Back to Journals » Journal of Inflammation Research » Volume 14
Exposure to Maternal Immune Activation Causes Congenital Unfolded Protein Response Defects and Increases the Susceptibility to Postnatal Inflammatory Stimulation in Offspring
Authors Shimizu Y ![]() , Tsukada T, Sakata-Haga H, Sakai D
, Tsukada T, Sakata-Haga H, Sakai D ![]() , Shoji H, Saikawa Y, Hatta T
, Shoji H, Saikawa Y, Hatta T
Received 1 December 2020
Accepted for publication 22 January 2021
Published 12 February 2021 Volume 2021:14 Pages 355—365
DOI https://doi.org/10.2147/JIR.S294238
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yo Shimizu,1 Tsuyoshi Tsukada,2 Hiromi Sakata-Haga,2 Daisuke Sakai,3 Hiroki Shoji,3 Yutaka Saikawa,1 Toshihisa Hatta2
1Department of Pediatrics, Kanazawa Medical University, Uchinada, Ishikawa, Japan; 2Department of Anatomy, Kanazawa Medical University, Uchinada, Ishikawa, Japan; 3Department of Biology, Kanazawa Medical University, Uchinada, Ishikawa, Japan
Correspondence: Toshihisa Hatta
Department of Anatomy, Kanazawa Medical University, Uchinada, Ishikawa, Japan
Tel +81-76-281-8113
Fax +81-76-218-8189
Email [email protected]
Background: A number of childhood diseases have been identified, such as severe infection or autoinflammatory disease, in which immune overreaction against inflammation is a possible underlying mechanism. Previous reports have demonstrated that fetal cells exposed to maternal immune activation (MIA) induced by polyriboinosinic-polyribocytidylic acid [poly(I:C)] exhibited hypersensitivity to inflammation in vitro. However, the details of this mechanism remain unclear. Therefore, this study aimed to reveal the reaction to inflammation in offspring exposed to MIA in the prenatal period, as well as its molecular mechanism, using a viral infection mouse model.
Materials and Methods: Pregnant mice at 12.5, 14.5, and 16.5 days post coitum were injected intraperitoneally with poly(I:C) 20 mg/kg body weight (BW) or saline. Offspring aged 3– 4 weeks received the second injection of 20 mg/kg BW or 4 mg/kg BW poly(I:C) or saline. Serum and tissues were collected at 2, 24, 48, and 72 h after the postnatal injection. The cytokine profile, histopathology of organs, and unfolded protein response (UPR) in offspring were examined.
Results: The serum levels of interleukin (IL)-6, IL-17, and interferon-γ were significantly higher in the MIA group, and acute liver necrosis was detected. Moreover, failure in UPR was observed in the MIA group compared with that in the control group.
Conclusion: Overall, MIA exposure in utero caused failure in UPR as well as immune overreaction to the second attack of inflammation in offspring. Our results suggested that prenatal exposure to MIA might contribute to the congenital inflammatory constitution after birth.
Keywords: maternal immune activation, liver necrosis, immune overreaction, unfolded protein response defects
Introduction
The prenatal environment has been identified as an important determinant for postnatal disorders.1 Moreover, maternal immune activation (MIA) is known to increase the risk of neurodevelopmental disorders, immune disorders, metabolic diseases, and cardiovascular diseases in offspring.2–6 Neurodevelopmental disorders such as autism spectrum disorder are associated with immune activation caused by viral infection, suggesting a possible relationship with the production of inflammatory cytokines such as interleukin (IL)-6 and IL-17.5,7–9 A previous study reported that a rodent MIA model induced using polyriboinosinic-polyribocytidylic acid [poly(I:C)], synthetic double-stranded RNA, or lipopolysaccharide (LPS) from mid-pregnancy led to different immune responses by the second attack of inflammation in offspring.2,3,10,11 MIA induced by LPS has been found to reduce the levels of IL-1, IL-6, and tumor necrosis factor (TNF)-α following LPS stimulation in vivo.10,11 Cells derived from offspring exposed to MIA, which was induced by poly(I:C), exhibited increased production of IL-1 and IL-12 following LPS stimulation in vitro.2,3 The levels of proinflammatory cytokines in offspring exposed to MIA were within the normal range if the offspring were not subjected to the second attack of inflammatory stimuli after birth. MIA induced by poly(I:C) might be associated with the constitution of hypersensitivity to inflammation. In severe cases of infection or autoinflammatory diseases, patients may have a dysfunctional immune system, which represents an immune hypersensitivity to inflammatory stimuli.12,13 Because of this constitution, these patients can be easily aggravated by inflammation caused by viral or bacterial infection. Viral infections are the most common cause of diseases in children and can cause various clinical manifestations, including mild cases with only cold symptoms and severe cases with multiple organ failure (encephalopathy, cardiomyopathy, liver failure, kidney failure, or respiratory failure) caused by cytokine storm.12,14–16 In influenza-associated encephalopathy, a representative disease of organ failure caused by viral infection, the influenza virus has not been detected in the brain. The immune overreaction against viral infection induces vascular permeability and apoptosis because of the cytokine storm. The primary aggravating factor for a viral infection is the overreaction to inflammation rather than direct damage caused by the virus.12,14–16 Immune responses are reported to be most sensitive during childhood. This is a possible aggravating factor of diseases, which further induces immune overreaction by inflammatory stimulation in offspring. Immune regulation is often determined by the number of monocytes or lymphocytes that participate in the immune response. However, the details of the mechanism have not been fully elucidated.3 Cell death is considered to occur via multiple pathways, in which endoplasmic reticulum (ER) stress is associated with the activation of inflammatory signaling pathways stimulated by proinflammatory cytokines. A previous study suggested that IL-6, IL-17, and IFN-γ are closely associated with cell death caused by unfolded protein response (UPR).17–19 Binding immunoglobulin protein (Bip) is a master regulator of ER homeostasis, which has been determined to inactivate the sensor proteins of UPR and block UPR in the normal state of the cell. Excess amounts of unfolding proteins dissociate Bip from each of the sensor proteins20 and then activate them. X-box-binding protein1 (Xbp-1), activating transcription factor 4 (Atf4), and activating transcription factor 6 (Atf6), which are translated by the stimulation of each sensor protein, activate UPR-related genes as well as ER chaperones.21 Moreover, it has not been investigated whether organ damage or dysfunction caused by postnatal inflammatory stimuli is more likely to occur in offspring exposed to MIA in the prenatal period.
Here we hypothesized that the inflammatory response was altered by MIA (first attack) in utero, whereas the second attack of viral infection promotes immune overreaction, leading to cytokine storms and organ failure in offspring. Using a viral infection model with poly(I:C), we examined the effects of MIA on the susceptibility to inflammation and UPR in offspring after birth and described the molecular mechanisms underlying this over-response against inflammation in vivo.
Materials and Methods
Animals
Pregnant C57BL/6J mice (Japan SLC Inc., Hamamatsu, Japan) were used. Mice were maintained under standard laboratory conditions with food and water available ad libitum. A female mouse was housed with a male mouse overnight, and the day when a vaginal plug was found in the morning was designated as day post-coitus (dpc) 0.5. All procedures in this study were performed in strict accordance with the guidelines of the Care and Use of Laboratory Animals of Kanazawa Medical University (Permit Number: 2012–25, 2013–16, 2014–33). All surgical procedures were performed under deep anesthesia by intraperitoneal (i.p.) injection of an anesthetic combination of midazolam (4 mg/kg), medetomidine (0.3 mg/kg), and butorphanol (5 mg/kg), and all efforts were made to minimize animal suffering.
MIA Model
In total, 40 mice with timed pregnancies were placed into two treatment groups: MIA group and saline (S) group. Pregnant mice received i.p. injection of either injection of poly(I:C) (Sigma Aldrich, St. Louis, MO) at 20 mg/kg body weight (BW) or saline 0.01 mL/g BW at 12.5, 14.5, and 16.5 dpc was then performed. Four dams were used per group. Pups were housed with their own dam and littermates until 3–4 weeks after birth. Two or three pups per litter were randomly selected regardless of gender; in total, eight–nine pups were evaluated per group. The pups were divided into groups based on postnatal poly(I:C) injection at doses of 20 mg/kg BW and 4 mg/kg BW or saline, and samples were collected (Suppl Figure S1, Suppl Table S1). The pups were anesthetized with midazolam (4 mg/kg), medetomidine (0.3 mg/kg), and butorphanol (5 mg/kg) intraperitoneally, and blood and tissue samples were collected at 2, 24, 48, and 72 h after i.p. injection of poly(I:C) or saline. All serum and tissue samples were collected only once, ie, when the animals were euthanized, and not repeatedly from the same animal. The injection volume of poly(I:C) and saline was unified into 0.01 mL/g. Four dams were used for each group.
Setting of Experimental Groups
Pups delivered from dams that were injected with poly(I:C) at a dose of 20 mg/kg BW at 12.5, 14.5, and 16.5 dpc were assigned as the MIA group (MIA), whereas those delivered from dams that were injected with saline at 12.5, 14.5, and 16.5 dpc were assigned as the saline group (S). Pups injected with poly(I:C) at a dose of 20 mg/kg BW and 4 mg/kg BW or with saline were designated as P20, P4, and S, respectively. Finally, owing to the combination of pre- and postnatal injections, the groups were designated as MIA/P20, MIA/P4, MIA/S, S/P20, S/P4, and S/S.
Multiplex Cytokine Bead Array Assay
At 2 and 24 h after the offspring were administered poly(I:C), blood was collected from the left ventricle and centrifuged to separate the serum. Serum was stored at −80°C until use. The levels of IL-1β, IL10, and TNF-α were measured using the Bio-Plex Pro Mouse Cytokine Th17 Panel by multiplex cytokine bead array assay (BIO-RAD, Hercules, CA) in accordance with the manufacturer’s instructions (sensitivity: IL-1β, 9.4 pg/mL; IL-10, 1.0 pg/mL; TNF-α, 1.4 pg/mL). Each sample was measured in duplicate using the Bio-Plex MAGPIX system (BIO-RAD).
Sandwich ELISA
At 2, 24, 48, and 72 h after the pups were administered poly(I:C), blood was collected from the left ventricle and centrifuged to separate the serum. Serum was stored at −80°C until use. IL-6 and IL-17 levels were measured using sandwich ELISA kits (BioLegend, Inc., San Diego, CA) in accordance with the manufacturer’s instructions (sensitivity: IL-6, 1.6 pg/mL; IL-17, 7.8 pg/mL). All plates were treated with 5% bovine serum albumin (BSA) (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan). Horseradish peroxidase (HRP) activity was determined using one-step Ultra TMB Substrates (Thermo Fisher Scientific, Waltham, MA). Each sample was measured in triplicate at 450 nm using 2014 EnVision (PerkinElmer, Waltham, MA).
Quantitative Real-Time Polymerase Chain Reaction (PCR)
The livers of the offspring were collected at 2 and 24 h after administration of poly(I:C) or saline and stored at −80°C until use. Total RNA of the livers was extracted using the RNeasy Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. The expression levels of Ifn-γ, toll-like-receptor (Tlr)-3, nuclear factor (NF)-κB p65, Bip, total Xbp-1, spliced Xbp-1, Atf4, and Atf6 were determined using a gene expression assay (Applied Biosystems, Foster City, CA). Approximately 1 μg of total RNA was reverse-transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed using cDNA with SYBR Green Premix Ex Taq II (Takara Bio Inc., Kusatsu, Japan), and the expression of GAPDH was measured as an internal control. The results were expressed as ratios of gene expression to those in the control group injected with saline. Reactions were performed in triplicate using a Thermal Cycler Dice (Takara Bio). Suppl Table S2 shows the primer sequences used for gene amplification. Before extracting RNA from the liver, we collected a small 10-mg sample from the liver of each pup and pooled the liver samples together for each dam. We used a total of four dams per experimental group and performed statistical analysis using each measurement per litter.
Western Blot Analysis
Protein extracted from the liver using PRO-PREP (iNtRon Biotechnology, Inc., Gyeonggi-do, Korea) was separated using 5%–20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Fujifilm Wako Pure Chemical Corporation) at 5 μg/lane and transferred onto polyvinylidene fluoride membranes using an iBlot device (Thermo Fisher Scientific). The membranes were blocked using 5% BSA (Fujifilm Wako Pure Chemical Corporation) in Tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) and incubated overnight at 4°C with the following primary antibodies (Ab) in Immunoshot1 (Cosmo Bio, Tokyo, Japan): inhibitory ΚB (IKB) α (ab32518) rabbit mAb (1:1000, 0.09 μg/µl) and phospho (p)-IKBα (ab133462) rabbit mAb (1:1000, 0.09 μg/µl) (Abcam, Cambridge, UK). The membranes were then washed using TBST and incubated at room temperature with HRP-conjugated secondary antibody (1:3000) for 2 h. The signal was then visualized by enhanced chemiluminescence (Thermo Fisher Scientific) using Fusion system (Vilber-Lourmat, Collegien, France). The band intensities from each blot membrane were quantified using ImageJ software. β-actin was used as an internal control. The relative expression was determined. Before protein extraction, we collected a 10-mg sample from the liver of each pup and pooled the samples together for each dam. We used a total of four dams per experimental group and performed the statistical analysis using each measurement per litter.
Histological Analysis
The liver, brain, heart, kidney, lung, and colon were immersed fixed in 4% paraformaldehyde 50 mM phosphate buffer (pH 7.4) and embedded in paraffin. Five-micrometer-thick serial sections were prepared. In total, three sections selected for every 100 μm from serial sections per pup were used for histopathological examinations. Sections were deparaffinized in xylene, rehydrated using graded alcohol and distilled water, and then stained with hematoxylin and eosin (HE). Pathological changes were observed using a light microscope. Focal or single necrosis of hepatocytes was determined. Degeneration of hepatocytes was identified as balloon degeneration characterized by swelling of the hepatocyte and vacuolization with clear cytoplasm. Moreover, we investigated inflammatory cell infiltration. First, the number of pups with liver necrosis or degeneration in each dam was examined (two or three pups per litter). Then, the incidence of liver necrosis and degeneration in each group was estimated by calculating the mean incidence per litter (four dams per group). Histopathological examination was performed in a blinded manner.
Immunohistochemistry
For immunostaining, sections were blocked with 5% BSA (Fujifilm Wako Pure Chemical Corporation) for 30 min at room temperature, incubated with anti-NF-κB p65 (1:1000) (Rock land, Philadelphia, PA) and anticleaved caspase-3 (1:100) (Santa Cruz Biotechnology, Dallas, TE) Abs in TBST overnight at 4°C, and then incubated with HRP-labeled secondary antibodies (Agilent, Santa Clara, CA). Immunoreactivities were visualized by 3.3′-diaminobenzidine (DAB) chromogenic reaction. The brown DAB deposits were observed and imaged using NDP slide scanner (Hamamatsu Photonics KK, Hamamatsu, Japan). Negative controls were incubated with normal IgG of the same concentration with each primary Ab.
Quantitative Analyses of NF-κB-Positive Nuclei
Serial sections with a thickness of 5 µm were scanned using an NDP slide scanner (Hamamatsu Photonics KK). In three randomly selected regions (196 mm2) in each group, the number of NF-κB-positive and NF-κB-negative nucleus were counted, and the positive ratio was calculated using QuPath software (version 0.2.0).22
Statistical Analysis
All statistical analyses were performed per litter. Data were presented as the mean ± standard error of the mean. Data were analyzed using the Mann–Whitney U-test with R 4.0.0 (http://www.r-project.org).
Results
Immune Overreaction Was Induced Following a Second Attack of Poly(I:C) After Birth in Offspring
The proinflammatory and anti-inflammatory cytokine levels at 2, 24, 48, and 72 h after poly(I:C) or saline administration in offspring were measured. The serum levels of IL-1β, IL10, and TNF-α at 2 h were not significantly different between the groups (Suppl Figure S2 A, B, and C). The serum level of IL-6 at 2 h was not different between the MIA and saline group, and the serum level of IL-6 at 24 h in the MIA/P20 and MIA/P4 groups was higher than that in the S/P20 and S/P4 groups (MIA/P20 vs S/P20: p < 0.05, MIA/P4 vs S/P4: p < 0.05) (Figure 1A). The serum level of IL-17 in the MIA/P20 group was significantly higher than that in the S/P20 group at 2 h (p < 0.05) but was not significantly different between the MIA/P20 and S/P20 groups at 24 h. The level in the MIA/P4 group was significantly higher than that in the S/P4 group at 24 h (p < 0.05) (Figure 1B). The level of Ifn-γ mRNA was not different between the groups at 2 h. Although the level of Ifn-γ at 24 h between the MIA/P20 and S/P20 groups was not significantly different, that in the MIA/P4 group was significantly higher than that in the S/P4 group at 24 h (p < 0.05) (Figure 1C). The serum levels of IL-6, IL-17, and Ifn-γ mRNA were not different between the MIA/S and S/S groups. ELISA analysis showed that the serum level of IL-6 in the MIA/P20 group was higher than that in the S/P20 group (MIA: 324 pg/mL vs S: 8 pg/mL) at 24 h (p < 0.05); the level decreased in both groups at 48 h. Moreover, it was no longer significantly different between the two groups at 24 h (Figure 1D). Thus, MIA exposure in late pregnancy at 12.5, 14.5, and 16.5 dpc can cause remarkable alterations in the serum levels of IL-6, IL-17, and Ifn-γ mRNA after the second attack of poly(I:C).
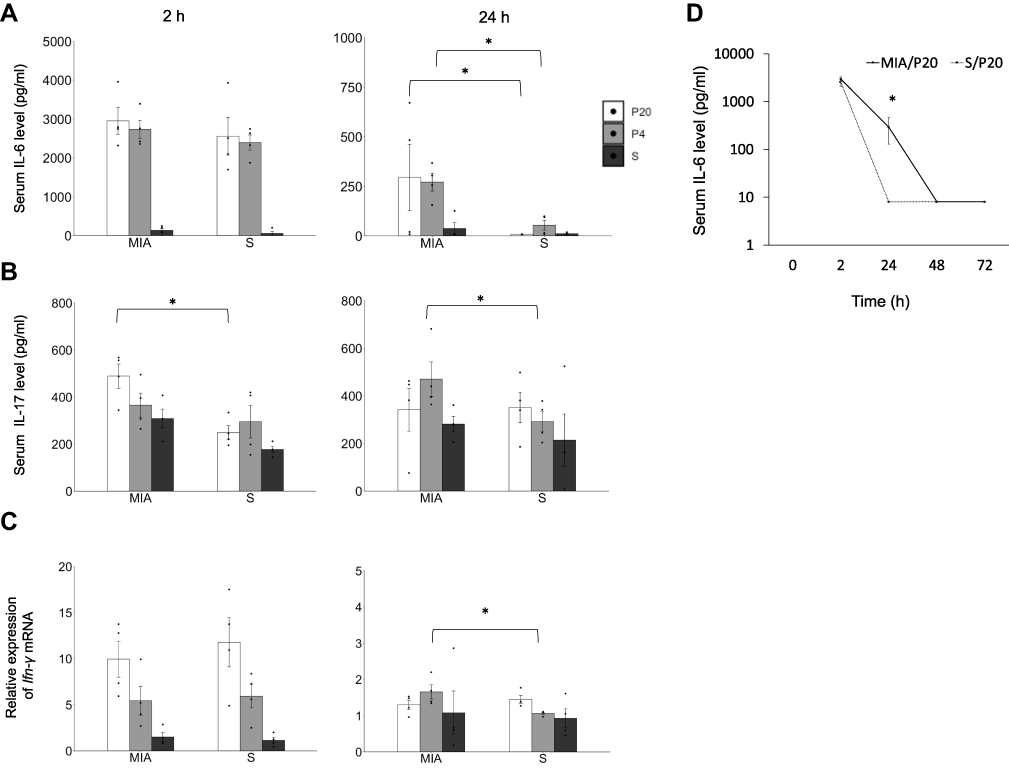 |
Figure 1 Immune overreaction induced by a second attack of poly(I:C) in offspring. (A) Serum IL-6 level at 2 and 24 h. (B) Serum IL-17 level at 2 and 24 h. (C) Relative expression level of Ifn-γ mRNA at 2 and 24 h. (D) Transition of IL-6 at 2, 24, 48, and 72 h in MIA/P20 and S/P20. *p < 0.05, Mann–Whitney U-test. Data are represented by mean ± SE. Number of dams = 4 in each group, number of pups = 8–9 in each group. P20, poly(I:C) 20 mg/kg; P4, poly(I:C) 4 mg/kg; S, saline. |
Histopathological Examination of Organs
To evaluate the effects of postnatal attack with poly(I:C) on various organs, histopathological analysis was performed using HE staining. Liver necrosis and degeneration were observed in the MIA/P20, MIA/P4, and S/P20 groups at 24 h (Figure 2A and B). The incidence rate of liver necrosis and degeneration at 24 h after the second attack was 100% in the MIA/P20 group, 50 ±20.4% in the MIA/P4 group, and 25 ± 14.4% in the S/P20 group (MIA/P20 vs S/P20; p < 0.05, MIA/P4 vs S/P4: p < 0.05) (Figure 2C). In the other groups, liver necrosis and degeneration were not observed at 2 and 24 h after the second attack. In other organs such as the brain, heart, kidney, lung, and colon, histopathological changes, including inflammatory injury such as immune cell infiltration, degeneration, or necrosis, were not detected in both the MIA and saline groups (Figure 2A). The activation of pro-apoptotic cleaved caspase-3 expression was detected around the necrosis area in the MIA/P20 and S/P20 groups (Figure 2D). These results suggest that the acute liver necrosis caused by the injection of high-dose poly(I:C) in the postnatal period was associated with prenatal exposure to MIA.
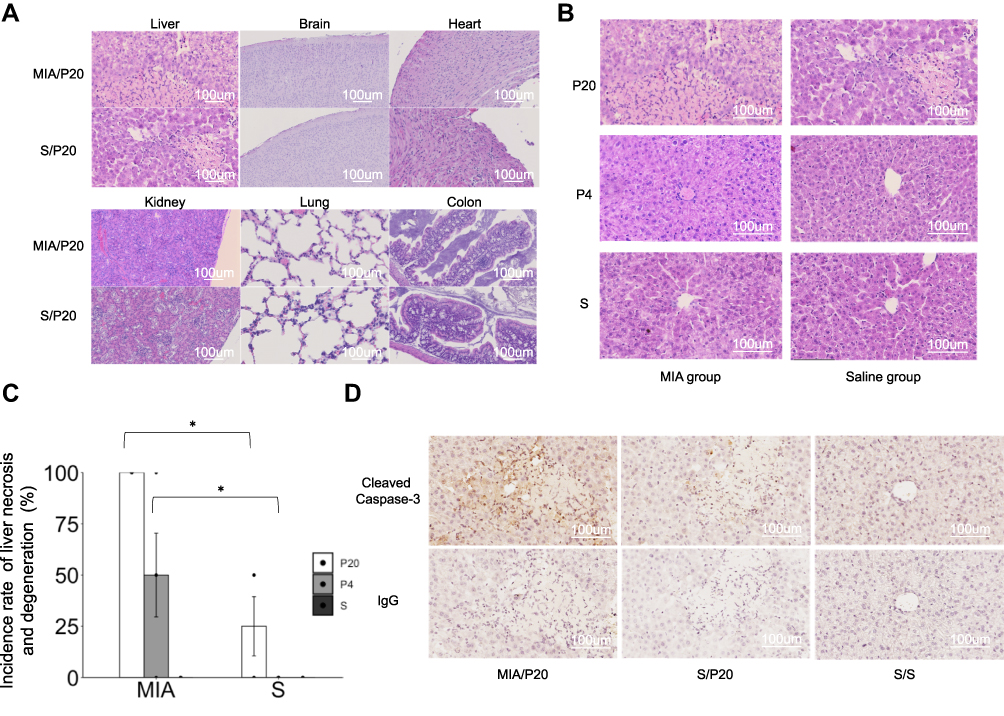 |
Figure 2 Histological examination of the organs. (A) HE-stained organs of MIA/P20 and S/P20 at 24 h. (B) HE-stained liver at 24 h in the MIA and saline groups. (C) Frequency of liver necrosis at 24 h in MIA and saline groups. (D) Immunostaining of cleaved caspase-3 in the liver at 24 h in MIA/P20, S/P20, and S/S. *p < 0.05, Mann–Whitney U-test. Data are represented by mean ± SE. Number of dams = 4 in each group; number of pups = 8–9 in each group. P20, poly(I:C) 20 mg/kg; P4, poly(I:C) 4 mg/kg; S, saline. |
Activation of the TLR-3-NF-κB Pathway Response in the Liver
The response of the TLR-3-NF-kB pathway in the liver was evaluated. The levels of Tlr-3 and NF-κb p-65 mRNA increased at 2 h after poly(I:C) administration in both the MIA and saline groups. The levels of Tlr-3 and NF-κb p-65 mRNA were not different between the MIA and saline groups at 2 and 24 h (Figure 3A), whereas the ratio of p-IkBα/IkBα in the liver at 2 h showed an increasing tendency in the MIA/P20 group compared with that in the S/P20 group (p < 0.05) (Figure 3B). Moreover, the number of NF-κB p65-positive nuclei at 2 h significantly increased in the MIA/P20 group compared with that in the S/P20 group (p < 0.05) (Figure 3C and D, Suppl Figure S3). These findings indicated that the TLR-3-NF-κB pathway was activated by the second attack of poly(I:C) postnatally in the MIA group.
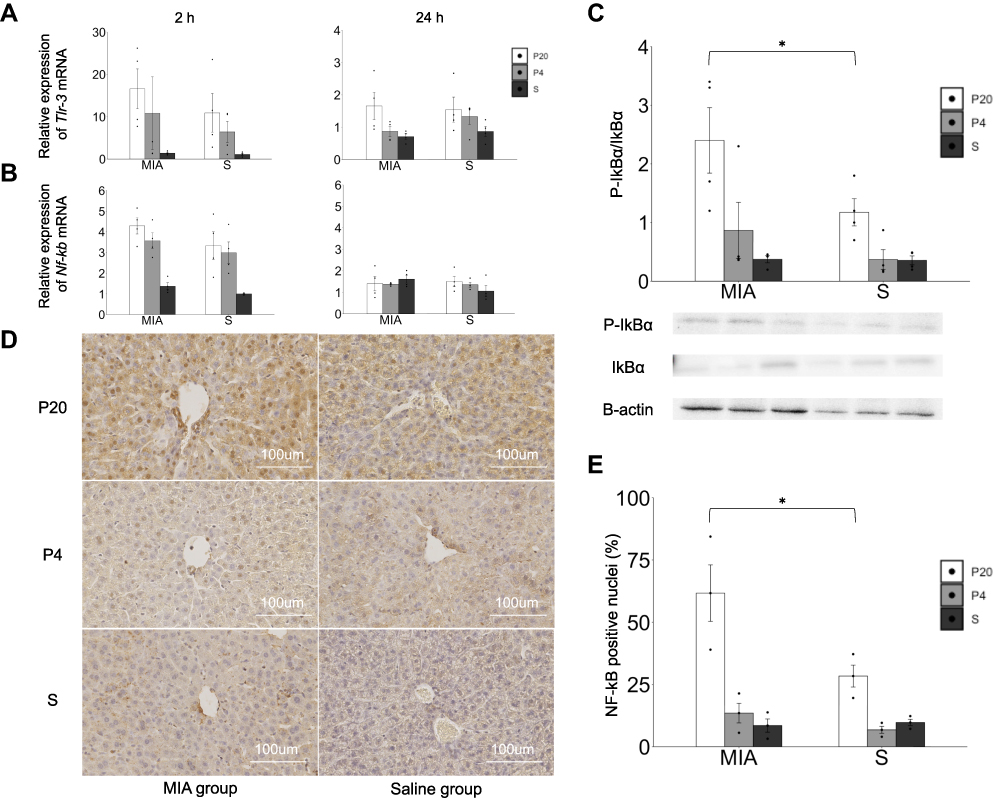 |
Figure 3 Activation of the TLR-3-NF-kB p65 pathway response in the liver. (A) Tlr-3 mRNA expression levels in the liver at 2 and 24 h. (B) Nf-κb p65 mRNA expression levels in the liver at 2 and 24 h. (C) P-IkBα/IkBα protein expression in the liver at 2 h. (D) Immunostaining of NF-κB p65 in the liver at 2 h. (E) Ratio of the NF-κB-positive nuclei in each area. *p < 0.05, Mann–Whitney U-test. Data are represented by mean ± SE. Number of dams = 4 in each group, number of pups = 8–9 in each group. P20, poly(I:C) 20 mg/kg; P4, poly(I:C) 4 mg/kg; S, saline. |
Response Failure of UPR Following a Second Attack in Offspring
The expressions of Bip, Xbp-1, Atf4, and Atf6 mRNAs in the liver were examined to clarify the involvement of UPR in the pathological changes in the liver. The levels of Bip mRNA in the liver in the MIA/P20 group at 2 h (p < 0.05) and 24 h (p < 0.05) were significantly lower than those in the S/P20 group (Figure 4A). Bip mRNA in the MIA/P4 group at 2h was significantly lower than that in the S/P4 group (p < 0.05), whereas there was no significant difference at 24 h between the groups. The level of Atf6 mRNA at 2 h in the MIA/P20 group was significantly lower than that in the S/P20 group (p < 0.05), whereas there were no significant difference at 24 h between the groups (Figure 4C). There was no statistically significant difference in the levels of Atf4, Xbp-1t, Xbp-1s, and Xbp1-s/t mRNA between the MIA and saline groups (Figure 4B, D–F). In the S/P20 and S/P4 groups, their levels were increased following poly(I:C) administration compared with that in the S/S group, which exhibited the normal response of UPR to the inflammatory stress.
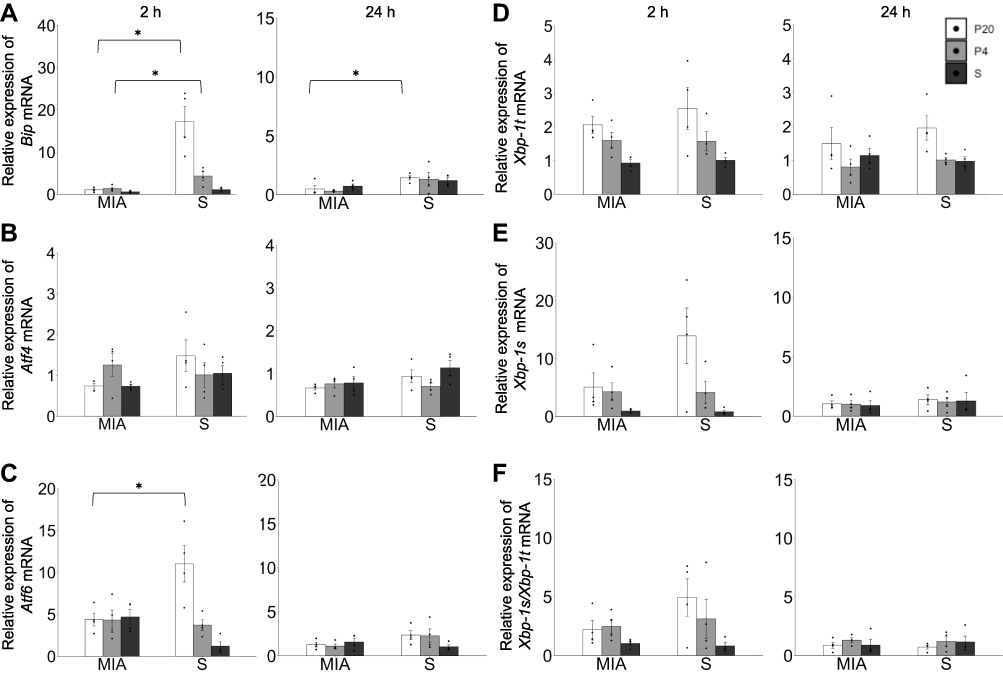 |
Figure 4 Response failure of unfolded protein response in the liver by a second attack in offspring. (A) Relative expression level of Bip mRNA at 2 and 24 h. (B) Relative expression level of Atf4 mRNA at 2 and 24 h. (C) Relative expression level of Atf6 mRNA at 2 and 24 h. (D) Relative expression of Xbp-1t mRNA at 2 and 24 h. (E) Relative expression level of Xbp-1s mRNA at 2 and 24 h. (F) Ratio of expression level of Xbp-1s/Xbp1t mRNA at 2 and 24 h. *p < 0.05, Mann–Whitney U-test. Data are represented by mean ± SE. Number of dams = 4 in each group, number of pups = 8–9 in each group. P20, poly(I:C) 20 mg/kg; P4, poly(I:C) 4 mg/kg; S, saline. |
Discussion
In the present study, offspring exposed to MIA prenatally exhibited immune overreaction induced by the second attack of poly(I:C), which is defined as a pseudovirus infection after birth. This suggests that MIA is an aggravating factor of viral infection in children. A previous in vitro study indicated that the immune overreaction in offspring exposed to MIA was associated with an increase in the number of macrophages of their dysfunction, but the details of the mechanism are yet to be clarified.3,10 This study showed that intrauterine exposure to maternal inflammation increased the susceptibility to inflammation after birth in offspring. The present histological examination revealed that liver necrosis was more frequently induced by postnatal administration of the maximum dose of poly(I:C) in the MIA group. Apoptosis-related protein caspase-3 was expressed in hepatocytes around the lesion of the liver necrosis in the MIA/P20 and S/P20 groups. In contrast, remarkable pathological changes such as necrosis or inflammatory cell infiltration were not identified in the brain, heart, kidney, lung, and colon but were identified in the liver. Although the reason why the liver was more affected than the other organs remains unclear, it may be associated with the large number of innate immune cells present and rich blood flow in the liver.11
Poly(I:C) is known to activate the TLR-3 signaling pathway. NF-κB p-65 has been identified as a major transcription factor that is released following the stimulation of TLR-3 or cytokines such as IL-6, IL-17, and IFN-γ. IKBα regulates NF-κB activation.23 Phosphorylated IkBα activates NF-κB, which is then translocated into the nuclei. In the present study, the activation of NF-κB in the liver resulted in increased activation of inflammatory signaling pathways, which possibly caused immune overreaction by TLR-3 stimulation and cytokine release. The occurrence of liver necrosis in the offspring exposed to MIA suggests that prenatal exposure to inflammation was associated with the overactivation of inflammatory signaling in the postnatal period.
We detected alterations in Bip and Atf6 expression, which are the main regulator and one of the major transcription factors of UPR, respectively. In the S/P20 and S/P4 groups, the levels of these major components of UPR are physiologically increased by inflammatory stimuli to maintain intracellular homeostasis by processing excess abnormal transcripts that cause ER stress.24,25 A previous study indicated Bip knock-out mouse-induced cell death and the activation of inflammatory signaling pathway in the liver.26 In MIA/P20 and MIA/P4, Bip and Atf6 expressions were low compared with those in S/P20 and S/P4. Thus, Bip and Atf6-dependent UPR did not respond properly to inflammation owing to prenatal MIA exposure. Xbp-1, another player of UPR, also showed a tendency of decreased response to the second attack in MIA/P20, although no statistical differences were observed in the expression levels among the experimental groups. However, the response of Atf4 against the second attack was not detected in both groups. These results indicated that MIA led to the response failure of UPR and uneven disruption of UPR signaling pathways in MIA-exposed offspring. Bip has two important roles in protecting cell survival. First, it is a main regulator of UPR. Second, it can operate as a typical heat shock protein 70 (Hsp70) chaperone-substrate that is involved in the recruitment of misfolded proteins.27 The reduction of Bip induces cell death because of the accumulation of unfolding proteins. Excessive or insufficient UPR leads to cell dysfunction and apoptosis and the development of illnesses such as liver diseases, neurodegenerative disease, cardiovascular disease, and metabolic disease.28 The mouse model of abnormal behavior caused MIA exposure in prenatally increased proinflammatory cytokine mRNA expression in the hippocampus and indicated an association with the peroxisome proliferator-activated receptor gamma (PPARγ)-mediated neuroprotective mechanism.29–31 Although pathological changes were not detected in the brain relating to the abnormal behavior, congenital alteration of susceptibility to inflammatory stimuli in the brain might have been revealed by modulating the timing, duration, and severity of the second attack. The inflammatory stimulation of poly(I:C) is also stimulated by all of the pathways of UPR,32 which is supported by the findings of this study on the increased expression of Bip, Atf6, Xbp-1s, and Xbp-1t in S/P20 and S/P4 compared to that in S/S (Figure 4). The expression of Atf4 is known to be lower in the liver compared to other organs.33,34 In this study, Atf6 or Xbp-1 showed an increase in UPR in the liver of the saline group, whereas Atf4 did not increase in both groups. These results suggested that the uneven distribution of UPR might be associated with the specificity of the organs or tissues. The present study suggested that the failure of UPR caused by the insufficient expression of Bip and Atf6 resulted in cell death in the liver of offspring in MIA/P20. UPR is triggered by inflammatory cytokines due to viral or bacterial infection. The activation of the UPR signaling pathway increases the levels of transcription factors such as NF-κB and regulates inflammation stimuli.24,25 Previous studies reported that the deletion of UPR-related proteins such as Bip, Atf6, and Xbp-1 resulted in dysregulation of inflammation and induced the enhancement of apoptosis-related molecules and immune response.26,35,36 Failure of UPR and increased and prolonged inflammation was observed in the MIA group. UPR defects caused NF-κB overexpression and immune overreaction. These results indicated that MIA exposure in the prenatal period caused susceptibility to inflammation after birth in offspring. Furthermore, what is the mechanism by which the levels of Bip and UPR sensor proteins were reduced in the MIA/P20 group? Epigenetics such as DNA methylation, histone modification, and non-coding RNAs often result in altered gene regulation without changing the DNA sequence. These alterations are associated with gene expression by changing the chromatin architecture, which can cause various diseases in offspring. A previous study reported that IL-6 and IL-17 activated DNA methyltransferase 1 and inhibited histone deacetylase, respectively.37,38 Thus, MIA induced by inflammation possibly caused epigenetic changes such as DNA methylation, histone modification, and non-coding RNAs in offspring in utero. From this perspective, the present study indicates that immune overreaction and UPR defects in the postnatal period may be associated with prenatal epigenetic modifications of Bip and UPR sensor protein promoters by activating the TLR-3 signaling pathway. Thus, further studies are needed to address this issue.
Finally, we propose that MIA might be one of the risk factors in patients with immune overreaction such as in the case of severe infection or autoinflammatory disease. The present findings will be helpful in detecting organ failure determinants in each disease. Overall, the findings of this study may contribute to the clinical treatment and prediction of pathological conditions.
Conclusion
The study findings indicated that MIA-exposed offspring acquire hypersensitivity constitution, UPR defects, and acute liver necrosis.
Acknowledgments
The authors would like to thank Mrs Tomoko Yasuda for her help in histological preparations, Mrs. Mayumi Mitani for technical support, and Dr Yasuo Iida for statistical analysis. The measurement of multiplex cytokine bead array assay was performed at the Regenerative Medicine Center of Kanazawa Medical University. This work was supported by JSPS KAKENHI Grants (Numbers 15K09713, 16H05364, 18K11659) and by Assistkakenn from Kanazawa Medical University (K2020-20).
Author Contributions
All authors made significant contributions to the work, especially in the conception, study design, execution, acquisition of data, analysis, and interpretation or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen T, Liu H-X, Yan H-Y, Wu D-M, Ping J. Developmental origins of inflammatory and immune diseases. Mol Hum Reprod. 2016;22(8):858–865. doi:10.1093/molehr/gaw036
2. Rose DR, Careaga M, Van de Water J, McAllister K, Bauman MD, Ashwood P. Long-term altered immune responses following fetal priming in a non-human primate model of maternal immune activation. Brain Behav Immun. 2017;63:60–70. doi:10.1016/j.bbi.2016.11.020
3. Onore CE, Schwartzer JJ, Careaga M, Berman RF, Ashwood P. Maternal immune activation leads to activated inflammatory macrophages in offspring. Brain Behav Immun. 2014;38:220–226. doi:10.1016/j.bbi.2014.02.007
4. Gaillard R, Rifas-Shiman SL, Perng W, Oken E, Gillman MW. Maternal inflammation during pregnancy and childhood adiposity. Obesity. 2016;24(6):1320–1327. doi:10.1002/oby.21484
5. Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A. 2012;109(31):12776–12781. doi:10.1073/pnas.1202556109
6. Deng Y, Song L, Nie X, Shou W, Li X. Prenatal inflammation exposure-programmed cardiovascular diseases and potential prevention. Pharmacol Ther. 2018;190:159–172. doi:10.1016/j.pharmthera.2018.05.009
7. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. doi:10.1523/JNEUROSCI.2178-07.2007
8. Tsukada T, Shimada H, Sakata-Haga H, Iizuka H, Hatta T. Molecular mechanisms underlying the models of neurodevelopmental disorders in maternal immune activation relevant to the placenta. Congenit Anom (Kyoto). 2019;59(3):81–87. doi:10.1111/cga.12323
9. Tsukada T, Simamura E, Shimada H, et al. The suppression of maternal-fetal leukemia inhibitory factor signal relay pathway by maternal immune activation impairs brain development in mice. PLoS One. 2015;10(6):e0129011. doi:10.1371/journal.pone.0129011
10. Surriga O, Ortega A, Jadeja V, Bellafronte A, Lasala N, Zhou H. Altered hepatic inflammatory response in the offspring following prenatal LPS exposure. Immunol Lett. 2009;123(1):88–95. doi:10.1016/j.imlet.2009.02.009
11. Hodyl NA, Krivanek KM, Lawrence E, Clifton VL, Hodgson DM. Prenatal exposure to a pro-inflammatory stimulus causes delays in the development of the innate immune response to LPS in the offspring. J Neuroimmunol. 2007;190(1–2):61–71. doi:10.1016/j.jneuroim.2007.07.021
12. DISEASES COI. Recommendations for Prevention and Control of Influenza in Children, 2018–2019. Pediatrics2018;142:24. doi:10.1542/peds.2018-2367
13. Ter HNM, Oswald M, Jeyaratnam J, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636–1644. doi:10.1136/annrheumdis-2015-207546
14. Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119(2):188–194. doi:10.1016/j.clim.2006.01.005
15. Kawashima H, Morichi S, Okumara A, Nakagawa S, Morishima T. Japan csgoi-aei. National survey of pandemic influenza A (H1N1) 2009-associated encephalopathy in Japanese children. J Med Virol. 2012;84(8):1151–1156. doi:10.1002/jmv.23317
16. Mori S-I, Nagashima M, Sasaki Y, et al. A novel amino acid substitution at the receptor-binding site on the hemagglutinin of H3N2 influenza A viruses isolated from 6 cases with acute encephalopathy during the 1997–1998 season in Tokyo. Arch Virol. 1999;144(1):147–155. doi:10.1007/s007050050491
17. Kim SR, Kim HJ, Kim DI, et al. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics. 2015;5(12):1343–1362. doi:10.7150/thno.11685
18. Fang P, Xiang L, Huang S, et al. IRE1α-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma.. Oncol Lett. 2018;16(4):4729–4736. doi:10.3892/ol.2018.9176
19. Sprooten J, Garg AD. Type I interferons and endoplasmic reticulum stress in health and disease. Int Rev Cell Mol Biol. 2020;350:63–118. doi:10.1016/bs.ircmb.2019.10.004
20. Soeda J, Cordero P, Li J, et al. Hepatic rhythmicity of endoplasmic reticulum stress is disrupted in perinatal and adult mice models of high-fat diet-induced obesity. Int J Food Sci Nutr. 2017;68(4):455–466. doi:10.1080/09637486.2016.1261086
21. So JS. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol Cells. 2018;41(8):705–716. doi:10.14348/molcells.2018.0241
22. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. doi:10.1038/s41598-017-17204-5
23. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2. doi:10.1038/sigtrans.2017.23
24. Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69(2):169–181. doi:10.1016/j.molcel.2017.06.017
25. Frakes AE, Dillin A. The UPR. Mol Cell. 2017;66(6):761–771. doi:10.1016/j.molcel.2017.05.031
26. Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54(1):229–239. doi:10.1002/hep.24368
27. Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26(11):1053–1062. doi:10.1038/s41594-019-0324-9
28. Kaneko M, Imaizumi K, Saito A, et al. ER Stress and Disease: toward Prevention and Treatment. Biol Pharm Bull. 2017;40(9):1337–1343. doi:10.1248/bpb.b17-00342
29. Zhao Q, Wang Q, Wang J, et al. Maternal immune activation-induced PPARγ-dependent dysfunction of microglia associated with neurogenic impairment and aberrant postnatal behaviors in offspring. Neurobiol Dis. 2019;125:1–13. doi:10.1016/j.nbd.2019.01.005
30. Han Y, Zhang L, Wang Q, et al. Minocycline inhibits microglial activation and alleviates depressive-like behaviors in male adolescent mice subjected to maternal separation. Psychoneuroendocrinology. 2019;107:37–45. doi:10.1016/j.psyneuen.2019.04.021
31. Han Y, Wang J, Zhao Q, et al. Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav Immun. 2020;87:568–578. doi:10.1016/j.bbi.2020.02.002
32. Lenna S, Chrobak I, Farina GA, et al. HLA-B35 and dsRNA induce endothelin-1 via activation of ATF4 in human microvascular endothelial cells. PLoS One. 2013;8(2):e56123. doi:10.1371/journal.pone.0056123
33. Wu H, Ng BS, Thibault G. Endoplasmic reticulum stress response in yeast and humans. Biosci Rep. 2014;34(4). doi:10.1042/BSR20140058
34. Hussain SG, Ramaiah KV. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun. 2007;355(2):365–370. doi:10.1016/j.bbrc.2007.01.156
35. Olivares S, Henkel AS. Hepatic Xbp1 Gene Deletion Promotes Endoplasmic Reticulum Stress-induced Liver Injury and Apoptosis. J Biol Chem. 2015;290(50):30142–30151. doi:10.1074/jbc.M115.676239
36. Yamamoto K, Takahara K, Oyadomari S, et al. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21(17):2975–2986. doi:10.1091/mbc.E09-02-0133
37. Hodge DR, Cho E, Copeland TD, et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics. 2007;4(6):387–398.
38. Zijlstra GJ, Ten Hacken NH, Hoffmann RF, van Oosterhout AJ, Heijink IH. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur Respir J. 2012;39(2):439–445. doi:10.1183/09031936.00017911
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.