Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Exploring the Hereditary Nature of Migraine
Authors Bron C, Sutherland HG, Griffiths LR
Received 26 January 2021
Accepted for publication 7 April 2021
Published 22 April 2021 Volume 2021:17 Pages 1183—1194
DOI https://doi.org/10.2147/NDT.S282562
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Charlene Bron, Heidi G Sutherland, Lyn R Griffiths
Queensland University of Technology (QUT), Centre for Genomics and Personalised Health, Genomics Research Centre, School of Biomedical Sciences, Institute of Health and Biomedical Innovation, Queensland, 4059, Australia
Correspondence: Lyn R Griffiths Tel +617 3138 6102
Fax +617 3138 6039
Email [email protected]
Abstract: Migraine is a common neurological disorder which affects 15– 20% of the population; it has a high socioeconomic impact through treatment and loss of productivity. Current forms of diagnosis are primarily clinical and can be difficult owing to comorbidity and symptom overlap with other neurological disorders. As such, there is a need for better diagnostic tools in the form of genetic testing. Migraine is a complex disorder, encompassing various subtypes, and has a large genetic component. Genetic studies conducted on rare monogenic subtypes, including familial hemiplegic migraine, have led to insights into its pathogenesis via identification of causal mutations in three genes (CACNA1A, ATP1A2 and SCN1A) that are involved in transport of ions at synapses and glutamatergic transmission. Study of familial migraine with aura pedigrees has also revealed other causal genes for monogenic forms of migraine. With respect to the more common polygenic form of migraine, large genome-wide association studies have increased our understanding of the genes, pathways and mechanisms involved in susceptibility, which are largely involved in neuronal and vascular functions. Given the preponderance of female migraineurs (3:1), there is evidence to suggest that hormonal or X-linked components can also contribute to migraine, and the role of genetic variants in mitochondrial DNA in migraine has been another avenue of exploration. Epigenetic studies of migraine have shown links between hormonal variation and alterations in DNA methylation and gene expression. While there is an abundance of preliminary studies identifying many potentially causative migraine genes and pathways, more comprehensive genomic and functional analysis to better understand mechanisms may aid in better diagnostic and treatment outcomes.
Keywords: migraine, migraine without aura, migraine with aura, familial hemiplegic migraine, X-linked, mitochondrial variants, epigenetics
A Letter to the Editor has been published for this article.
Introduction
Migraine is a common neurovascular disorder which affects 15–20% of the population.1–5 Given the prevalence of the disorder, migraine has a large socioeconomic impact through treatment costs and loss of productivity, estimated at USD$19.6 billion, €27 billion and AUD$35.7 billion per annum in America, Europe and Australia, respectively.4–6 Migraine is characterised by severe headache, typically including unilateral throbbing, nausea, vomiting, photophobia and/or phonophobia.1–5,7,8 There are two common subtypes of migraine, which are determined by the presence or absence of aura.9 Migraine without aura (MO) is the most common, affecting around 70% of migraineurs, while approximately 30% suffer migraine with aura (MA).10,11 In MA, the migraine is accompanied by aura symptoms, largely manifested as visual disturbances, but can also include sensory and speech-related symptoms, muscle weakness and motor disturbances in some cases.7,12
The Hereditary Nature of Migraine
It has long been established that there is a significant genetic component to migraine.13,14 Studies have in addition shown an increased familial risk for common migraine, which is considered to be polygenic, with heritability estimates of 34–64%.8,12,15 Investigations into migraine families and twin studies have shown that a first-degree relative of an MO proband is twice as likely to have MO and 1.4 times as likely to have MA.12 However, a first-degree relative of an MA proband is four times more likely to have MA, but has no increased risk of MO.12 Furthermore, there are some rare subtypes of migraine which are considered to be monogenic, predominantly caused by autosomal dominant mutations in genes encoding ion channels. Owing to the complex nature of common migraine, determining all genes and their interaction with environmental factors that contribute to the disorder remains a challenge.16 Initial studies focused on genes involved in neuronal, vascular and hormonal functions using candidate gene association studies (CGAS), testing for differences in frequency of genetic polymorphisms across particular genes between migraine cases and controls to determine whether they influence susceptibility to the disorder.17–20 An extensive study of 155 ion transporter genes in 841 migraineurs and 884 controls21 identified 21 promising genes associated with migraine; however, these results failed to be replicated in a larger cohort of thousands, suggesting that the majority were potentially false positives.22 This could be for several reasons: CGAS tend to have modest sample sizes which are underpowered to detect genetic variants that have small effects; they also use a hypothesis-driven set of genes and polymorphisms for testing,23,24 and while the genes selected may have a likely role in the disease pathway, they may not present a genetic risk, or only be a cofactor of the cause. Many of these issues have been tackled through advances in technology which allow for fast and cost-effective genotyping. Genome-wide association studies (GWAS) use microarray platforms to test up to millions of single-nucleotide polymorphisms (SNPs) in one experiment. As GWAS are not hypothesis driven, they can identify novel genes and pathways, and with large sample sizes and statistical correction for multiple testing are less likely to produce the false positives of CGAS. The largest GWAS performed to date included 59,674 migraineurs and 316,078 healthy controls to identify 44 SNPs mapped to 38 loci associated with common polygenic migraine.25,26 This study largely supported previous GWAS performed by Anttila et al25 and Chasman et al27 identifying neuronal variants (Figure 1). Freilinger et al28 added a number of genes associated with vascular pathways and, finally, the Gormley study introduced a number of genes related to ion homeostasis.26
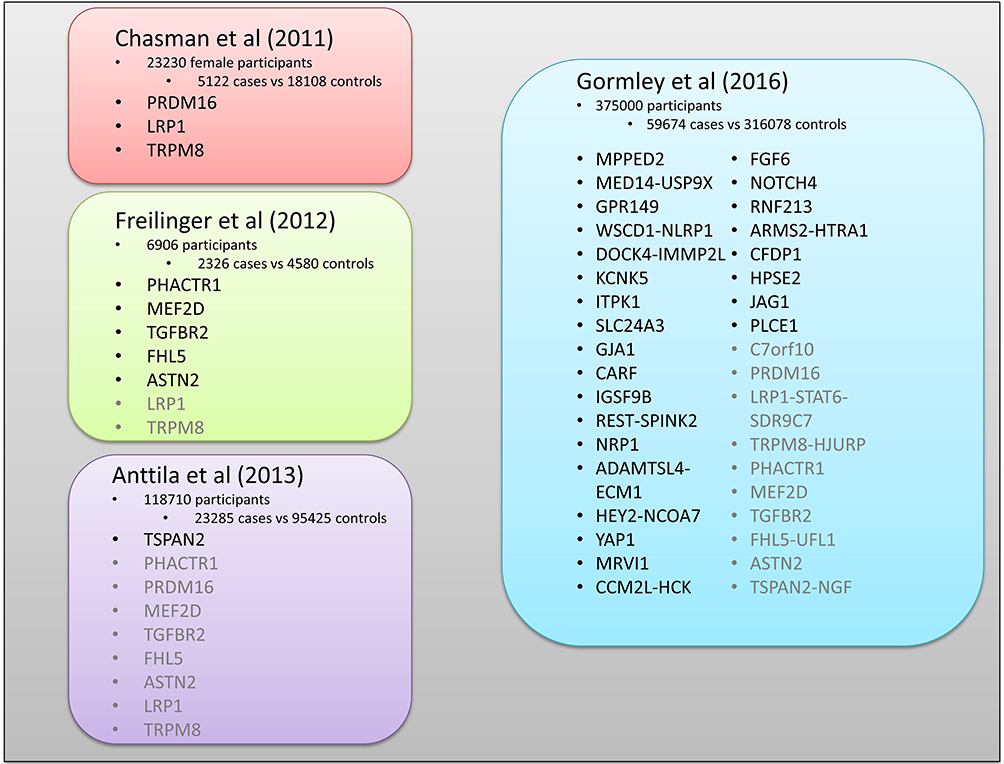 |
Figure 1 GWAS-identified genes associated with migraine. The figure shows the major studies identifying the genes associated with polygenic forms of migraine, which largely fall into either neuronal or vascular function. Genes displayed in black text were new discoveries of each study, while the grey text highlights those that have been replicated for existing studies.25–28 |
Monogenic Forms of Migraine
An alternative approach used to study the complex genetic pathways of this neurological disorder is to examine monogenic subtypes of migraine,12,27 and there have been a number of multigenerational studies performed on MA pedigrees29 and unrelated hemiplegic migraine probands.30,31
Familial Migraine with Aura
A study of particular families which show strong inheritance of MA showed that a frameshift mutation of the KCNK18/TRESK gene segregated with migraine diagnosis, suggesting that it was causal.29 This was further corroborated in a study by Pettingill et al, which demonstrated through the use of CRISPR-Cas9 that frameshift mutations of TRESK are associated with migraine phenotypes.32 The TRESK gene is part of a two-pore domain family of potassium channels that regulate cellular excitability, and are themselves regulated by calcium-dependent phosphatase calcineurin.29,33 A study by Rainero et al has also shown that mutations in TRESK can be found in non-related probands in both MO and MA populations.34 In addition, mouse models have shown that knocking out TRESK modifies nociceptive afferentation.35 While some variants have been found in this gene that are causal of MA, other studies have shown loss of function variants in both migraine and control populations.33,36 This complex relationship may be explained by the findings that some TRESK frameshift mutations produce altered protein fragments which can affect the function of other ion channels.37
Rasmussen et al used an approach where they first identified migraine-associated modules through analysis of RNAseq from brain and vascular tissues, and then performed analysis of the genes from these modules in whole genome sequencing (WGS) data from migraine families with Mendelian-like segregating patterns. They found that rare variants in genes involved in five pathways were over-represented: thyrotropin-releasing hormone receptor signalling, oxytocin receptor-mediated signalling, Alzheimer’s disease amyloid secretase, serotonin receptor signalling, and heterotrimeric G-protein signalling pathway-Gq alpha and Go alpha-mediated pathways, and furthermore identified the ATXN1, FAM153B and CACNA1B genes as being the most frequently mutated among migraine families.38
Hemiplegic Migraine
Hemiplegic migraine (HM) is a rare and severe MA subtype, with patients also having symptoms of hemiparalysis accompanying the migraine or motor disturbances.12 A typical aura attack can last anywhere from 10 minutes to a few hours, with paraesthesia and speech disturbances.39 In addition to this, 66% of HM patients report that they suffer from normal MO (without paralysis) on occasion.40 There is variability within cases; some with symptoms that last days or weeks, and some with increased severity that can result in coma or seizures.41–43 HM has both sporadic (SHM) and familial forms (FHM), with the latter typically inherited in an autosomal dominant fashion with a 70–90% penetrance.44 To date, three main genes have been implicated in the cause of FHM, all of which encode for ion channels (Table 1). FHM1 is caused by mutations in the CACNA1A gene on chromosome 19p13, which encodes for the α1A subunit of P/Q-type voltage-gated calcium channels.45 These particular channels are responsible for the control of calcium in the neuron as a response to membrane depolarisation.46 The second gene associated with FHM is the ATP1A2 gene (FHM2) on chromosome 1q23, which encodes for a glial sodium/potassium pump.47 The third FHM gene is SCN1A (FHM3) located on chromosome 2q24, which encodes a neuronal voltage-gated sodium channel.48 The symptoms of the three FHM subtypes are similar, although the functional effect and mechanism of FHM-causing mutations for each gene vary (Table 1). A fourth causal gene for FHM, the proline-rich transmembrane protein 2 (PRRT2), was suggested by Riant et al.49 PRRT2 is located on chromosome 16p11.2 and encodes for a type II transmembrane protein which interacts with SNARE complexes to regulate voltage-gated calcium channels in glutamatergic neurons.50–53 Various studies have suggested that PRRT2 could be considered a fourth FHM gene,49,54–56 although there is some controversy on that front, and Riant et al suggested that PRRT2 may only act as a modifier because of complex heterogeneity and pleiotropy of phenotypes.49 Suzuki et al have also shown that homozygous mutations in the SLC4A4 gene can cause syndromic HM, but as yet it has not been included as a known FHM gene.57
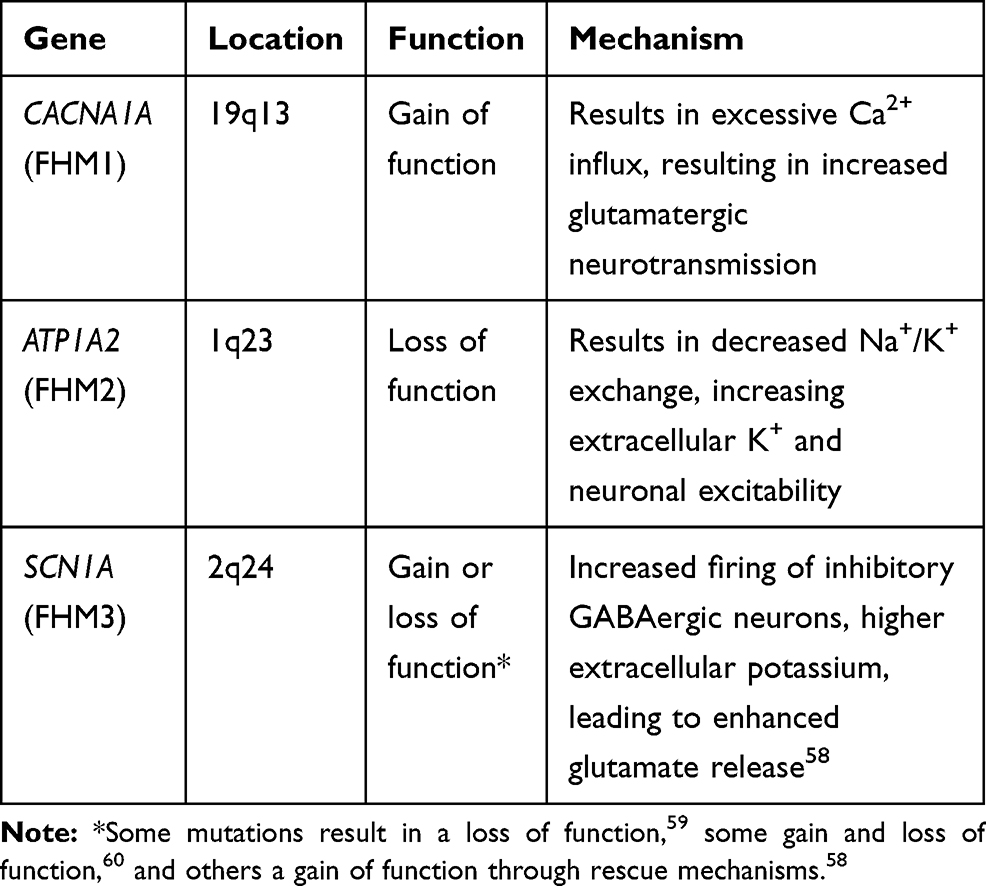 |
Table 1 Known FHM Causal Genes and Functional Effects of Mutations Identified in FHM |
Aside from their role as the three FHM genes, these genes have also been implicated in many other disorders. Specific types of mutations in CACNA1A have been linked to spinocerebellar ataxia type 6, episodic ataxia, progressive cerebellar ataxia and epilepsy; ATP1A2 has been linked with cerebellar ataxia and epilepsy, but also confusion, coma and mental retardation; and finally, SCN1A is associated with more severe epilepsy syndromes, such as myoclonic epilepsy in infants and epilepsy with febrile seizures.2 Currently, migraine and FHM are diagnosed through extensive patient interview, family history and pedigrees.7,61 Patients are diagnosed based on the specifications of the International Classification of Headache Disorders (ICHD), which outlines the age of disorder onset, the severity and frequency of the attacks, and the presence of aura symptoms.7 However, this form of diagnosis is complicated owing to the variability between patients and symptoms.41–43 This has also contributed to the difficulty in identifying genes associated with classical migraine for diagnosis. The common genetic mechanisms behind these disorders often lead to clinical presentation with similar symptoms, making molecular diagnosis through limited gene sequencing difficult. In recent years, the advent of next generation sequencing (NGS) technologies has allowed for better screening for neurological diseases, which has, in turn, increased diagnostic rates. NGS allows for millions of small fragments of DNA to be sequenced at once, allowing for entire gene sets or even the whole genome to be investigated, rather than targeted exons or regions.15,62,63 Several research groups have reported increased diagnostic rates for several neurological diseases, through the use of whole exome sequencing (WES)64–66 and targeted gene panels.67–69 WES analysis has shown that not all suspected HM cases have exonic mutations in the known FHM genes,30,31 or genes that cause overlapping disorders, which suggests that additional genes or other factors as yet unknown are causative of HM,12 some of which may be revealed through WES or whole genome sequencing and structural analysis.
Migraine and Circadian Rhythms
Environmental factors such as sleep and stress have been cited as leading triggers for migraine attacks.70,71 This can be attributed to the fact that there are several hormones and neurotransmitters that play a role in both sleep disruption and migraine;71 there is evidence to show that dopamine, serotonin, gamma-aminobutyric acid and gonadotropin-releasing hormone changes are involved in both disorders.71–73 There are also studies showing that migraine and sleep disorders tend to co-segregate within family pedigrees.74,75 Brennan et al showed that a mutation in CSNK1D, which has been shown to cause familial advanced sleep phase syndrome, was found in two large independent migraine pedigrees. The study also demonstrated that mice with a transgene CKIδ-T44A mutation display increased sensitisation to nitroglycerin infusions, particularly in female mice. In addition, they showed that the CKIδ-T44A mice have reduced thresholds for cortical spreading depression (CSD) compared to the wild type.74
Farahani et al have also shown a link between the circadian clock and migraine; genotyping of 200 migraineurs and 200 healthy controls showed that a variant in the RAR-related orphan receptor (RORA), which regulates both inflammation and circadian rhythm, was associated with circadian rhythm dysfunction and migraine.76 Further research by Baksa et al suggests that variations in the circadian locomotor output cycles kaput (CLOCK) gene may influence the risk of migraine in response to financial stress.77 In addition to circadian rhythm, Kim et al posited that various sleep disorders can be linked to migraine; subjects with insomnia, for example, have significantly higher prevalence of migraine compared to those without.78 Kilic et al similarly showed that insufficient glycogen-derived energy (or lack of sleep) can cause synaptic stress and lower the threshold for CSD, suggesting that this lack of synaptic energy may predispose subjects to migraine attack.79 Thus, genes related to sleep disorders may be a promising target for migraine diagnosis and treatment, although further investigations with larger cohorts are required.
Role of X-Chromosome Loci in Migraine
While studies have shown that there are genetic components to all forms of migraine, there is also evidence to suggest that gender plays a fundamental role in the disorder. Numerous migraine studies demonstrate a preponderance of migraine in females.12,16,27,80–82 A population study by Stewart et al83 showed that in 1992, 18% of women suffered from migraine compared to 6% of men, setting the incidence rate at 3:1. This unequal distribution of gender in migraine sufferers is common to both MO and MA,81,83–87 suggesting either a hormonal link to migraine or possibly an X-chromosomal link if there is a dominant inheritance pattern. The presence of X-linked migraine loci is supported by results from linkage analysis by Nyholt et al81,85,88 in several studies showing localisation of migraine susceptibility to the X chromosome. Further refinement through haplotype analysis suggested that a migraine susceptibility locus may be localised to the Xq24-28 region.89 In addition, a study by Maher et al90 using the genetically isolated Norfolk Island population showed evidence of migraine susceptibility loci at Xq27 and Xq12. However, genes and variants within these regions that may contribute to migraine risk are yet to be identified. Another study of a migraine family and the X chromosome conducted by Weiser et al82 suggests that a migraine susceptibility locus resides at Xp22. A case–control study performed by Quintas et al reported a link between migraine and the synapsin I (SYN1) gene, which is on the X chromosome.91 Utilising a cohort of 188 migraine patients and 286 healthy controls, and genotyping 119 SNPs in genes related to pathways involving synaptic vesicle molecular machinery or neurotransmission, the study determined that in females the rs5906437 GC genotype acts as a protective factor for migraine, while the rs5906435 G allele conferred increased migraine susceptibility,91 but the same effect was not observed in males. A larger sample size may be necessary to determine whether this is a true reflection of a gender-specific effect.91
Role of the Mitochondrial Genome in Migraine
Another avenue of exploration of migraine susceptibility is through the genetic analysis of mitochondrial DNA (mtDNA). As mtDNA does not undergo recombination and it is passed exclusively through the maternal line, it allows mutations in the mtDNA sequence to be tracked across multiple generations. Through the use of family studies, researchers have been able to find clear causal variants in various mitochondrial diseases.92,93 There are various justifications for an investigation into the mitochondrial genome in relation to migraine. The primary role of the mitochondria is to create ATP via the electron transport chain, providing all the energy required for cellular processes.94 Mitochondrial disorders result in decreased ATP production which, in turn, causes oxidative stress. Given that the muscles and brain are highly dependent on oxidative metabolism, it stands to reason that these tissues will be the most adversely affected by any variations in mtDNA.94 It has also been shown that decreased ATP resulting from mitochondrial disease can decrease the threshold for CSD, which has been extensively linked to migraine (Figure 2).95–97 Furthermore, mitochondria play an essential role in calcium ion homeostasis, which is a necessary requirement for normal neuronal function.98 There is also an abundance of studies showing that migraine is often a by-product or comorbidity of many mitochondrial disorders.99,100 Imaging techniques have been used to show that migraine patients show disturbances in mitochondrial metabolism in specific regions of the brain.101 There are also various studies linking mitochondrial dysfunction with other neurological disorders, such as Alzheimer’s disease,102 Parkinson’s disease103 and amyotrophic lateral sclerosis.104 Given the role of mitochondria in these and other disorders, it is reasonable to explore potential genetic aspects in relation to migraine. A systematic review by Smeitink et al105 suggested a tentative genetic link between migraine and several mitochondrial disorders, such as mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and maternally inherited diabetes and deafness (MIDD). More specifically, these mitochondrial diseases have a particular mutation in the mitochondrial transfer of RNA leucine; patients with the m.3243A>G mutation seem to have a significantly higher prevalence of migraine than the general population.106 Cohort studies showed that in the general population there was an 18–29% prevalence of migraine.107−109 However, similar research using the m.3243A>G mutation population showed migraine prevalence to be 38–89%, with migraine being the only visible symptom in some patients.110,111 These studies suggest that there may be a link between mitochondrial dysfunction and migraine prevalence, even just as a starting point of the neurological cascade of events that cause migraine. Conversely, a GWAS of mitochondrial migraine using the HUNT study cohort did not find any common genetic variants associated with migraine.112 The study used a cohort of 18,309 participants, with 4021 migraine cases and 14,288 healthy controls, examining 775 variants, but none was significant after correction for multiple testing.112 This was also reflected in the haplogroups, which showed no significant association with migraine.112 The study acknowledges that there may be a limitation based on the questionnaire used for selection criteria, and only focused on current migraine participants (the last 12 months); this may have introduced past migraineurs into the control group, which would skew the results.112 Several aspects require further study, for example, distant maternal relatives will share mtDNA but will not necessarily share other genetic risk factors, such as nuclear variants or environmental factors.113 At this stage, there are no known methods to adjust for close maternal relatedness in mitochondrial association studies, and this may have an impact on the results.112 Another aspect to consider is the presence of heteroplasmy, as most mitochondrial diseases can be impacted by the presence of heteroplasmy. While mitochondrial dysfunction in migraine patients merits investigation, the role of rare variants, nuclear-encoded mitochondrial variants and environmental factors needs further exploration.
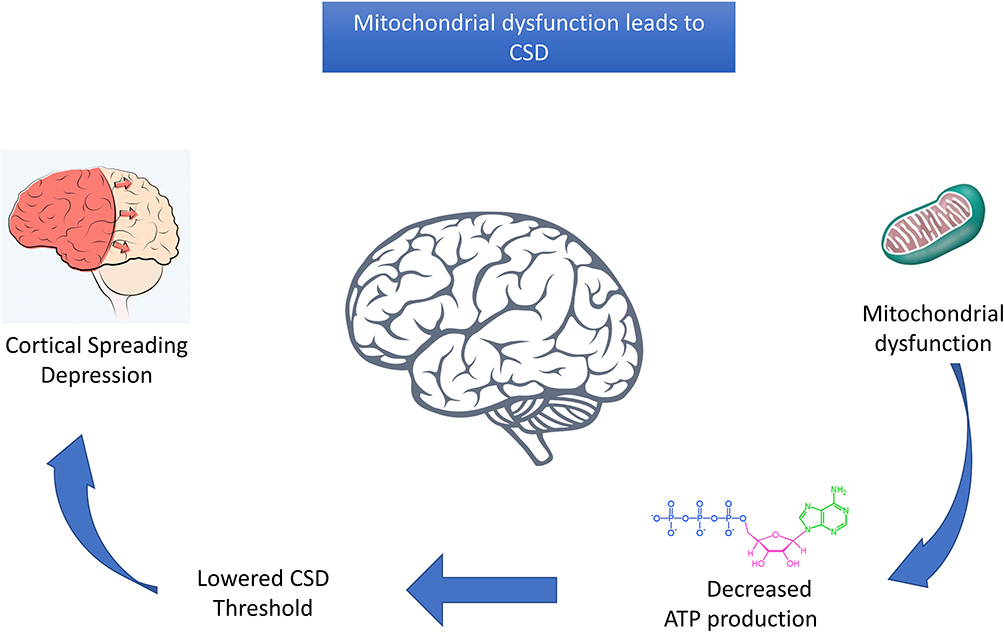 |
Figure 2 Mitochondrial dysfunction leads to CSD. The image demonstrates how mechanical disruptions to the mitochondrion’s energy production could cause migraine by lowering the threshold for propagation of CSD. |
Migraine Epigenetics
While there is clearly a genetic component to migraine susceptibility, there is also significant evidence to suggest that environmental factors also contribute to the disorder.12,114,115 Monozygotic twin studies have shown that MO heritability is up to 60% genetic, while 40% can be attributed to environmental factors.114,115 Commonly held environmental contributors to migraine range from stress, lack of sleep and eating habits, to the changes in hormones during menstruation, pregnancy and menopause.116 The hormonal theory has received great traction in the field given that migraines are three times more common in women than men.117 Furthermore, many women experience migraines more often during menstruation and pregnancy, but have significantly fewer migraines when not influenced by the hormonal changes associated with these conditions,118 and the changes in hormones during menopause have been linked with decreased migraine frequency.116 These variations in hormones can lead to changes in neuronal activity and inflammation; these can result in remodelling of the chromatin, which then affects gene expression.119 Alternatively, rat studies have shown that hormonal treatments can alter the activity of the trigeminal nociceptive pathway via nuclear receptors, which adjust the epigenetic programming of targeted genes.120,121 Glucose transport Glut4 is regulated by oestrogen receptor β through maintaining low levels of DNA methylation at the promoter.122 Using oestrogen receptor-β agonists in mice increased GABA synthesis,123 while oestrogen receptor-α activation enhanced expression of SLC1a3 and Eaat1, changing the balance between inhibitory and excitatory neurotransmission;124 all of which has been linked to FHM1 through transgenic mouse models.125 Animal studies have also shown that female mice with an FHM mutation are more susceptible to CSD.126,127 There is also the comorbidity factor; migraine has been shown to have comorbidity with depression, epilepsy and stroke, all of which have been shown to have an epigenetic component.128,129 Further evidence suggests that epigenetics may contribute to CSD by changing neuronal plasticity and neuroprotection.130–132 Stress is a common modulator for migraine attack, which is highly sensitive to epigenetic programming. Rat models have shown that early life stress can have lasting effects on behaviour, as a result of increased DNA methylation of Nr4a3.133 Research in humans as also shown that stress during early life and adulthood can be linked to a variety of epigenetic alterations at stress effector genes such as BDNF, NR3C1 and CRH, all of which affect structural and functional aspects of synaptic plasticity and stress reactivity.134 Some of the genetic factors linked to migraine have also been linked to epigenetic mechanisms; for example, functional polymorphisms in MTHFR (a gene involved in the pathway for generating the methyl donor required for DNA methylation) have been found to be associated with migraine in some studies.135,136 MTHFR is not the only gene related to epigenetic processes that has been linked to migraine pathophysiology; genes such as MTDH, MEF2D and PRDM16 have also been implicated in the disorder.25,27,28 Winsvold et al showed that changes in episodic to chronic migraine can be associated with changes in the DNA methylation profile compared to healthy controls; they further showed that the most strongly associated CpG site was at the SH2D5 gene, which is involved in the regulation of synaptic plasticity.137 Terlizzi et al show changes in methylation for catechol-O-methyltransferase (COMT), zinc finger protein 234 (ZNF234) and suppressor of cytokine signalling 1 (SOCS1) in chronic migraine.138 Gerring et al identified 63 differentially methylated regions in genes largely involved in solute transport and cellular homeostasis (SLC2A9, SLC38A4 and SLC6A5).139 Each of these studies has various limitations and weaknesses, such as small sample sizes and the lack of consideration of medication regimes, as many drugs can influence the epigenetic profile of the subjects. Thus, while several genes have been implicated in relation to epigenetics and migraine, research is still required to fully understand the impact of environmental factors on migraine risk.
Conclusions
Migraine is a complex neurological disorder with a genetic basis, although we do not fully understand all the genes and the mechanisms behind the disorder. Genetic studies of migraine are further complicated by variation in patient symptoms and comorbidity with other complex neurological diseases. Linkage studies and sequencing in family pedigrees, as well as GWAS to identify common variants associated with migraine, have shed light on some of the suspected genes and pathways involved in migraine, although there is still a need for further research using larger participant numbers. The advent of more cost-effective NGS approaches has allowed researchers to more readily explore population cohorts and family pedigrees for rare variants. Migraine is genetically complex and is likely to have a combination of factors contributing to risk and cause; unravelling these will lead to better diagnosis and treatment for the disorder.
Abbreviations
MO, migraine without aura; MA, migraine with aura; CGAS, candidate gene association studies; GWAS, genome-wide association studies; SNP, single-nucleotide polymorphism; WGS,whole genome sequencing; HM, hemiplegic migraine; SHM, sporadic hemiplegic migraine; FHM, familial hemiplegic migraine; NGS, next generation sequencing; WES, whole exome sequencing; CSD, cortical spreading depression; mtDNA, mitochondrial DNA; MELAS, mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes; MIDD, maternally inherited diabetes and deafness.
Disclosure
Professor Lyn R Griffiths reports grants from the US Migraine Research Foundation, TEVA, and the Australian National Health and Medical Research Council, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Burch RC, Loder S, Loder E, Smitherman TA. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache. 2015;55(1):21–34. doi:10.1111/head.12482
2. Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AM. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14(1):65–80. doi:10.1016/S1474-4422(14)70220-0
3. Headache Australia. Migraine. 2018. Available from: https://headacheaustralia.org.au/migraine/.
4. Lipton RB, Bigal ME, Diamond M, Freitag F, Reed ML, Stewart WF. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68(5):343. doi:10.1212/01.wnl.0000252808.97649.21
5. Lipton RB, Stewart WF, Diamond S, Diamond ML, Reed M. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646–657. doi:10.1046/j.1526-4610.2001.041007646.x
6. Headache Australia. Prevalence and cost of headache. 2018. Available from: https://headacheaustralia.org.au/what-is-headache/prevalence-and-cost-of-headache/.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38(1):1–211. doi:10.1177/0333102417738202
8. Eising E, Huisman SMH, Mahfouz A, et al. Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: a GWAS-based study using the Allen Human Brain Atlas. Hum Genet. 2016;135(4):425–439. doi:10.1007/s00439-016-1638-x
9. Russell MB, Olesen J. A nosographic analysis of the migraine aura in a general population. Brain. 1996;119(2):355–361. doi:10.1093/brain/119.2.355
10. Queiroz LP, Friedman DI, Rapoport AM, Purdy RA. Characteristics of migraine visual aura in Southern Brazil and Northern USA. Cephalalgia. 2011;31(16):1652–1658. doi:10.1177/0333102411430263
11. Sjaastad O, Bovim G, Stovner LJ. Common migraine (“migraine without aura”): localization of the initial pain of attack. Funct Neurol. 1993;8(1):27–32.
12. de Vries B, Haan J, van den Maagdenberg AMJM, Ferrari MD. Migraine; Genetics. In: Aminoff MJ, Daroff RB, editors. Encyclopedia of the Neurological Sciences (Second Edition). Oxford: Academic Press; 2014:42–46.
13. Mulder EJ, Van Baal C, Gaist D, et al. Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. 2003;6(5):422–431. doi:10.1375/136905203770326420
14. Svensson DA, Larsson B, Waldenlind E, Pedersen NL. Shared rearing environment in migraine: results from twins reared apart and twins reared together. Headache. 2003;43(3):235–244. doi:10.1046/j.1526-4610.2003.03047.x
15. Hansen RD, Christensen AF, Olesen J. Family studies to find rare high risk variants in migraine. J Headache Pain. 2017;18(1):32. doi:10.1186/s10194-017-0729-y
16. Chalmer MA, Esserlind AL, Olesen J, Hansen TF. Polygenic risk score: use in migraine research. J Headache Pain. 2018;19(1):29. doi:10.1186/s10194-018-0856-0
17. Corominas R, Sobrido MJ, Ribasés M, et al. Association study of the serotoninergic system in migraine in the spanish population. Am J Med Genetics Part B. 2010;153B(1):177–184.
18. Fernandez F, Lea RA, Colson NJ, Bellis C, Quinlan S, Griffiths LR. Association between a 19 bp deletion polymorphism at the dopamine beta-hydroxylase (DBH) locus and migraine with aura. J Neurol Sci. 2006;251(1):118–123. doi:10.1016/j.jns.2006.09.013
19. Kowa H, Yasui K, Takeshima T, Urakami K, Sakai F, Nakashima K. The homozygous C677T mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for migraine. Am J Med Genet. 2000;96(6):762–764. doi:10.1002/1096-8628(20001204)96:6<762::AID-AJMG12>3.0.CO;2-X
20. Todt U, Netzer C, Toliat M, et al. New genetic evidence for involvement of the dopamine system in migraine with aura. Human Genetics. 2009;125(3):265–279. doi:10.1007/s00439-009-0623-z
21. Nyholt DR, LaForge KS, Kallela M, et al. A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet. 2008;17(21):3318–3331. doi:10.1093/hmg/ddn227
22. de Vries B, Anttila V, Freilinger T, et al. Systematic re-evaluation of genes from candidate gene association studies in migraine using a large genome-wide association data set. Cephalalgia. 2016;36(7):604–614. doi:10.1177/0333102414566820
23. Hagen K, Stovner LJ, Zwart JA. Potentials and Pitfalls in analytical headache epidemiological studies—lessons to be learned from the Head-HUNT study. Cephalalgia. 2007;27(5):403–413. doi:10.1111/j.1468-2982.2007.01302.x
24. van Oosterhout WPJ, Weller CM, Stam AH, et al. Validation of the web-based LUMINA questionnaire for recruiting large cohorts of migraineurs. Cephalalgia. 2011;31(13):1359–1367. doi:10.1177/0333102411418846
25. Anttila V, Winsvold BS, Gormley P, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;45(8):912–917. doi:10.1038/ng.2676
26. Gormley P, Anttila V, Winsvold BS, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48(8):856–866. doi:10.1038/ng.3598
27. Chasman DI, Schurks M, Anttila V, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43(7):695–698. doi:10.1038/ng.856
28. Freilinger T, Anttila V, de Vries B, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44(7):777–782. doi:10.1038/ng.2307
29. Lafrenière RG, Cader MZ, Poulin J-F, et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med. 2010;16(10):1157–1160. doi:10.1038/nm.2216
30. Hiekkala ME, Vuola P, Artto V, et al. The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: a clinical and genetic study in Finnish migraine families. Cephalalgia. 2018;38(12):1849–1863. doi:10.1177/0333102418761041
31. Sutherland HG, Maksemous N, Albury CL, et al. Comprehensive exonic sequencing of hemiplegic migraine-related genes in a cohort of suspected probands identifies known and potential pathogenic variants. Cells. 2020;9:11. doi:10.3390/cells9112368
32. Pettingill P, Weir GA, Wei T, et al. A causal role for TRESK loss of function in migraine mechanisms. Brain. 2019;142(12):3852–3867. doi:10.1093/brain/awz342
33. Andres-Enguix I, Shang L, Stansfeld PJ, et al. Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci Rep. 2012;2(1):237. doi:10.1038/srep00237
34. Rainero I, Rubino E, Gallone S, et al. KCNK18 (TRESK) genetic variants in Italian patients with migraine. Headache. 2014;54(9):1515–1522. doi:10.1111/head.12439
35. Dobler T, Springauf A, Tovornik S, et al. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol. 2007;585(Pt 3):867–879. doi:10.1113/jphysiol.2007.145649
36. Liu P, Xiao Z, Ren F, et al. Functional analysis of a migraine-associated TRESK K+ channel mutation. J Neurosci. 2013;33(31):12810–12824. doi:10.1523/JNEUROSCI.1237-13.2013
37. Royal P, Andres-Bilbe A, Ávalos Prado P, et al. Migraine-associated TRESK mutations increase neuronal excitability through alternative translation initiation and inhibition of TREK. Neuron. 2019;101(2):232–245.e236. doi:10.1016/j.neuron.2018.11.039
38. Rasmussen AH, Kogelman LJA, Kristensen DM, Chalmer MA, Olesen J, Hansen TF. Functional gene networks reveal distinct mechanisms segregating in migraine families. Brain. 2020;143(10):2945–2956. doi:10.1093/brain/awaa242
39. Ducros A. Familial hemiplegic migraine. In: Jes Olesen PT-H, Michael K, Welch A, editors. The Headaches.
40. Ferrari MD, Goadsby JP. Migraine as a Cerebral Ionopathy with Impaired Central Sensory Processing. 2007.
41. Haan J, Terwindt GM, Bos PL, Ophoff RA, Frants RR, Ferrari MD. Familial hemiplegic migraine in the Netherlands. Dutch Migraine Genetics Research Group. Clin Neurol Neurosurg. 1994;96(3):244–249. doi:10.1016/0303-8467(94)90076-0
42. Terwindt GM, Ophoff RA, Haan J, Frants RR, Ferrari MD. Familial hemiplegic migraine: a clinical comparison of families linked and unlinked to chromosome 19.DMG RG. Cephalalgia. 1996;16(3):153–155. doi:10.1046/j.1468-2982.1996.1603153.x
43. Haan J, Terwindt GM, Ophoff RA, et al. Is familial hemiplegic migraine a hereditary form of basilar migraine? Cephalalgia. 1995;15(6):477–481. doi:10.1046/j.1468-2982.1995.1506477.x
44. Ducros A, Denier C, Joutel A, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345(1):17–24. doi:10.1056/NEJM200107053450103
45. Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia Type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543–552. doi:10.1016/S0092-8674(00)81373-2
46. Ackerman MJ, Clapham DE. Ion channels—basic science and clinical disease. N Engl J Med. 1997;336(22):1575–1586. doi:10.1056/NEJM199705293362207
47. Fusco MD, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33(2):192–196. doi:10.1038/ng1081
48. Dichgans M, Freilinger T, Eckstein G, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet (London, England). 2005;366(9483):371–377. doi:10.1016/S0140-6736(05)66786-4
49. Riant F, Roze E, Barbance C, et al. PRRT2 mutations cause hemiplegic migraine. Neurology. 2012;79(21):2122. doi:10.1212/WNL.0b013e3182752cb8
50. Condliffe SB, Corradini I, Pozzi D, Verderio C, Matteoli M. Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons*. J Biol Chem. 2010;285(32):24968–24976. doi:10.1074/jbc.M110.145813
51. Fruscione F, Valente P, Sterlini B, et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. 2018;141(4):1000–1016. doi:10.1093/brain/awy051
52. Rossi P, Sterlini B, Castroflorio E, et al. A novel topology of Proline-rich Transmembrane Protein 2 (PRRT2): HINTS FOR AN INTRACELLULAR FUNCTION AT THE SYNAPSE. J Biol Chem. 2016;291(12):6111–6123. doi:10.1074/jbc.M115.683888
53. Valente P, Castroflorio E, Rossi P, et al. PRRT2 is a key component of the Ca2+-dependent neurotransmitter release machinery. Cell Rep. 2016;15(1):117–131. doi:10.1016/j.celrep.2016.03.005
54. Castiglioni C, López I, Riant F, Bertini E, Terracciano A. PRRT2 mutation causes paroxysmal kinesigenic dyskinesia and hemiplegic migraine in monozygotic twins. Eur J Paediatric Neurol. 2013;17(3):254–258. doi:10.1016/j.ejpn.2012.10.010
55. Dale RC, Gardiner A, Antony J, Houlden H. Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine. Dev Med Child Neurol. 2012;54(10):958–960. doi:10.1111/j.1469-8749.2012.04394.x
56. Suzuki-Muromoto S, Kosaki R, Kosaki K, Kubota M. Familial hemiplegic migraine with a PRRT2 mutation: phenotypic variations and carbamazepine efficacy. Brain Dev. 2020;42(3):293–297. doi:10.1016/j.braindev.2019.12.007
57. Suzuki M, Van Paesschen W, Stalmans I, et al. Defective membrane expression of the Na(+)-HCO(3)(-) cotransporter NBCe1 is associated with familial migraine. Proc Natl Acad Sci U S A. 2010;107(36):15963–15968. doi:10.1073/pnas.1008705107
58. Fan C, Wolking S, Lehmann-Horn F, et al. Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia. 2016;36(13):1238–1247. doi:10.1177/0333102415608360
59. Kahlig KM, Rhodes TH, Pusch M, et al. Divergent sodium channel defects in familial hemiplegic migraine. Proce Natl Acad Sci. 2008;105(28):9799. doi:10.1073/pnas.0711717105
60. Cestèle S, Labate A, Rusconi R, et al. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia. 2013;54(5):927–935. doi:10.1111/epi.12123
61. Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population‐based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125(6):1379–1391. doi:10.1093/brain/awf132
62. Hu Y, Jiang H, Wang Q, Xie Z, Pan S. Identification of a novel nonsense mutation p.Tyr1957Ter of CACNA1A in a Chinese family with episodic Ataxia 2. PLoS One. 2013;8(2):e56362. doi:10.1371/journal.pone.0056362
63. Oh S-K, Baek J-I, Weigand KM, et al. A missense variant of the ATP1A2 gene is associated with a novel phenotype of progressive sensorineural hearing loss associated with migraine. Eur J Human Genetics. 2015;23(5):639–645. doi:10.1038/ejhg.2014.154
64. Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatric Neurol. 2020;24:15–23. doi:10.1016/j.ejpn.2019.12.008
65. Dunn PJ, Maksemous N, Smith RA, Sutherland HG, Haupt LM, Griffiths LR. Investigating diagnostic sequencing techniques for CADASIL diagnosis. Hum Genomics. 2020;14(1):2. doi:10.1186/s40246-019-0255-x
66. McRae JF, Clayton S, Fitzgerald TW, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–438.
67. Maksemous N, Roy B, Smith RA, Griffiths LR. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol Genetics Genomic Med. 2016;4(2):211–222. doi:10.1002/mgg3.196
68. Maksemous N, Smith RA, Haupt LM, Griffiths LR. Targeted next generation sequencing identifies novel NOTCH3 gene mutations in CADASIL diagnostics patients. Hum Genomics. 2016;10(1):38. doi:10.1186/s40246-016-0093-z
69. Maksemous N, Smith RA, Sutherland HG, et al. Targeted next generation sequencing identifies a genetic spectrum of DNA variants in patients with hemiplegic migraine. Cephalalgia Rep. 2019;2:2515816319881630. doi:10.1177/2515816319881630
70. Russell MB, Olesen J. Increased familial risk and evidence of genetic factor in migraine. BMJ (Clin Res Ed). 1995;311(7004):541–544. doi:10.1136/bmj.311.7004.541
71. Viticchi G, Altamura C, Falsetti L, et al. Poor sleep quality in patients affected by migraine: a population study. Neurol Sci. 2020;41(Suppl 2):495–496. doi:10.1007/s10072-020-04673-2
72. Cologno D, Cicarelli G, Petretta V, d’Onofrio F, Bussone G. High prevalence of Dopaminergic Premonitory Symptoms in migraine patients with Restless Legs Syndrome: a pathogenetic link? Neurol Sci. 2008;29(1):166. doi:10.1007/s10072-008-0915-4
73. Drummond PD. Tryptophan depletion increases nausea, headache and photophobia in migraine sufferers. Cephalalgia. 2006;26(10):1225–1233. doi:10.1111/j.1468-2982.2006.01212.x
74. Brennan KC, Bates EA, Shapiro RE, et al. Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci Transl Med. 2013;5(183):183ra156, 181–111. doi:10.1126/scitranslmed.3005784
75. Xu Y, Padiath QS, Shapiro RE, et al. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434(7033):640–644. doi:10.1038/nature03453
76. Farahani S, Solgi L, Bayat S, et al. RAR-related orphan receptor A: one gene with multiple functions related to migraine. CNS Neurosci Ther. 2020;26(12):1315–1321. doi:10.1111/cns.13453
77. Baksa D, Gonda X, Eszlari N, et al. Financial stress interacts with CLOCK gene to affect migraine. Front Behav Neurosci. 2020;13:284. doi:10.3389/fnbeh.2019.00284
78. Kim J, Cho S-J, Kim W-J, Yang KI, Yun C-H, Chu MK. Impact of migraine on the clinical presentation of insomnia: a population-based study. J Headache Pain. 2018;19(1):86. doi:10.1186/s10194-018-0916-5
79. Kilic K, Karatas H, Dönmez-Demir B, et al. Inadequate brain glycogen or sleep increases spreading depression susceptibility. Ann Neurol. 2018;83(1):61–73. doi:10.1002/ana.25122
80. Maher BH, Lea RA, Benton M, et al. An X chromosome association scan of the norfolk island genetic isolate provides evidence for a novel migraine susceptibility locus at Xq12. PLoS One. 2012;7(5):e37903.
81. Nyholt DR, Dawkins JL, Brimage PJ, Goadsby PJ, Nicholson GA, Griffiths LR. Evidence for an X-linked genetic component in familial typical migraine. Hum Mol Genet. 1998;7(3):459–463. doi:10.1093/hmg/7.3.459
82. Wieser T, Pascual J, Oterino A, Soso M, Barmada M, Gardner KL. A novel locus for familial migraine on Xp22. Headache. 2010;50(6):955–962. doi:10.1111/j.1526-4610.2010.01673.x
83. Stewart WF, Lipton RB, Celentano DD, Reed ML. Prevalence of migraine headache in the United States. Relation to age, income, race, and other sociodemographic factors. JAMA. 1992;267(1):64–69. doi:10.1001/jama.1992.03480010072027
84. Launer LJ, Terwindt GM, Ferrari MD. The prevalence and characteristics of migraine in a population-based cohort: the GEM study. Neurology. 1999;53(3):537–542. doi:10.1212/WNL.53.3.537
85. Nyholt DR, Curtain RP, Griffiths LR. Familial typical migraine: significant linkage and localization of a gene to Xq24-28. Hum Genet. 2000;107(1):18–23.
86. Rasmussen BK, Olesen J. Migraine with aura and migraine without aura: an epidemiological study. Cephalalgia. 1992;12(4):
87. Rozen TD, Swanson JW, Stang PE, McDonnell SK, Rocca WA. Increasing incidence of medically recognized migraine headache in a United States population. Neurology. 1999;53(7):1468–1473. doi:10.1212/WNL.53.7.1468
88. Nyholt DR, Lea RA, Goadsby PJ, Brimage PJ, Griffiths LR. Familial typical migraine: linkage to chromosome 19p13 and evidence for genetic heterogeneity. Neurology. 1998;50(5):1428–1432. doi:10.1212/WNL.50.5.1428
89. Nyholt DR, Curtain RP, Griffiths LR. Familial typical migraine: significant linkage and localization of a gene to Xq24–28. Hum Genet. 2000;107(1):18–23.
90. Maher BH, Lea RA, Benton M, et al. An X chromosome association scan of the Norfolk Island genetic isolate provides evidence for a novel migraine susceptibility locus at Xq12. PLoS One. 2012;7(5):e37903–e37903. doi:10.1371/journal.pone.0037903
91. Quintas M, Neto JL, Sequeiros J, et al. Going deep into synaptic vesicle machinery genes and migraine susceptibility – a case-control association study. Headache. 2020;60(10):2152–2165. doi:10.1111/head.13957
92. Elson JL, Andrews RM, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Analysis of European mtDNAs for recombination. Am J Hum Genet. 2001;68(1):145–153. doi:10.1086/316938
93. Piganeau G, Eyre-Walker A. A reanalysis of the indirect evidence for recombination in human mitochondrial DNA. Heredity. 2004;92(4):282–288. doi:10.1038/sj.hdy.6800413
94. Sparaco M, Feleppa M, Lipton RB, Rapoport AM, Bigal ME. Mitochondrial dysfunction and migraine: evidence and hypotheses. Cephalalgia. 2006;26(4):361–372. doi:10.1111/j.1468-2982.2005.01059.x
95. Barbiroli B, Montagna P, Cortelli P, et al. Abnormal brain and muscle energy metabolism shown by 31P magnetic resonance spectroscopy in patients affected by migraine with aura. Neurology. 1992;42(6):1209–1214. doi:10.1212/WNL.42.6.1209
96. Barbiroli B, Montagna P, Cortelli P, et al. Complicated migraine studied by phosphorus magnetic resonance spectroscopy. Cephalalgia. 1990;10(5):263–272. doi:10.1046/j.1468-2982.1990.1005263.x
97. Welch KM, Levine SR, D’Andrea G, Schultz LR, Helpern JA. Preliminary observations on brain energy metabolism in migraine studied by in vivo phosphorus 31 NMR spectroscopy. Neurology. 1989;39(4):538–541. doi:10.1212/WNL.39.4.538
98. Yang J-L, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst). 2008;7(7):1110–1120. doi:10.1016/j.dnarep.2008.03.012
99. Kraya T, Deschauer M, Joshi PR, Zierz S, Gaul C. Prevalence of headache in patients with mitochondrial disease: a cross-sectional study. Headache. 2018;58(1):45–52. doi:10.1111/head.13219
100. Vollono C, Primiano G, Della Marca G, Losurdo A, Servidei S. Migraine in mitochondrial disorders: prevalence and characteristics. Cephalalgia. 2018;38(6):1093–1106. doi:10.1177/0333102417723568
101. Sarchielli P, Tarducci R, Presciutti O, et al. Functional 1H-MRS findings in migraine patients with and without aura assessed interictally. NeuroImage. 2005;24(4):1025–1031. doi:10.1016/j.neuroimage.2004.11.005
102. Sheng B, Wang X, Su B, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120(3):419–429. doi:10.1111/j.1471-4159.2011.07581.x
103. Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012;121(5):830–839. doi:10.1111/j.1471-4159.2012.07734.x
104. Hervias I, Beal MF, Manfredi G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve. 2006;33(5):598–608. doi:10.1002/mus.20489
105. Smeitink J, Koene S, Beyrath J, Saris C, Turnbull D, Janssen M. Mitochondrial Migraine: disentangling the angiopathy paradigm in m 3243A>G Patients. 2019;46(1):52–62.
106. Nesbitt V, Pitceathly RD, Turnbull DM, et al. The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m 3243A> G mutation—implications for diagnosis and management. 2013;84(8):936–938.
107. de Laat P, Koene S, van den Heuvel LP, Rodenburg RJ, Janssen MC, Smeitink JA. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A > G mutation. J Inherit Metab Dis. 2012;35(6):1059–1069. doi:10.1007/s10545-012-9465-2
108. Mancuso M, Orsucci D, Angelini C, et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J Neurol. 2014;261(3):504–510. doi:10.1007/s00415-013-7225-3
109. Nesbitt V, Pitceathly R, Rahman S, et al. P80 The MRC centre for translational research in neuromuscular disease: mitochondrial disease patient cohort study UK. Neuromuscular Disorders. 2011;21:S29–S30. doi:10.1016/S0960-8966(11)70099-2
110. Ciafaloni E, Ricci E, Shanske S, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31(4):391–398. doi:10.1002/ana.410310408
111. Guo S, Esserlind AL, Andersson Z, et al. Prevalence of migraine in persons with the 3243A>G mutation in mitochondrial DNA. Eur j Neurol. 2016;23(1):175–181. doi:10.1111/ene.12832
112. Børte S, Zwart JA, Skogholt AH, et al. Mitochondrial genome-wide association study of migraine - the HUNT Study. Cephalalgia. 2020;40(6):625–634. doi:10.1177/0333102420906835
113. Andersen MM, Balding DJ. How many individuals share a mitochondrial genome? PLoS Genet. 2018;14(11):e1007774. doi:10.1371/journal.pgen.1007774
114. Russell MB, Ulrich V, Gervil M, Olesen J. Migraine without aura and migraine with aura are distinct disorders. a population-based twin survey. Headache. 2002;42(5):332–336. doi:10.1046/j.1526-4610.2002.02102.x
115. Ulrich V, Gervil M, Kyvik KO, Olesen J, Russell MB. The inheritance of migraine with aura estimated by means of structural equation modelling. J Med Genetics. 1999;36(3):225–227.
116. Freeman EW, Sammel MD, Lin H, Gracia CR, Kapoor S. Symptoms in the menopausal transition: hormone and behavioral correlates. Obstet Gynecol. 2008;111:1. doi:10.1097/01.AOG.0000295867.06184.b1
117. Colson NJ, Lea RA, Quinlan S, MacMillan J, Griffiths LR. Investigation of hormone receptor genes in migraine. Neurogenetics. 2005;6(1):17–23. doi:10.1007/s10048-004-0205-0
118. Granella F, Sances G, Pucci E, Nappi RE, Ghiotto N, Nappi G. Migraine with aura and reproductive life events: a case control study. Cephalalgia. 2000;20(8):701–707. doi:10.1046/j.1468-2982.2000.00112.x
119. Eising E, van den Maagdenberg AM, Ferrari MD. Epigenetic mechanisms in migraine: a promising avenue? BMC Med. 2013;11:26.
120. Flake NM, Bonebreak DB, Gold MS. Estrogen and inflammation increase the excitability of rat temporomandibular joint afferent neurons. J Neurophysiol. 2005;93(3):1585–1597. doi:10.1152/jn.00269.2004
121. Liverman CS, Brown JW, Sandhir R, Klein RM, McCarson K, Berman NEJ. Oestrogen increases nociception through ERK activation in the trigeminal ganglion: evidence for a peripheral mechanism of allodynia. Cephalalgia. 2009;29(5):520–531. doi:10.1111/j.1468-2982.2008.01755.x
122. Rüegg J, Cai W, Karimi M, et al. Epigenetic regulation of glucose transporter 4 by estrogen receptor β. Mol Endocrinol. 2011;25(12):2017–2028. doi:10.1210/me.2011-1054
123. Tan XJ, Dai YB, Wu WF, et al. Reduction of dendritic spines and elevation of GABAergic signaling in the brains of mice treated with an estrogen receptor β ligand. Proc Natl Acad Sci U S A. 2012;109(5):1708–1712. doi:10.1073/pnas.1121162109
124. Lee ES, Sidoryk M, Jiang H, Yin Z, Aschner M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem. 2009;110(2):530–544. doi:10.1111/j.1471-4159.2009.06105.x
125. Tottene A, Conti R, Fabbro A, et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61(5):762–773. doi:10.1016/j.neuron.2009.01.027
126. Eikermann-Haerter K, Dileköz E, Kudo C, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119(1):99–109. doi:10.1172/JCI36059
127. van den Maagdenberg AMJM, Pietrobon D, Pizzorusso T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41(5):701–710. doi:10.1016/S0896-6273(04)00085-6
128. McWilliams LA, Goodwin RD, Cox BJ. Depression and anxiety associated with three pain conditions: results from a nationally representative sample. Pain. 2004;111(1):77–83. doi:10.1016/j.pain.2004.06.002
129. Oedegaard KJ, Neckelmann D, Mykletun A, et al. Migraine with and without aura: association with depression and anxiety disorder in a population-based study. The HUNT study. Cephalalgia. 2006;26(1):1–6. doi:10.1111/j.1468-2982.2005.00974.x
130. Guo JU, Ma DK, Mo H, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14(10):1345–1351. doi:10.1038/nn.2900
131. Passaro D, Rana G, Piscopo M, Viggiano E, De Luca B, Fucci L. Epigenetic chromatin modifications in the cortical spreading depression. Brain Res. 2010;1329:1–9. doi:10.1016/j.brainres.2010.03.001
132. Rana G, Donizetti A, Virelli G, et al. Cortical spreading depression differentially affects lysine methylation of H3 histone at neuroprotective genes and retrotransposon sequences. Brain Res. 2012;1467:113–119. doi:10.1016/j.brainres.2012.05.043
133. Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–854. doi:10.1038/nn1276
134. Hunter RG. Epigenetic effects of stress and corticosteroids in the brain. Front Cell Neurosci. 2012;6:18. doi:10.3389/fncel.2012.00018
135. Rubino E, Ferrero M, Rainero I, Binello E, Vaula G, Pinessi L. Association of the C677T Polymorphism in the MTHFR gene with migraine: a meta-analysis. Cephalalgia. 2009;29(8):818–825. doi:10.1111/j.1468-2982.2007.01400.x
136. Schürks M, Rist PM, Kurth T. MTHFR 677C>T and ACE D/I polymorphisms in migraine: a systematic review and meta-analysis. Headache. 2010;50(4):588–599. doi:10.1111/j.1526-4610.2009.01570.x
137. Winsvold BS, Palta P, Eising E, et al. Epigenetic DNA methylation changes associated with headache chronification: a retrospective case-control study. Cephalalgia. 2018;38(2):312–322. doi:10.1177/0333102417690111
138. Terlizzi R, Bacalini MG, Pirazzini C, et al. Epigenetic DNA methylation changes in episodic and chronic migraine. Neurol Sci. 2018;39(Suppl 1):67–68. doi:10.1007/s10072-018-3348-8
139. Gerring ZF, McRae AF, Montgomery GW, Nyholt DR. Genome-wide DNA methylation profiling in whole blood reveals epigenetic signatures associated with migraine. BMC Genomics. 2018;19(1):69. doi:10.1186/s12864-018-4450-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.