin, cut into 5 μm sections, and stained with hematoxylin and eosin for histological analysis. For immunohistochemistry, the sections were deparaffinized in xylene, rehydrated in graded ethanol, and then immersed into citrate buffer (pH 6.0) for 10 minutes at 96°C for antigen retrieval. Endogenous peroxidase was eliminated by incubation with 3% H2O2 for 15 minutes, and then the slices were blocked with 1% bovine serum albumin for 30 minutes. Next, the slices were incubated with Nlrp3 (1:100 dilution; Santa Cruz Biotechnology Inc.) at 4°C overnight. After three washes with PBS, the slices were incubated with a biotin-conjugated secondary antibody for 30 minutes at room temperature, and signals were visualized using a diaminobenzidine (DAB) Peroxidase Substrate Kit.
Statistical analysis
Data are expressed as mean ± standard error of the mean of at least three independent experiments. One-way ANOVA followed by Bonferroni’s multiple comparisons post hoc test or Student’s t-test was used to compare the different treatment groups. Graphics and analyses were performed using PRISM 5.0 (GraphPad Software, Inc., La Jolla, CA, USA), and a P<0.05 was considered statistically significant.
Results
NiONP characterization
Transmission electron microscopy analysis showed that the size of the primary NiONPs was 18.6±5.5 nm, and that most of the particles were crystalline spheres with polyhedral morphologies that tended to aggregate/agglomerate in aqueous solution (Figure 1). The hydrodynamic diameters of NiONPs in DMEM with 10% FBS, DMEM, and saline, as determined by DLS, were 313.2±12.6 nm, 302.6±10.3 nm, and 327.3±19.4 nm, respectively. The zeta potentials of NiONPs in these three types of liquid media were −10.2±0.6 mV, −10.3±0.3 mV, and −1.9±0.2 mV, respectively (Table 1).
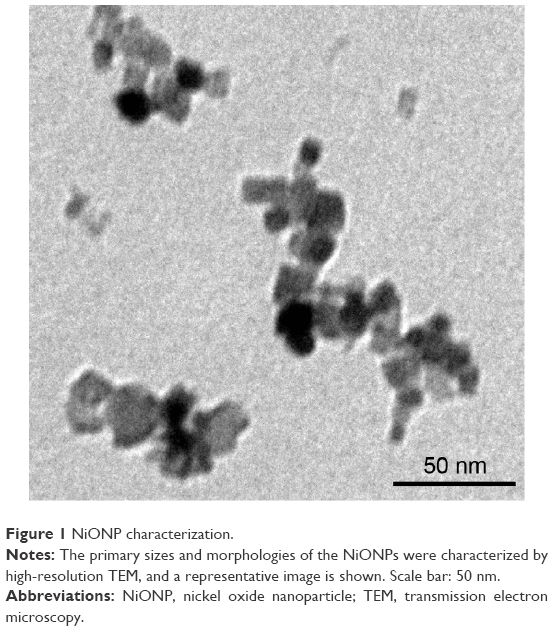 | Figure 1 NiONP characterization. |
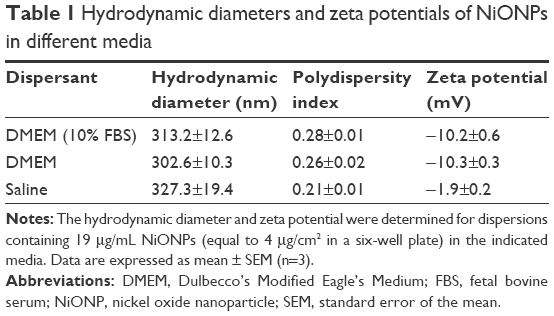 | Table 1 Hydrodynamic diameters and zeta potentials of NiONPs in different media |
Intratracheal instillation of NiONPs induces pulmonary inflammation
To investigate the effect of instillation of NiONPs on the inflammatory response, we first analyzed the BALF. The total cell and neutrophil counts were significantly increased from 3 days to 28 days in the NiONP-instilled groups (Figure 2A and B). The total number of AMs in the BALF was increased, but the proportion was decreased in the NiONP-exposed groups from 3 days to 28 days compared with the vehicle control group (Figure 2C). NiONP exposure induced ALP release compared with the vehicle control group from 3 days to 28 days, as shown in Figure 2D. The level of LDH, a marker of lung cell injury, in the BALF, and the amount of total protein, a marker of airway microvascular permeability, were significantly elevated at 3 days after NiONP exposure and remained elevated to 28 days (Figure 2E and F). MPO, a marker of neutrophil infiltration, was significantly increased after NiONP instillation compared with the vehicle control group during the observational period (Figure 2G and H). Concordantly, the MPO activity in the lung tissues was also significantly increased after NiONP instillation (Figure 2I). Histopathological analysis of the lung tissues showed that exposure to NiONP resulted in persistent and progressive pulmonary inflammatory responses. Neutrophil infiltration and AMs were observed in the alveolar spaces and peribronchial and perivascular regions in the NiONP-instilled group. Cell debris and alveolar wall thickening were observed at 7 days up to the end of the observational period. Foamy macrophages and alveolar proteinosis were discovered at 28 days after instillation (Figure 2J). Overall, these findings indicate that intratracheal instillation of NiONPs induces persistent pulmonary inflammation.
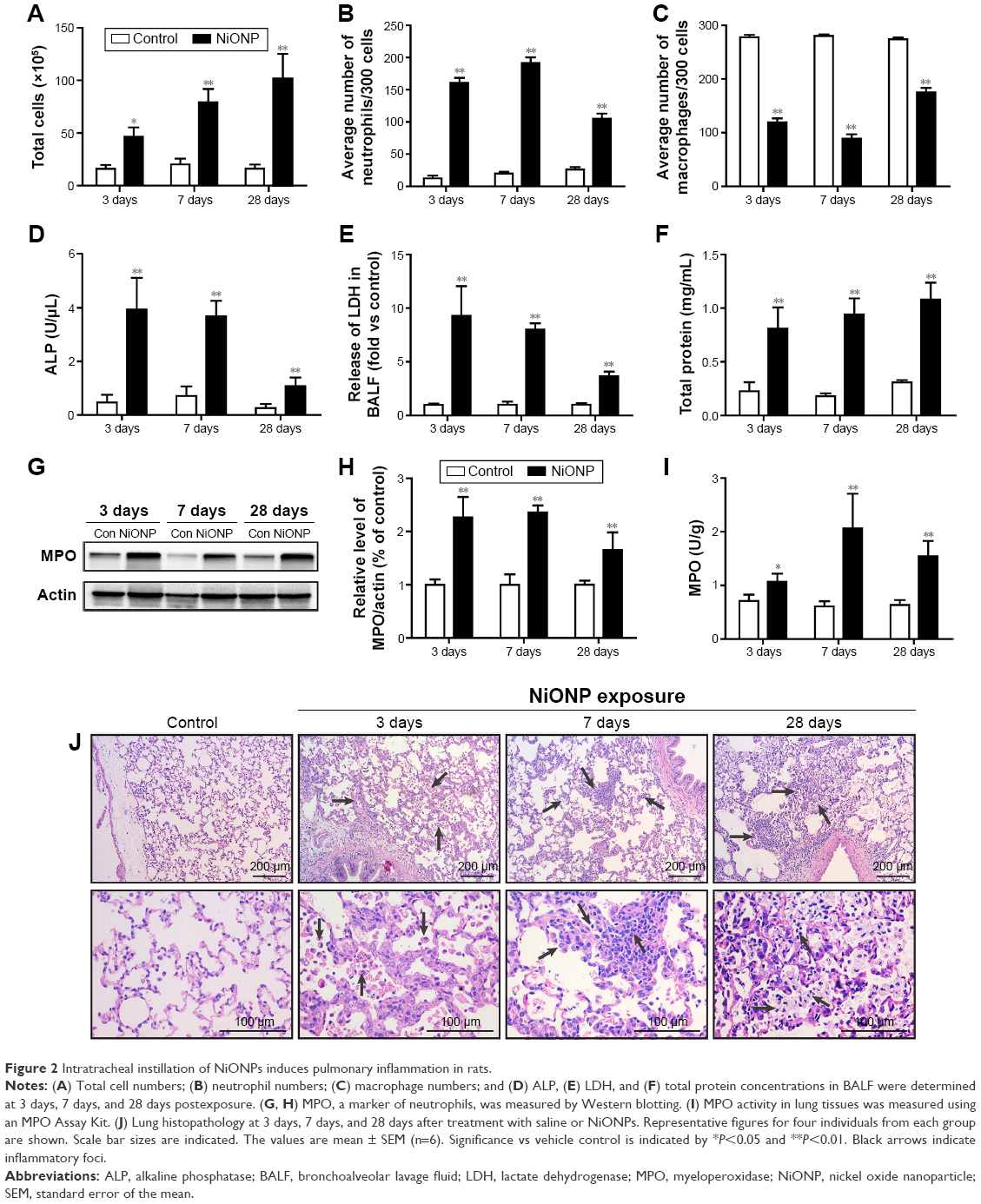 | Figure 2 Intratracheal instillation of NiONPs induces pulmonary inflammation in rats. |
NiONPs activate NLRP3 inflammasome and induce inflammatory cytokines secretion in rat lungs
The NLRP3 inflammasome has been confirmed to play key roles in the development of several chronic pulmonary diseases and in acute lung injuries. Thus, the effects of NiONP exposure on NLRP3 inflammasome activation and cytokine secretion were investigated. The levels of pro-inflammatory cytokines were measured in BALF supernatants, and IL-1β and IL-18 were found to be significantly increased at 3 days after exposure to NiONP and to remain elevated up to 28 days (Figure 3A and B). Moreover, the IL-1β mRNA level in the AMs, isolated from the NiONP exposed rats, was increased from 3 days to 28 days (Figure 3C). The IL-18 mRNA level was increased at 3 days after NiONP exposure, but returned to the control level at 7 days, and it increased again at 28 days (Figure 3D). Western blot analysis of the lung tissues revealed that the Nlrp3, pro-caspase-1 (p45), caspase-1 (p20), and IL-1β protein levels were increased in the NiONP exposed groups from 3 days to 28 days compared with the vehicle control group (Figure 3E–I). In addition, immunohistochemistry staining showed that Nlrp3 was present in AMs in the control group tissues. The Nlrp3 level was significantly increased in the NiONP-instilled group, which demonstrated further increases at all observed time points. Interestingly, the expression of NLRP3 was not only strongly positive in AMs but also seen in bronchial epithelial cells and alveolar type II cells after NiONP exposure (Figure 3J). Overall, these findings indicate that NLRP3 inflammasome is involved in NiONP-induced pulmonary inflammation and injury in vivo.
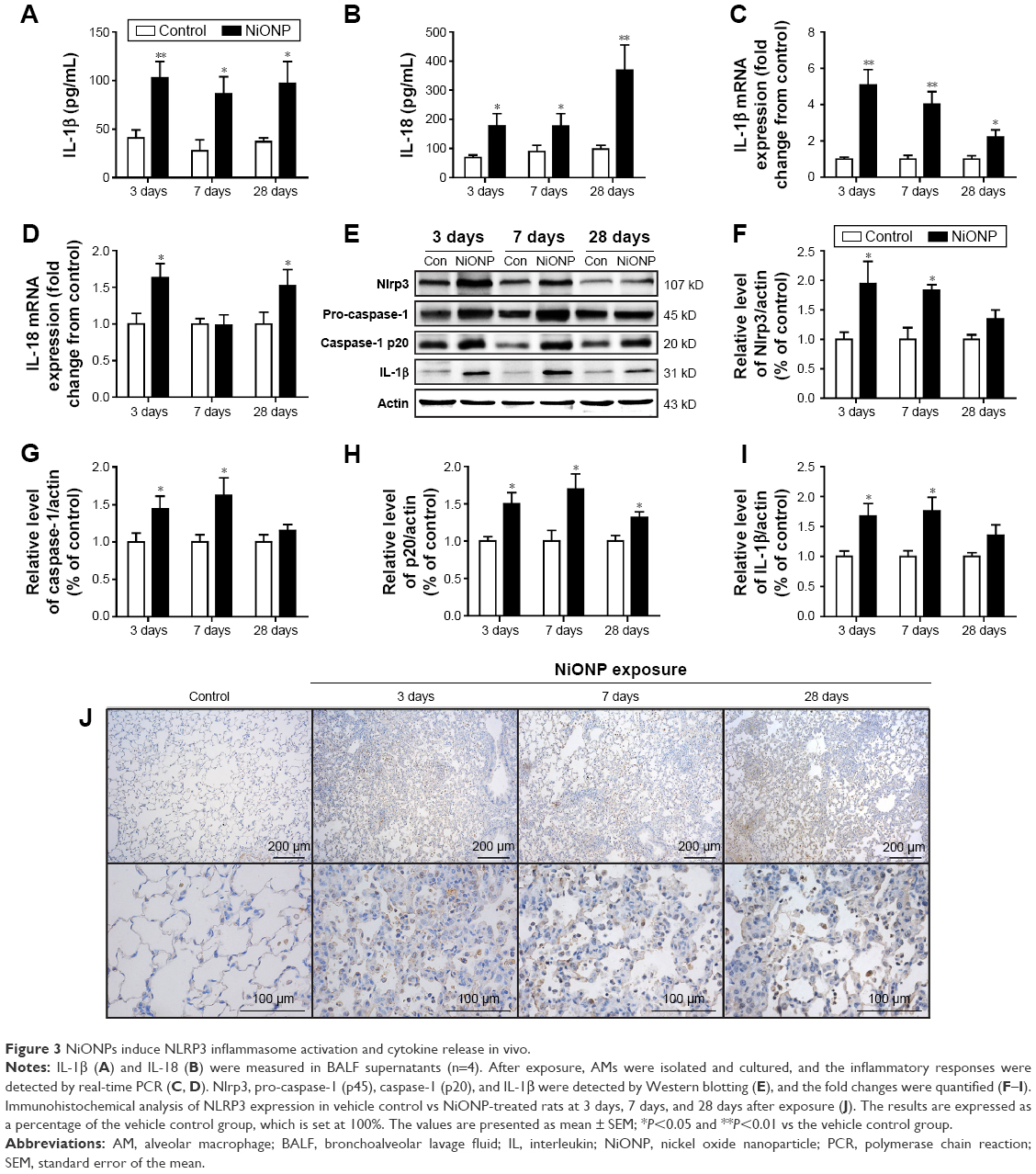 | Figure 3 NiONPs induce NLRP3 inflammasome activation and cytokine release in vivo. |
NiONPs induce cytotoxicity and cytokines secretion in RAW264.7 cells
In our earlier study, we have demonstrated that NiONPs enter into human bronchial epithelial cells and release Ni2+ inside of these cells.21 In the current study, to evaluate NiONP-induced cellular Ni2+ accumulation in RAW264.7 cells, cells were stained with Newport Green, a specific Ni2+ probe. As shown in Figure 4A, the cells exposed to NiONPs demonstrated a green fluorescence signal, the intensity of which increased in a dose-dependent manner. To evaluate the cytotoxicity of NiONPs, we first exposed RAW264.7 cells to various doses of NiONPs (0 μg/cm2, 1 μg/cm2, 2 μg/cm2, and 4 μg/cm2) for 12 hours or to 2 μg/cm2 of NiONPs for different time periods (0 hours, 6 hours, 12 hours, and 24 hours). We found that NiONPs inhibited cell viability in a dose- and time-dependent manner (Figure 4B and C). To investigate the inflammatory potential of NiONPs, we examined cytokine release in the cell culture supernatants. The treatment of LPS-primed macrophages resulted in a dose- and time-dependent increase in IL-1β release (Figure 4D and E). Notably, NiONP exposure did not increase TNF-α release in the primed or unprimed mouse macrophages (Figure 4F and G). Furthermore, the treatment of mouse macrophages with NiONPs did not alter the IL-1β or TNF-α mRNA level (Figure 4H and I), suggesting that NiONP-induced IL-1β maturation and release is independent of its transcription.
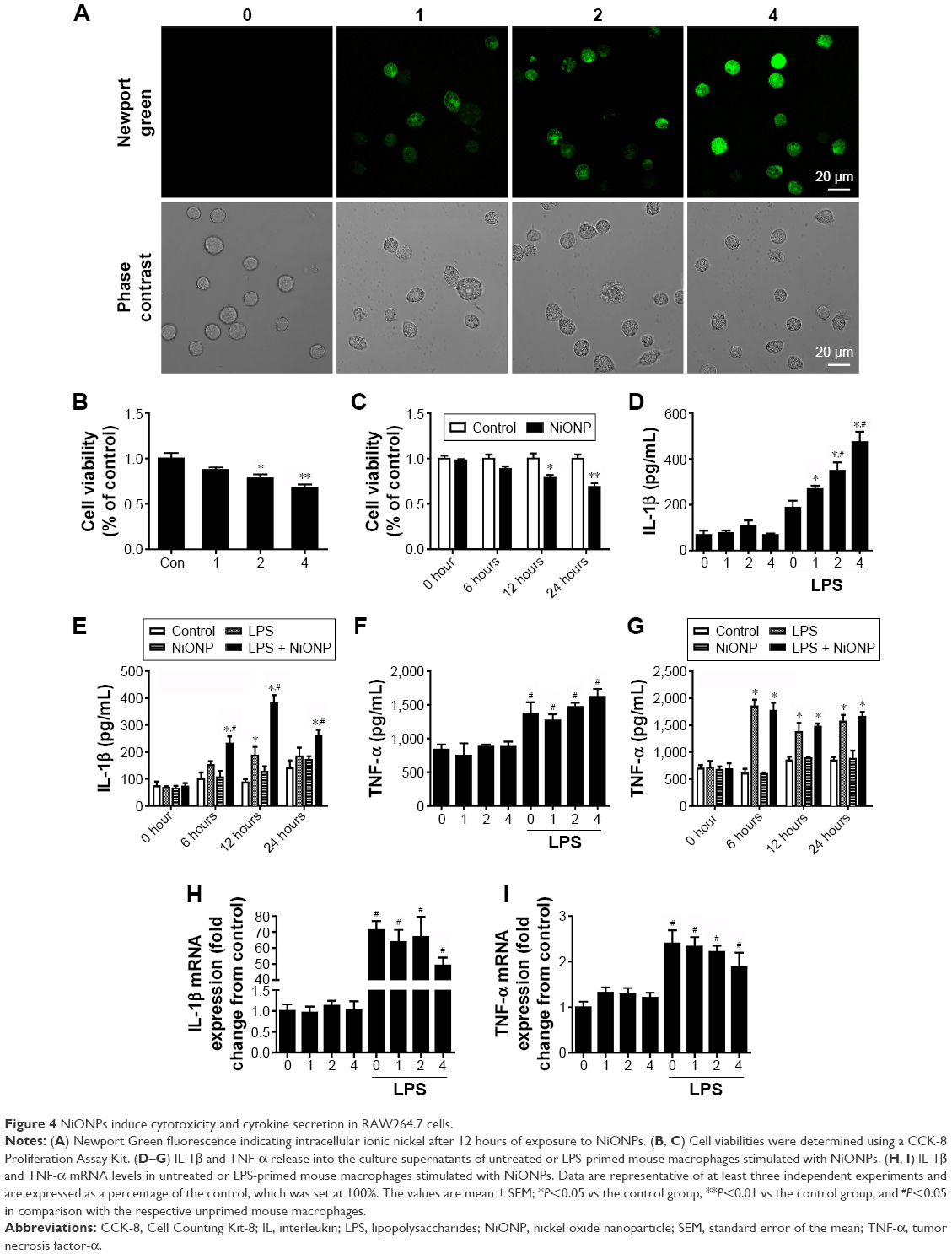 | Figure 4 NiONPs induce cytotoxicity and cytokine secretion in RAW264.7 cells. |
NiONP-induced cytokine secretion is dependent on caspase-1 activation in RAW264.7 cells
To confirm that NiONPs stimulate caspase-1 activation, RAW264.7 cells were exposed to different doses of NiONPs for 12 hours. Western blot analysis showed that the pro-caspase-1 and pro-IL-1β protein levels did not change following the NiONP treatment compared with those in the primed and unprimed groups (Figure 5A and B). However, the NiONP treatment increased the expression of the active form of caspase-1 (p20) as well as the release of mature IL-1β in the cell culture supernatants (Figure 5A and C). Moreover, caspase-1 activity in the cell lysates was increased by the NiONP treatment (Figure 5D). The secreted IL-1β in the culture supernatants was measured by ELISA and was found to be significantly increased in a dose-dependent manner after NiONP stimulation (Figure 5E). To further explore the role of caspase-1 activation in NiONP-induced IL-1β secretion, we evaluated caspase-1 activity and IL-1β secretion in the presence of a specific caspase-1 inhibitor z-yvad-fmk. We observed that this caspase-1 inhibitor strongly reduced the NiONP-mediated increases in the p20 subunit level, caspase-1 activity, and IL-1β secretion (Figure 5F–K). Thus, these results demonstrated that the NiONP-mediated IL-1β release was dependent on caspase-1 activation.
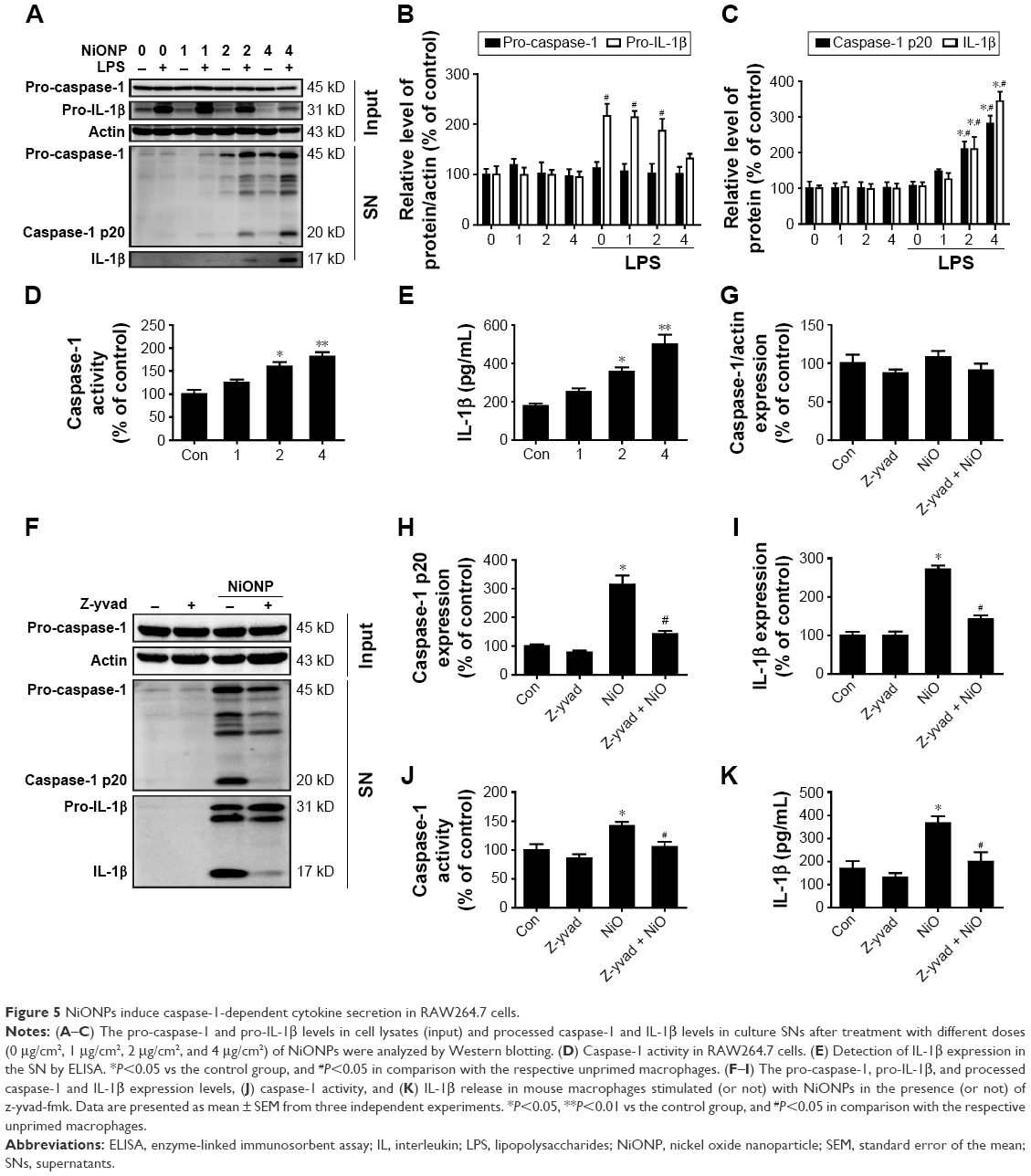 | Figure 5 NiONPs induce caspase-1-dependent cytokine secretion in RAW264.7 cells. |
NiONP-induced IL-1β release is dependent on Nlrp3/caspase-1 activation
To ascertain whether the NLRP3 inflammasome is involved in NiONP-induced IL-1β secretion, the Nlrp3 expression was downregulated in LPS-primed macrophages using siRNA (Figure 6A–C). Expression of the active form of caspase-1 (p20) and the release of mature IL-1β induced by NiONPs were completely abolished by Nlrp3 siRNA (Figure 6D and E). Similarly, Nlrp3 knockdown decreased caspase-1 activity in the cell lysates and the IL-1β level in culture supernatants treated with NiONPs (Figure 6F and G). These data suggested that the NiONP-mediated increase in IL-1β release was dependent on Nlrp3/caspase-1 activation.
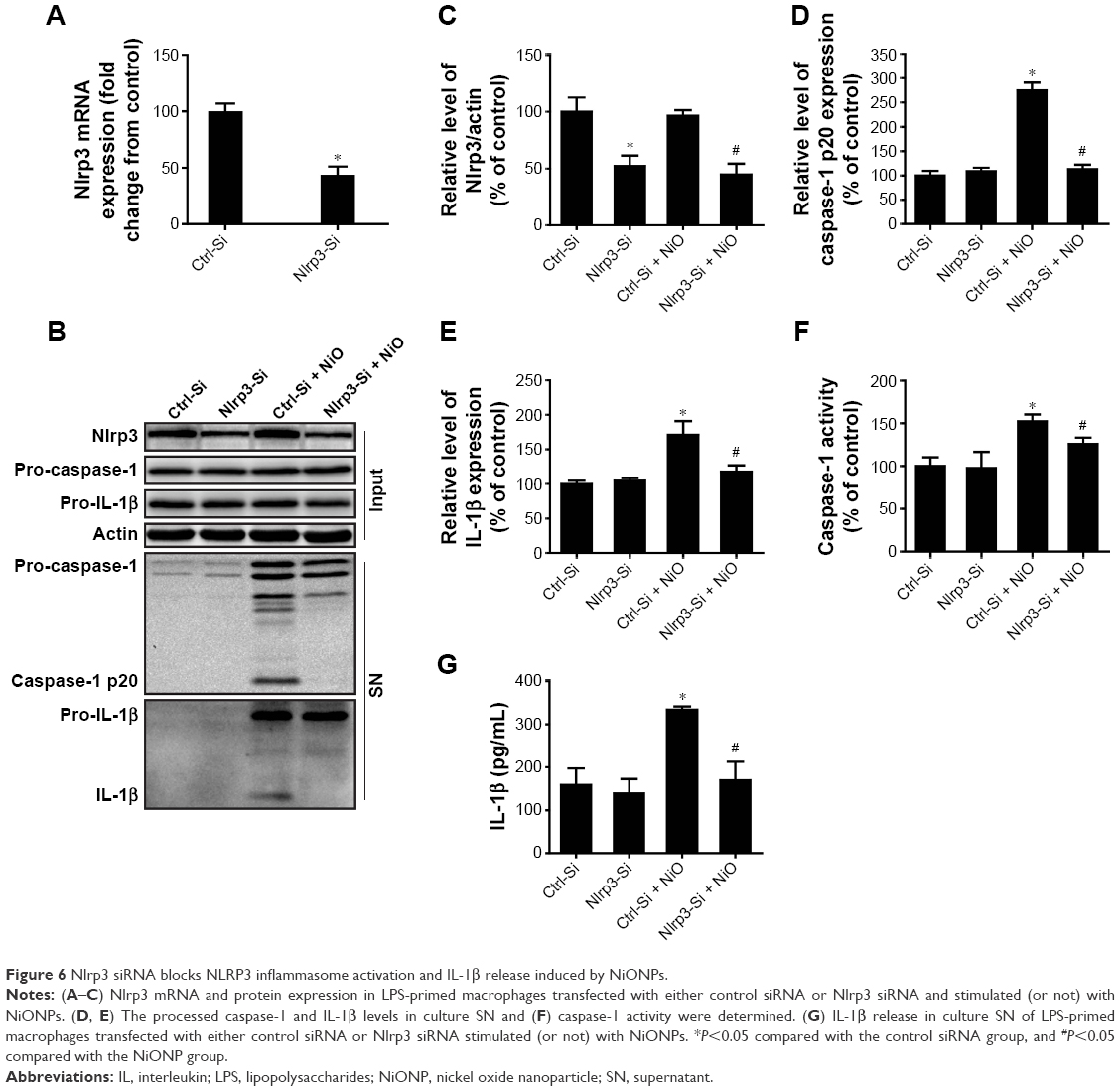 | Figure 6 Nlrp3 siRNA blocks NLRP3 inflammasome activation and IL-1β release induced by NiONPs. |
NiONP-induced IL-1β release and caspase-1 activation require phagocytosis and ROS production
Phagocytosis plays a crucial role in initiating macrophage-derived inflammatory responses.23 To assess whether the phagocytosis is required in NiONP-induced NLRP3 inflammasome activation, NiONP uptake was blocked in macrophages by pretreatment with cytochalasin D (Figure 7A). Interestingly, disruption of actin-mediated phagocytosis completely suppressed the IL-1β production, caspase-1 (p20) expression, and caspase-1 activity induced by NiONPs (Figure 7B–D). It is well known that the stimulation of macrophages with NPs induces ROS production.24,25 Consistent with earlier studies, we found that NiONPs induce a dose-dependent ROS production (Figure 7E). Phagocytosis of particles or fibers has been shown to activate the NLRP3 inflammasome via ROS production.26 To examine whether ROS are involved in the NiONP-induced IL-1β production, we stimulated RAW264.7 cells with NiONPs in the presence of a broad ROS scavenger, NAC. Our experimental data showed that NAC significantly decreased the extent of ROS production (Figure 7F). Notably, the presence of NAC significantly suppressed the IL-1β secretion, caspase-1 (p20) expression, as well as caspase-1 activity induced by NiONPs (Figure 7G and H). These data suggested that ROS production contributes to NiONP-induced NLRP3 inflammasome activation. Overall, the results indicated that NiONP-induced NLRP3 inflammasome activation coincidently requires phagocytosis and ROS production.
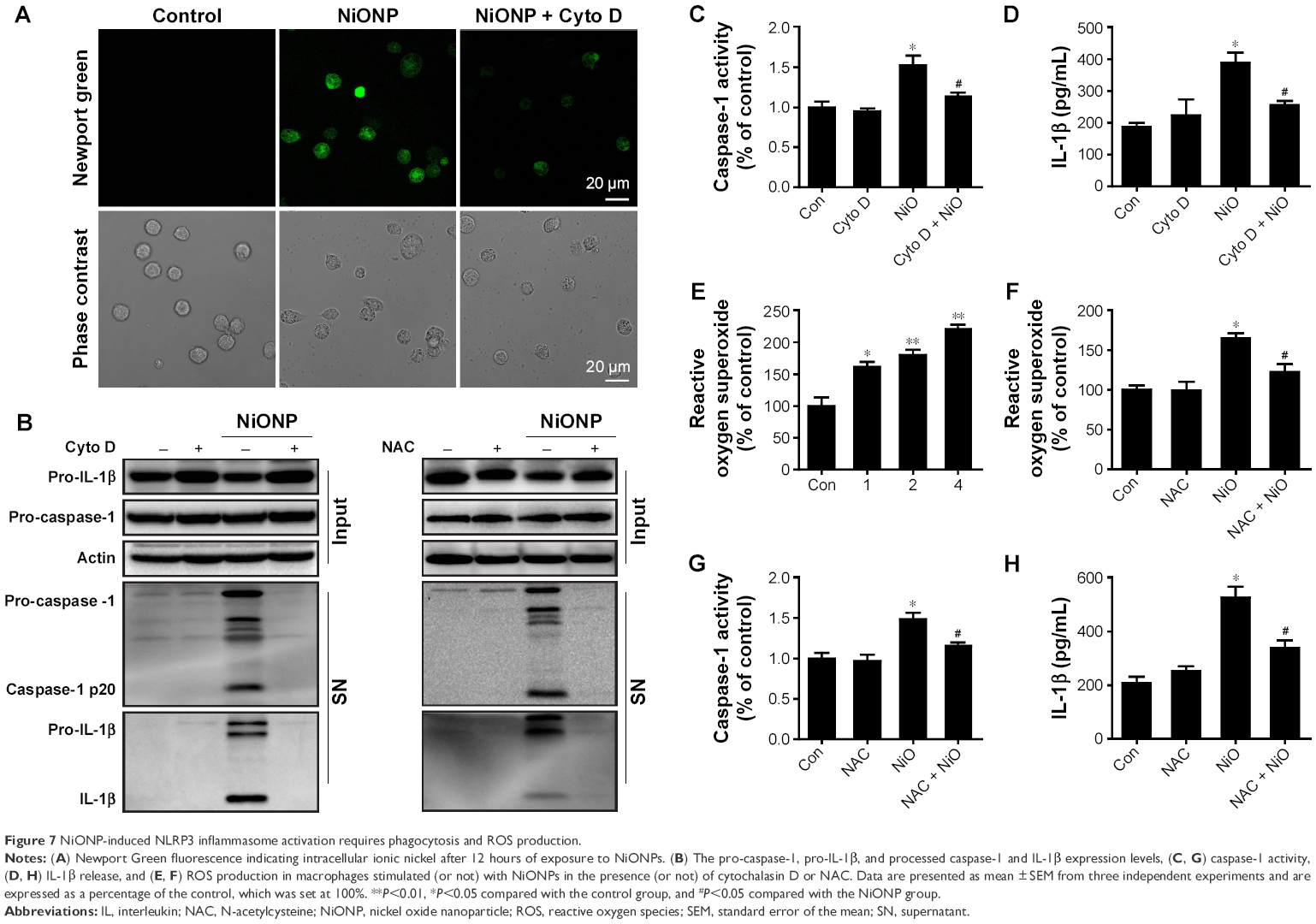 | Figure 7 NiONP-induced NLRP3 inflammasome activation requires phagocytosis and ROS production. |
Discussion
NiONPs are widely used NPs, and concerns about their health effects have been raised. However, the underlying cellular and molecular mechanisms still remain unclear. The current study is the first to confirm that NiONP exposure induces pulmonary inflammation by activating the NLRP3 inflammasome in vivo and in vitro. We demonstrated that NiONP exposure induced pulmonary inflammation and injury, which was accompanied by inflammatory cell infiltration and cytokine secretion. We also showed that an elevation in Nlrp3 expression resulted in increased caspase-1 activity, promoting IL-1β and IL-18 secretion in NiONP-induced pulmonary inflammation. Moreover, we determined that Nlrp3 knockdown decreased the NiONP-mediated increases in caspase-1 activity and mature IL-1β secretion. Furthermore, we revealed that NiONP-induced IL-1β release and caspase-1 activation were mediated by phagocytosis and ROS production.
NPs, including metal catalysts such as Ni2+, are being increasingly used for industrial applications. NPs tend to aggregate/agglomerate in aqueous solutions. The hydrodynamic diameters of the NiONP aggregates/agglomerates mainly range from 300 nm to 400 nm, in accordance with our earlier study.21 Agglomeration was likely due to particle concentration, ionic strength, pH, and media composition.27 Occupational exposure to Ni2+ NPs could cause inflammation and injury to multiple organs and tissues.28 Inhalation is the major exposure route of Ni2+ NPs; AMs and airway epithelial cells are the most important target cells.10,29 Agglomerates of NiONPs have been shown to induce persistent inflammatory effects and the increased expression of cytokines, such as IL-1β and monocyte chemotactic protein-1.30 In this study, intratracheal instillation of NiONPs resulted in persistent pulmonary inflammation, which was accompanied by neutrophil, eosinophil, and macrophage infiltration and cytokine secretion. Interestingly, we found that the total number of macrophages in the BALF was increased, while the proportion of macrophages in the total cells was downregulated after NiONP exposure. These results indicated that NiONP-induced inflammatory infiltration was mainly mediated by neutrophils. The increased inflammatory cell influx is considered to be a physiological response to clearly exogenous substances, including bacteria and particles. Studies have revealed that AMs engulf particles and regulate the immune response.31 In addition, cell debris and alveolar wall thickening were observed after NiONP exposure, and foamy macrophages and alveolar proteinosis were discovered at 28 days after instillation. Moreover, NiONP exposure increased ALP activity, which is associated with type II alveolar cell damage and stimulation.32 Interestingly, the change in various inflammatory response parameters such as the number of neutrophils and macrophages, ALP release, and LDH level are not linear. These observed changes may be related to the time course of different parameters and the dynamic nature of the cytokine network after NP exposure.10 However, some studies have shown that NiONP exposure induces only mild and reversible inflammation.33,34 Zhang et al35 found that NiO (20 μg suspended in 50 μL of PBS) exposure induced less neutrophil cell counts, monocyte chemotactic protein-1, and IL-6 levels in BALF compared with the same amount of CuO and Cr2O3 exposure in C57 BL/6 mice. We suppose that the discrepancy in these results may be due to the different properties and the ability to induce oxidative stress of NPs. In addition, the uses of different cell types, animal models, exposure doses, different dispersion statuses, and durations of exposure in experiments could also be possible explanations for these inconsistent results.36
Inhalation of NPs induces a local response in the lungs that is initiated by AMs and airway epithelial cells.37 These cells recognize NPs as exogenous materials and trigger a chain reaction of inflammatory responses. Pro-inflammatory mediators play vital roles in the airways and lungs during the early inflammatory response. It has been demonstrated that instilled silica increases IL-1β release in BALF and neutralizes IL-1β attenuated silica-induced pulmonary inflammation and fibrosis.38 An earlier study has reported that the levels of pro-inflammatory cytokines, such as IL-1β and IFN-γ, are significantly increased in BALF following NiONP exposure.39 In the current study, we found that NiONP exposure significantly increased IL-1β protein expression in BALF and mRNA expression in AMs. Interestingly, our data showed that IL-18 mRNA level was increased at 3 days, but returned to the control level at 7 days, and it increased again at 28 days after NiONP exposure. We postulate that this result is related to the change of cell viability in AMs. Furthermore, NiONPs induced IL-1β secretion in a dose- and time-dependent manner in macrophages. These results indicate that NiONPs induce cytokine secretion in vivo and in vitro and that cytokines are involved in pulmonary inflammatory responses.
Recent studies have revealed that the NLRP3 inflammasome is central to the pathogenesis of a wide variety of chronic inflammatory diseases, including diabetes, gout, silica, chronic obstructive pulmonary disease, and asthma.14,40,41 This inflammasome consists of a complex of proteins that oligomerize and activate the caspase-1 cascade, which induces production of the active pro-inflammatory cytokines, IL-1β and IL-18, when triggered by various environmental, pathogenic, or endogenous danger signals. Full activation of the NLRP3 inflammasome generally requires a two-step mechanism. The first step is a priming step that is initiated by various pathogen-associated molecular patterns and damage-associated molecular patterns, which further leads to NF-κB activation and increased expression of pro-IL-1β, pro-IL-18, and the components of the inflammasome. The second step is the activation step, involving the assembly of the inflammasome complex, production of active caspase-1, and further processing of pro-IL-1β and pro-IL-18 to their mature forms.12,42 The specific mechanism of activation is not known, but NLRP3 inflammasome has been shown to contain an abundance of activators. CNTs,43 titanium oxide NPs,44 silver NPs, and silica26 have been demonstrated to be recognized by NLRP3 inflammasome, leading to secretion of the pro-inflammatory cytokine IL-1β in both in vitro and in vivo models. In this study, we found that Nlrp3, pro-caspase-1, and active caspase-1 (p20) expression was increased in the lung tissues after NiONP exposure, suggesting that NiONP exposure not only increases caspase-1 expression but also enhances caspase-1 activity. To further confirm this result, we exposed RAW264.7 cells to different doses of NiONPs for 12 hours in vitro. The cell model has been applied to investigate NP-induced pulmonary toxicity and inflammasome activation.45,46 This study has also shown that NiONPs induce active caspase-1 expression and IL-1β release in macrophages. Notably, knockdown of Nlrp3 completely abolishes caspase-1 activity and mature IL-1β release induced by NiONPs. Moreover, the caspase-1 inhibitor strongly decreased the NiONP-mediated increases in caspase-1 activity and IL-1β secretion. Overall, these data suggest that NiONP-induced pulmonary inflammation and cytokine secretion is dependent on NLRP3 inflammasome and caspase-1 activation.
However, the precise upstream mechanism of NLRP3 inflammasome activation is incompletely understood. Accumulating evidences indicate that NPs trigger an inflammation and toxicity following endocytosis into the target cells.47 Meunier et al17 showed that phagocytosis of fibrous particles is required as an initial step for inflammasome activation. The actin cytoskeleton plays a vital role in phagocytizing long materials or particles.48 In this study, we showed that disruption of actin-mediated phagocytosis by cytochalasin D completely inhibited NiONP uptake and suppressed the IL-1β production. These results indicate that NiONP-induced NLRP3 inflammasome activation depends upon uptake process. Earlier in vitro studies demonstrated that NiONP enhanced ROS production in human airway epithelial and breast cancer.6 Recent studies have suggested that ROS produced during phagocytosis play a crucial role in mediating NLRP3 inflammasome activation. In the current study, we found that preventing NiONP-induced ROS production through NAC appeared to successfully inhibit NLRP3 inflammasome activation and IL-1β maturation. This finding suggests that the accumulation of ROS induced by NiONP triggers assembly of NLRP3 inflammasome and inflammatory cytokine release. In addition, particle–membrane interaction, potassium flux, and lysosomal destabilization caused by various environmental signals or NPs seem to be essential intermediate steps in the activation of NLRP3 inflammasome.49–51 Further study is necessary to identify whether these mechanisms are involved in NiONP-induced NLRP3 inflammasome activation.
Conclusion
We propose an intriguing mechanism, whereby NiONPs induce pulmonary inflammation by activating the NLRP3 inflammasome and promoting the release of pro-inflammatory cytokines. NiONP exposure causes sustained pulmonary inflammation accompanied by inflammatory cell infiltration, alveolar proteinosis, and cytokine secretion. The pulmonary inflammation induced by NiONPs is dependent on Nlrp3–caspase-1 pathway activation. Furthermore, we also speculate that NiONP-induced IL-1β production coincidently requires phagocytosis and ROS production. Overall, these results may contribute to a better understanding of NiONP-mediated lung toxicity and offer new strategies to combat the pulmonary toxicity induced by NiONPs.
Acknowledgments
This work was supported by the Outstanding Young Scientist Project of the National Natural Science Foundation of China (No 81422039) and the Grant for Outstanding Young Scientist from Chongqing Science and Technology Commission (No CSTC2013JCYJJQ10002).
Disclosure
The authors report no conflicts of interest in this work.
References
Savolainen K, Alenius H, Norppa H, Pylkkänen L, Tuomi T, Kasper G. Risk assessment of engineered nanomaterials and nanotechnologies – a review. Toxicology. 2010;269(2–3):92–104. | ||
Horie M, Nishio K, Fujita K, et al. Ultrafine NiO particles induce cytotoxicity in vitro by cellular uptake and subsequent Ni(II) release. Chem Res Toxicol. 2009;22(8):1415–1426. | ||
Ogami A, Morimoto Y, Myojo T, et al. Pathological features of different sizes of nickel oxide following intratracheal instillation in rats. Inhal Toxicol. 2009;21(10):812–818. | ||
Pietruska JR, Liu X, Smith A, et al. Bioavailability, intracellular mobilization of nickel, and HIF-1alpha activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol Sci. 2011;124(1):138–148. | ||
Mu Y, Jia D, He Y, Miao Y, Wu HL. Nano nickel oxide modified non-enzymatic glucose sensors with enhanced sensitivity through an electrochemical process strategy at high potential. Biosens Bioelectron. 2011;26(6):2948–2952. | ||
Siddiqui MA, Ahamed M, Ahmad J, et al. Nickel oxide nanoparticles induce cytotoxicity, oxidative stress and apoptosis in cultured human cells that is abrogated by the dietary antioxidant curcumin. Food Chem Toxicol. 2012;50(3–4):641–647. | ||
Denkhaus E, Salnikow K. Nickel essentiality, toxicity, and carcinogenicity. Crit Rev Oncol Hematol. 2002;42(1):35–56. | ||
Magaye R, Zhao J, Bowman L, Ding M. Genotoxicity and carcinogenicity of cobalt-, nickel- and copper-based nanoparticles. Exp Ther Med. 2012;4(4):551–561. | ||
Morimoto Y, Hirohashi M, Ogami A, et al. Expression of cytokine-induced neutrophil chemoattractant in rat lungs following an intratracheal instillation of micron-sized nickel oxide nanoparticle agglomerates. Toxicol Ind Health. 2014;30(9):851–860. | ||
Gillespie PA, Kang GS, Elder A, et al. Pulmonary response after exposure to inhaled nickel hydroxide nanoparticles: short and long-term studies in mice. Nanotoxicology. 2010;4(1):106–119. | ||
Capasso L, Camatini M, Gualtieri M. Nickel oxide nanoparticles induce inflammation and genotoxic effect in lung epithelial cells. Toxicol Lett. 2014;226(1):28–34. | ||
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. | ||
De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32(8):373–379. | ||
Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther Adv Respir Dis. 2015;9(4):188–197. | ||
Sun B, Wang X, Ji Z, Li R, Xia T. NLRP3 inflammasome activation induced by engineered nanomaterials. Small. 2013;9(9–10):1595–1607. | ||
Yang EJ, Kim S, Kim JS, Choi IH. Inflammasome formation and IL-1beta release by human blood monocytes in response to silver nanoparticles. Biomaterials. 2012;33(28):6858–6867. | ||
Meunier E, Coste A, Olagnier D, et al. Double-walled carbon nanotubes trigger IL-1beta release in human monocytes through Nlrp3 inflammasome activation. Nanomedicine. 2012;8(6):987–995. | ||
Sun B, Wang X, Ji Z, et al. NADPH oxidase-dependent NLRP3 inflammasome activation and its important role in lung fibrosis by multiwalled carbon nanotubes. Small. 2015;11(17):2087–2097. | ||
Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. | ||
Hamilton RF Jr, Buford M, Xiang C, Wu N, Holian A. NLRP3 inflammasome activation in murine alveolar macrophages and related lung pathology is associated with MWCNT nickel contamination. Inhal Toxicol. 2012;24(14):995–1008. | ||
Duan WX, He MD, Mao L, et al. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol Appl Pharmacol. 2015;286(2):80–91. | ||
Morimoto Y, Hirohashi M, Ogami A, et al. Pulmonary toxicity following an intratracheal instillation of nickel oxide nanoparticle agglomerates. J Occup Health. 2011;53(4):293–295. | ||
Morishige T, Yoshioka Y, Tanabe A, et al. Titanium dioxide induces different levels of IL-1beta production dependent on its particle characteristics through caspase-1 activation mediated by reactive oxygen species and cathepsin B. Biochem Biophys Res Commun. 2010;392(2):160–165. | ||
Xia W, Song HM, Wei Q, Wei A. Differential response of macrophages to core-shell Fe3O4@Au nanoparticles and nanostars. Nanoscale. 2012;4(22):7143–7148. | ||
Heim J, Felder E, Tahir MN, et al. Genotoxic effects of zinc oxide nanoparticles. Nanoscale. 2015;7(19):8931–8938. | ||
Morishige T, Yoshioka Y, Inakura H, et al. The effect of surface modification of amorphous silica particles on NLRP3 inflammasome mediated IL-1beta production, ROS production and endosomal rupture. Biomaterials. 2010;31(26):6833–6842. | ||
Allouni ZE, Cimpan MR, Høl PJ, Skodvin T, Gjerdet NR. Agglomeration and sedimentation of TiO2 nanoparticles in cell culture medium. Colloids Surf B Biointerfaces. 2009;68(1):83–87. | ||
Phillips JI, Green FY, Davies JC, Murray J. Pulmonary and systemic toxicity following exposure to nickel nanoparticles. Am J Ind Med. 2010;53(8):763–767. | ||
Kadoya C, Lee BW, Ogami A, et al. Analysis of pulmonary surfactant in rat lungs after inhalation of nanomaterials: fullerenes, nickel oxide and multi-walled carbon nanotubes. Nanotoxicology. 2016;10(2):194–203. | ||
Morimoto Y, Ogami A, Todoroki M, et al. Expression of inflammation-related cytokines following intratracheal instillation of nickel oxide nanoparticles. Nanotoxicology. 2010;4(2):161–176. | ||
Patel B, Gupta N, Ahsan F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur J Pharm Biopharm. 2015;89:163–174. | ||
Cobben NA, Drent M, Jacobs JA, et al. Relationship between enzymatic markers of pulmonary cell damage and cellular profile: a study in bronchoalveolar lavage fluid. Exp Lung Res. 1999;25(2):99–111. | ||
Morimoto Y, Hirohashi M, Ogami A, et al. Inflammogenic effect of well-characterized fullerenes in inhalation and intratracheal instillation studies. Part Fibre Toxicol. 2010;7:4. | ||
Morimoto Y, Oyabu T, Ogami A, et al. Investigation of gene expression of MMP-2 and TIMP-2 mRNA in rat lung in inhaled nickel oxide and titanium dioxide nanoparticles. Ind Health. 2011;49(3):344–352. | ||
Zhang H, Ji Z, Xia T, et al. Use of metal oxide nanoparticle band gap to develop a predictive paradigm for oxidative stress and acute pulmonary inflammation. ACS Nano. 2012;6(5):4349–4368. | ||
Sager T, Wolfarth M, Keane M, Porter D, Castranova V, Holian A. Effects of nickel-oxide nanoparticle pre-exposure dispersion status on bioactivity in the mouse lung. Nanotoxicology. 2016;10(2):151–161. | ||
Hiraiwa K, van Eeden SF. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflamm. 2013;2013:619523. | ||
Guo J, Gu N, Chen J, et al. Neutralization of interleukin-1 beta attenuates silica-induced lung inflammation and fibrosis in C57BL/6 mice. Arch Toxicol. 2013;87(11):1963–1973. | ||
Cho WS, Duffin R, Poland CA, et al. Differential pro-inflammatory effects of metal oxide nanoparticles and their soluble ions in vitro and in vivo; zinc and copper nanoparticles, but not their ions, recruit eosinophils to the lungs. Nanotoxicology. 2012;6(1):22–35. | ||
Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. | ||
Amaral FA, Costa VV, Tavares LD, et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012;64(2):474–484. | ||
Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. | ||
Hussain S, Sangtian S, Anderson SM, et al. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part Fibre Toxicol. 2014;11:28. | ||
Yazdi AS, Guarda G, Riteau N, et al. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proc Natl Acad Sci U S A. 2010;107(45):19449–19454. | ||
Sandberg WJ, Låg M, Holme JA, et al. Comparison of non-crystalline silica nanoparticles in IL-1beta release from macrophages. Part Fibre Toxicol. 2012;9:32. | ||
Reisetter AC, Stebounova LV, Baltrusaitis J, et al. Induction of inflammasome-dependent pyroptosis by carbon black nanoparticles. J Biol Chem. 2011;286(24):21844–21852. | ||
Hara K, Shirasuna K, Usui F, et al. Interferon-tau attenuates uptake of nanoparticles and secretion of interleukin-1beta in macrophages. PLoS One. 2014;9(12):e113974. | ||
Misawa T, Takahama M, Kozaki T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. | ||
Hari A, Zhang Y, Tu Z, et al. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci Rep. 2014;4:7281. | ||
Rivers-Auty J, Brough D. Potassium efflux fires the canon: potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 2015;45(10):2758–2761. | ||
Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–856. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.