Back to Journals » Journal of Inflammation Research » Volume 16
Exploring the Connectivity of Neurodegenerative Diseases: Microglia as the Center
Authors Wang Y ![]() , Cui L, Zhao H, He H, Chen L, Song X
, Cui L, Zhao H, He H, Chen L, Song X ![]() , Liu D, Qiu J, Sun Y
, Liu D, Qiu J, Sun Y ![]()
Received 15 September 2023
Accepted for publication 5 December 2023
Published 12 December 2023 Volume 2023:16 Pages 6107—6121
DOI https://doi.org/10.2147/JIR.S440377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yan Wang,1– 4,* Limei Cui,2– 4,* He Zhao,1– 4,* Huhuifen He,1– 4 Liang Chen,2– 4 Xicheng Song,2– 4 Dawei Liu,2– 4 Jingjing Qiu,2– 4 Yan Sun2– 4
1The Second Medical College, Binzhou Medical University, Yantai, Shandong, People’s Republic of China; 2Department of Otolaryngology and Head and Neck Surgery, Yantai Yuhuangding Hospital, Qingdao University, Yantai, Shandong, People’s Republic of China; 3Shandong Provincial Clinical Research Center for Otorhinolaryngologic Diseases, Yantai, Shandong, People’s Republic of China; 4Yantai Key Laboratory of Otorhinolaryngologic Diseases, Yantai, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Sun, Department of Otorhinolaryngology & Head and Neck Surgery, Yuhuangding Hospital Affiliated to Qingdao University, No. 20 East, Yuhuangding Road, Yantai, Shandong, 264000, People’s Republic of China, Email [email protected]
Abstract: Degenerative diseases affect people’s life and health and cause a severe social burden. Relevant mechanisms of microglia have been studied, aiming to control and reduce degenerative disease occurrence effectively. This review discussed the specific mechanisms underlying microglia in neurodegenerative diseases, age-related hearing loss, Alzheimer’s disease, Parkinson’s disease, and peripheral nervous system (PNS) degenerative diseases. It also reviewed the studies of microglia inhibitors (PLX3397/PLX5622) and activators (lipopolysaccharide), and suggested that reducing microglia can effectively curb the genesis and progression of degenerative diseases. Finally, microglial cells’ anti-inflammatory and pro-inflammatory dual role was considered the critical communication point in central and peripheral degenerative diseases. Although it is difficult to describe the complex morphological structure of microglia in a unified manner, this does not prevent them from being a target for future treatment of neurodegenerative diseases.
Keywords: microglia, neurodegenerative diseases, neuroinflammation
Introduction
Ageing is a risk factor for neurodegenerative diseases. The aging process contributes to the development of various degenerative diseases characterized by significant impairment in tissue or cellular functional disease.1 Degenerative disease is a series of diseases caused by organ and tissue aging. Most chronic degenerative human diseases are inherently associated with increasing age.2 The most common degenerative diseases are neurodegenerative disorders, such as age-related hearing loss (ARHL), Alzheimer’s disease (AD), Parkinson’s disease (PD), retinal degenerative diseases, etc.3–5 The hallmark of these changes is the morphological alterations observed in nerve cells due to aging and other detrimental factors, including inflammation and cancer. Ultimately, these changes impact nerve function by leading to neuronal degeneration.6 Some degenerative diseases are peripheral neurodegenerative diseases, such as pain neuron degeneration and osteoarthritis.7,8 Although many studies are conducted on degenerative diseases, few study has explored the underlying commonality of these degenerative diseases. Therefore, exploring the commonality of central and peripheral mechanisms is vital in finding novel treatments, which is the objective of this review.
Over time, the theory of microglia as the main target of degenerative diseases has been suggested. Microglial cells have unique mechanisms, appearing as star cells in a hot circle. The first point to consider is that microglia are macrophages that reside in the brain and play a significant role in immunomodulation.9 They are activated in response to various damaging factors such as aging, tissue damage, abnormal stimulation, neurotoxins, infection, etc. Moreover, they exhibit prompt responsiveness toward biological damage.10 Second, unlike most immune cells, such as white blood cells, microglial cells, although their early response is protective,11,12 can rapidly transform into pro-inflammatory cells and initiate a series of neuroinflammatory responses, in which case they can physically attack healthy neurons through phagocytosis or the secretion of marcelling inflammatory cytokines.13 This neuroinflammatory phenomenon is commonly seen throughout the disease course. With time, regulating pro- and anti-inflammatory signals has dynamic changes, such as converting the M1 phenotype with pro-inflammatory activity into the M2 phenotype with anti-inflammatory activity.14
The dual role of microglia in neurodegenerative diseases involves both pro-inflammatory and anti-inflammatory mechanisms. For instance, in Alzheimer’s disease (AD), microglia can phagocytose Aβ fragments to exert a protective effect; however, when phagocytosis is activated, it can lead to a pro-inflammatory response.15 M1/M2 microglia can exhibit dual effects in peripheral degenerative diseases by releasing exosomes with distinct properties.16,17 Additionally, the microglia-astroglia-glial axis serves as a potential avenue for information transmission between glial cells, allowing this dual effect to occur. Our research primarily investigates how this interaction can be harnessed to suppress the chronic activation of microglia. Thus, this positive feedback circulatory process leads to degenerative diseases. Therefore, inhibiting the chronic activation of microglia to prevent pro-inflammatory mediator production and synaptic phagocytosis can efficiently reduce neurodegenerative disease development.
The function of microglia is complex, and the morphology of various states is different, so it is difficult to put together a discussion. Therefore, to understand the function of microglia in the context of disease, it is important to understand their neuroinflammatory connectedness. We will discuss several examples of microglia function in the context of neuroinflammation, including central and peripheral diseases.(Figure 1).
 |
Figure 1 By discussing and summarizing the known mechanisms of microglia connections in central and peripheral degenerative diseases, the mechanism of the occurrence and development of degenerative diseases is speculated, which provides new ideas and guidance for exploring other degenerative diseases. |
Microglia and Neurodegenerative Disorders
Microglia and Age-Related Hearing Loss
Age-Related Hearing Loss (ARHL) or presbycusis represents a frequently seen sensory impairment among older adults. With cochlear aging (peripheral hearing organ), hearing loss may develop and is associated with increased sensory nerve cell degeneration risk in the cochlea and worsening of age-related hearing loss, which seriously affects patient’s quality of life. Considering the serious aging problem worldwide and the social burden due to ARHL,18 treatments alleviating age-related hearing loss should be developed. AHRL accounts for the main hearing impairment with features of pathological variations of central and peripheral auditory systems. Animal models with early-onset ARHL have mild cochlear inflammation, suggesting that inflammation is related to ARHL genesis and progression.19
Meanwhile, Seicol et al found that the central cochlear nucleus is activated during the aging of microglial cells,20 and the increase of the iconic antibody IBA-1 is significant. Microglial cells proliferated and differentiated, and C1q expression was significantly increased, indicating that ARHL occurrence involved the activation of neuroinflammation and the complement system. Therefore, more treatments targeting the innate immune and complement pathways can offer novel ideas for developing new therapeutic approaches. In addition, the experiment showed that microglial production and increased C1q deposition in middle-aged mice were small,21 indicating that this time window may be the best period for ARHL treatment. We speculated that administering microglia inhibitors in middle-aged ARHL mouse models may delay or treat ARHL.
Concurrently, we further studied the pathway mechanism of presbycusis. Current studies showed that microglia activated by injury factors could release complement factor C1q, which critically affects the classical complement system as an initiation factor for the complement pathway.20 Additionally, C3, located downstream of C1q, also critically affects the complement pathway. Meanwhile, C3 is mainly produced via activated astrocytes,22 which also target C3aR on the microglial cell surface. In addition, studies on microglia suggested that the complement pathway can be used as a novel therapeutic target. This complement system activation increases the risk of further activation, causing normal tissue death.23 These may be impaired in degenerative diseases, providing a mechanism for degeneration. Thus, C1q secreted by microglia and inflammatory factors IL-β and TNF-α activates astrocytes and the complement system, which further affects surrounding healthy neurons or tissues, leading to the aggravation of degenerative diseases. Therefore, the complement pathway is considered a new therapeutic direction, with C3 as an important therapeutic target factor (Figure 2).
 |
Figure 2 During age-related hearing loss progression, activated microglia can release inflammatory factors that destroy the blood–brain barrier and recruit peripheral macrophages into the center. Meanwhile, the released inflammatory factors can also directly act on neurons and cause neuronal degeneration. Concurrently, complement pathway activation may be another key factor in enhancing neuroinflammation. The flowchart was created using Biorender.com. |
Microglia and Retinal Degenerative Diseases
Retinal degeneration (RD) is a series of retinal diseases characterized by gradual death and nerve cell loss, such as photoreceptor cells, retinal ganglion cells, or retinal pigment epithelium.24 Currently, no effective interventions have targeted the apoptotic pathway of degenerative cells in treating RD. Moreover, microglia-mediated inflammatory responses have a critical effect on RD progression. Within the normal retina, microglial cells are usually at rest. As the only innate retinal immune cells, their functions include monitoring and maintaining the retina’s microenvironment. However, in the degenerative retina, microglial cells can be activated by injury factors, such as aging, and regulate the homeostasis of the retinal microenvironment through “double action”. Additionally, as defense cells, activated microglial cells produce various neuroprotective factors, anti-inflammatory cytokines, phagocytosis, etc., to improve and maintain the retinal microenvironment. By contrast, microglial over-activation can produce various pro-inflammatory cytokines, worsen the retinal microenvironment, and aggravate the apoptosis of nerve cells.25 Therefore, regulating the activation of degenerative retinal microglial cells is an important strategy to protect nerve cells and delay RD progression. Moreover, microglial cells can clear synapses and other neural elements through complement regulation and delay abnormal neurogenesis and visual function degeneration during retinal senescence and degeneration.26 CX3CR1 and its ligand CX3CL1 regulate microglia through RAS-guided signal transduction pathways to exert their effects on neurons and retinal cell tissues.27 Continuous exploration and research on CX3CR1 and its ligand CX3CL1 from multiple perspectives will provide important research directions and ideas for retinal degenerative diseases and provide a novel research target to prevent and treat retinal degenerative diseases.28 This suggests that microglia can impact the occurrence and progression of RD through the Ras pathway, and inhibiting excessive microglia activation may be an effective strategy to delay RD development.
Microglia and Alzheimer’s Disease
Alzheimer’s Disease (AD) accounts for major disease-causing dementia in the world.29 Many studies showed that aging is the most important factor related to the pathology of neurodegenerative disorders, such as AD.30 Consequently, understanding microglial alterations during aging stimulation is necessary to prevent and treat these diseases. Microglia also have a dual role within AD. Generally, microglia can protect the brain because they are defense cells maintaining tissue homeostasis through phagocytosis and clearing amyloid-beta peptide (Aβ) extracellular space, thereby preventing AD.31 With Aβ accumulation, microglial cells continuously exert Aβ aggregate phagocytosis and removal functions. Still, Aβ aggregates are compacted into dense core plaques before isolation from the neurons when microglia functions exceed their ability to remove them.32 This protection involves microglial activation into the disease-associated microglia (DAM) state, determined by apoE and acting in triggering receptor expressed on myeloid cells 2 (TREM2).33 Moreover, genetic factors and aging may lead to abnormal microglial cell function and may not prevent AD genesis and progression. Toxic amyloid protein (tau) pathological accumulation, especially within injured or stressed neurons, induces the overactivation and inflammation of microglial cells, where phagocytic synapses are released, neuron-damaging neurotoxic factors are secreted, and tau pathological transmission is transmitted.34 The vast majority of cells in the human body can secrete exosomes. Exosomes have become an important vehicle for signaling between cells. Hirohide Asai et al used a model of adenovirus showed microglia spread tau via exosome secretion, and inhibiting exosome synthesis significantly reduced tau propagation in vitro and in vivo.35 Clarifying the relationship between harmful proteins such as tau and microglia metabolism will help people better understand the crosstalk mechanism between neurons and microglia. Microglia have a dual role in the AD pathogenic model. Harmful microglia populations appear in the disease’s later stages, consistent with synaptic elimination and neuronal degeneration.36
Moreover, microglia may represent a new target to delay or halt AD evolution due to recent progress in exploring underlying major mechanisms, exosome, neurodegeneration, plasticity regulation, and synaptic pruning microglia depletion (Figure 3).
 |
Figure 3 In Alzheimer’s disease, microglia phagocytosis clears Aβ aggregates. This protective mechanism involves microglial activation in the DAM state, determined by TREM2, assisted by apoE. Eventually, it acts on neurons and causes neuronal degeneration. |
Microglia and Parkinson’s Disease
Parkinson’s disease (PD) can be caused by abnormal accumulation of alpha-synaptic nucleoproteins, or alpha-syn aggregates, within dopaminergic neurons of the substantia nigra.37 Many hypotheses exist on PD genesis and progression, and neuroinflammation has an important effect on the neurodegenerative pathogenic mechanism of PD. Microglial cell-induced neuroinflammation is particularly associated with PD pathogenic mechanism.38 Such neuroinflammatory neurodegeneration is related to microglial activation and up-regulated pro-inflammatory factors.39 Chronic microglia activation in patients with PD may produce increased pro-inflammatory and cytotoxic cytokine levels, such as IL-6, TNF-α, ROS, etc., resulting in disease exacerbation. These molecules can mediate effective antigen presentation by CD4+T cells through the major histocompatibility complex(MHC) class II pathway, causing cell differentiation and growth, followed by slow degeneration and, eventually, neuron death.40,41
Moreover, microglia are major intracerebral resident immune cells that normally phagocytose dead cells while clearing misfolded alpha-syn aggregates during PD.42 However, alpha-syn activates microglia, leading to pro-inflammatory factor release. Recently, microglia can effectively generate exosomes, part of antigen presentation and production mechanism.43 These microglial cell-derived exosomes can critically affect alpha-syn transmission, inducing alpha-synaptic nucleoprotein aggregation within neurons,44 thereby inducing degeneration of the substantia striatum, indicating such exosomes play an important role in PD pathology. Similar to the TREM2 mechanism within AD, TREM2 significantly affects the transformation of M1 microglial cells with pro-inflammatory phenotypes into M2 phenotypes, which benefits PD immunopathogenesis.45,46 Therefore, chronic microglial activation in patients with PD may produce excessive pro-inflammatory and cytotoxic factors, thereby exacerbating the disease. By contrast, we identified the neuroprotective effect of microglial cells, which plays a major role in clearing alpha-syn nucleoproteins released by neurons.
In conclusion, PD is closely associated with microglia, and the chronic activation of microglia is also responsible for the development of chronic neuroinflammation in PD. Simultaneously, injury-induced microglia can impact the progression of PD through its polarization (M1/M2). Furthermore, the secretion of microglial exosomes also presents novel prospects for diagnosing and treating PD. (Figure 4).
 |
Figure 4 Microglia will engulf abnormal neurons and alpha-syn aggregates, whereas over-activation will become a DAM state, causing neuroinflammation and eventually leading to PD. This also reflects the dual role of microglia. The flowchart was created using Biorender.com. |
Microglia and Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) accounts for an adult-onset neurodegenerative disorder characterized by rapid progression showing selective destruction of upper/lower motor neurons, leading to limb muscle tissue atrophy and weakness in varying levels, spasm, conformation disorders, and dysphagia.47 Neuroinflammation, such as T-cell infiltration or microglial activation, represents a neuropathological marker for ALS. Traditionally, immune function in patients with ALS was considered dominant in CNS inflammation, but neuroinflammation has been currently considered as the major characteristic shared by different classical neurodegenerative diseases.48 Neurochemical or morphological studies showed that ALS microglial cells are activated and proliferated.49 Microglial activation was observed in relevant brain domains in patients with ALS, and microglia proliferated with increasing disease severity. Neuronal damage models showed that microglial cell activation also plays a dual role in ALS, promoting neuronal protection and damage. Moreover, microglial cells exhibit functional changes during activation, including cell count, morphology, cytokine and growth factor generation, and surface receptor levels. This indicates that microglial cell activation status changes due to signals in damaged neurons and adjacent glial cells. Like macrophages, microglial cells may display alternative activation or inactivation of the M2 phenotype or classical activation of the M1 phenotype in ALS.50,51 M2 microglial cells may suppress pro-inflammatory cytokine production, contribute to inflammation regression using endocytic clearance, produce increased IGF-1, IL-4, and IL-10, accelerate tissue repair, and promote Th2 function, enhancing neuron survival. Exosomes derived from microglia have the potential to disseminate pathogenic factors. For instance, the secretion of SOD1 by NSC-34 Motor Neuron-Like Cells in hSOD34G1A overexpressing mice can promote inflammation and impact neuronal survival (Figure 5).52
 |
Figure 5 In ALS, microglial cells not only play a role through different polarization states but also present antigens to different T cells to affect neurons. The flowchart was created using Biorender.com. |
Therefore, the dual role of microglial and T cells may protect neurons by regulating beneficial M2 microglial inflammatory responses in chronic neurodegenerative models of ALS, leading to slower disease progression. Similarly, microglial exosomes also evidence a well-coordinated and complicated dialog between ALS neurons, microglial cells, and T cells.
Microglia and Multiple Sclerosis
Multiple sclerosis (MS) is a chronic inflammatory CNS disorder, and its pathological and clinical manifestations were initially identified by Jean-Martin Charcot in 1961.53 Unlike other degenerative diseases, the cause of neuron mortality during MS was secondary to decreased reactive T-cell activation due to decreased function of targeted myelin.54 Nonetheless, such neurodegenerative diseases exhibit the characteristics of gradual neuron loss associated with cognitive impairment and motor dysfunction, which are related to inflammatory and immune responses. The neuroinflammatory process in MS is similar to other neurodegenerative diseases. The pathology of MS is better understood in studies of other neurodegenerative disorders.
Microglia plays a vital role in molecular recognition during MS. In cases of aging, irritation, bacteria, or injury sign, microglial cells are rapidly activated and migrate toward the injury site. However, activated microglial cells also exhibit neurotoxic effects in MS, despite their altered state and function to protect the CNS.55,56 Microglial activation is not limited to the lesion area, and patients with MS have more pronounced microglial activation in normal white matter compared with healthy controls.57 In addition, clusters of activated microglia, known as microglia nodules, are gradually deposited during axon degeneration, oligodendrocyte activation, or complement pathway product activation.58,59 In MS, activated C3 and C1q levels are up-regulated within the hippocampi, which are localized in synapses in lysosomes and microglia, indicating microglial cell-induced synaptic phagocytosis and removal.60 These microglia nodules exhibit characteristics of insufficient demyelination and leukocyte infiltration, finally developing into active demyelinating MS lesions. Furthermore, microglial cells mainly exhibit pro-inflammatory phenotypes within early active lesions, which begin to be expressed and are related to inflammatory factor secretion, oxidative damage, phagocytosis, T-cell stimulation, and antigen presentation. Apart from acute lesions, activated microglia are observed in the lesion extension of chronic and slow-progressing MS lesions, which continuously produce inflammatory factors causing chronic damage. Thus, these results render microglial cells in the core of MS pathology, and microglia are stimulated by damage factors and regulated by the immune system to produce pro-inflammatory behavior, which is critical in MS pathogenesis.
Therefore, effective utilization of the dual role of microglia to regulate their polarization characteristics and fully use their regulatory effect of the complement pathway to limit and reduce potential reactions promoting that promote degeneration, protect the CNS from damage, and promote local recovery (Figure 6).
 |
Figure 6 In MS, microglia can affect neurons through phagocytosis through microglial cluster formation that destroys myelin sheaths. The flowchart was created using Biorender.com. |
Therefore, from the above examples of microglia, it is not difficult to find that although the function of microglia in each disease state is complex, but the dual role of microglial cells under different activation states plays an important role in central nervous system degenerative diseases.
Microglia and Peripheral Nervous System (PNS) Degenerative Diseases
Aging is an inevitable biological process characterized by neurological dysfunction, such as emotional and cognitive decline. Aging is associated with neuroinflammation, neurodegeneration, amyloid plaque deposition, dementia, and neuropsychiatric diseases in CNS.61 In addition, aging affects the PNS to varying degrees. For example, pain neuron excitability in the lumbar dorsal horn was significantly enhanced in older rats than younger ones.62 Therefore, aging may also be a key factor leading to degenerative abnormalities in signal transduction between pain-sensing neurons. Microglial cell activation can effectively regulate the excitability of pain-sensing neurons within the spinal dorsal horn to cope with the occurrence of PNS diseases, such as sciatic nerve injury.63,64 Our understanding of the effect of microglial cells on CNS degenerative disorders, particularly their dual role, further explores the important function of microglial cells on PNS degenerative diseases.
Chronic pain in osteoarthritis (OA), characterized by pain sensitization, is a common pain and disabling disorder worldwide. Additionally, it represents PNS degenerative disease with a high incidence. OA involves peripheral and central mechanisms, and spinal microglial cell and synovial macrophage hyperactivity are the basis for the pain sensitization mechanism of OA in CNS and PNS.65 Hahm et al concluded that analgesic tolerance of OA pain control induced by repeated electrical stimulation and changes in microglial cell activation are related.66 Microglial cells are also present in the caudate subnucleus of the spinal cord of the trigeminal nerve (Vc) and the upper part of the cervical spinal cord (C1/C2). Thus, microglial cell activation within Vc and C1/C2 can be related to the pathology of oral and facial pain hypersensitivity.67,68 Studies on accelerated susceptibility to aging (SAMP8) mice and anti-aging control (SAMR1) mice showed that microglial cells play an important role in the periphery through the mutual transformation of M1 and M2,69 thereby causing the development of degenerative diseases. IL-10 and TNF-α modulate pro-inflammatory circulation and polarity change in activated microglial cells into M1/M2 macrophages.70 In addition, reduced IL-10 levels similarly promote microglial polarization into M1 in the pro-inflammatory environment.71 Consequently, the mechanism of microglia phenotype change may not be independent but interrelated, and it is a key factor in maintaining M1/M2 polarized balance to promote aberrant oral and facial pain while delaying OA pain. In summary, aging injury-induced age-related neuroinflammation affects activated microglial cell polarity variation and enhances neuronal excitability at microglial aggregation sites, such as Vc and C1/C2. These findings may provide useful information in exploring the influence of aging damaging factors on the development of PNS neurodegenerative disorders.
In summary, the microglial cell polarization state has a critical effect on PNS degenerative diseases. How to effectively control microglial activation may be the key to treating degenerative diseases in the future.
Relationship Between Microglial Inhibition and Activation and Degenerative Diseases
Microglia and CSF1R Inhibitors
Microglia modulate the architecture of adult neurons, and signals from colony-stimulating factor 1 receptor (CSF1R) determine adult microglia regulation.72 CSF1 is important in modulating macrophage growth and differentiation.73 Moreover, mice lacking CSF1 or CSF1R showed decreased density of macrophages in several tissues, and neurons showed demyelination and other neuronal degeneration symptoms.74 Excessive pruning and even eliminating synapses are common essential characteristics of numerous disorders with excessive microglial proliferation and differentiation, such as AD, ARHL, etc.75,76 CSF1R inhibitors, which can cross the blood–brain barrier, rapidly remove microglial cells in the CNS while inhibiting microglia continuously. In addition, studies showed that CSF1R inhibitor treatment significantly increased the density of dendritic spines and synaptic points in all brain regions tested in microglia-lacking adult mice.77 Concurrently, chronic microglial cell depletion can increase the stability of perineuronal networks and enhance local inhibitory/excitatory circuit connections with excitatory neurons.78 Therefore, neurons can better play the signaling function by inhibiting microglia, reducing the occurrence and development of degenerative diseases.
PLX339779 and PLX562280 are the most well-known CSF1R inhibitors. Mice fed with both showed whole-body microglia inhibition,81,82 and their incidence of degenerative diseases greatly reduced, such as AD,75,83 further confirming the critical effect of microglia on degenerative diseases. Meanwhile, Elmore et al measured measuring PLX3397 levels within the brain tissue and showed continued microglia cell elimination during a medication withdrawal. However, trace amounts of microglia were detected 1 day after drug withdrawal and recovered to normal levels on the 7th day of recovery,84 indicating that CSF1R inhibitors continuously act on microglia, leading to its clearance in the brain and effectively alleviate degenerative diseases without causing permanent damage. By contrast, several studies suggested that activating microglia to produce neuroinflammation can promote the development of degenerative disorders, such as PD.
Microglia and Activators
Neuroinflammatory response is related to neurodegenerative diseases, and microglial cells are resident immune cells within the CNS, whose activation promotes neuroinflammation in neurodegenerative diseases. Lipopolysaccharide (LPS), as an excellent pro-inflammatory agent, provides a good basis for studying neurodegenerative diseases.85,86 LPS is closely related to microglial activation. Microglial cells are key nervous system-specific immune cells with various phenotypes, which can metastasize to maintain tissue homeostasis. Additionally, microglial cells exhibit common and specific characteristics in phenotypic polarization relative to peripheral macrophages. LPS is a typical bacterial-derived endotoxin that directly activates microglia through toll-like receptor 4,87 which may cause and amplify neuroinflammation and influence various neurodegenerative diseases. Microglial activation can sustain the increase in immunoreactivity with the microglia-specific antibody iba-1.88 Kondo et al evaluated glial activation after LPS administration and observed morphologic changes in microglia 2 days after LPS administration, with partial recovery within 1 week but long-lasting.89 Thus, the role of LPS in microglial activation is long-lasting. LPS stimulates the polarization of microglial cells for producing various inflammatory factors, including TNF-α and IL-β, while activating neuronal degeneration and loss of neuronal function due to persistent neuroinflammation, eventually leading to degenerative diseases.
Coincidentally, the CSF1R antagonist impaired the potential coupling between microglia and astrocytes in mice subjected to microglial pharmacological ablation using PLX5622. In contrast, priming microglial cells with LPS increased synaptic transmission after induction, further enhancing communication with astroglia.90 Signaling between glial cells to promote neuroinflammation is the key to developing affective disorders in many neurodegenerative diseases, suggesting that microglia are crucial for developing degenerative diseases (Figure 7).
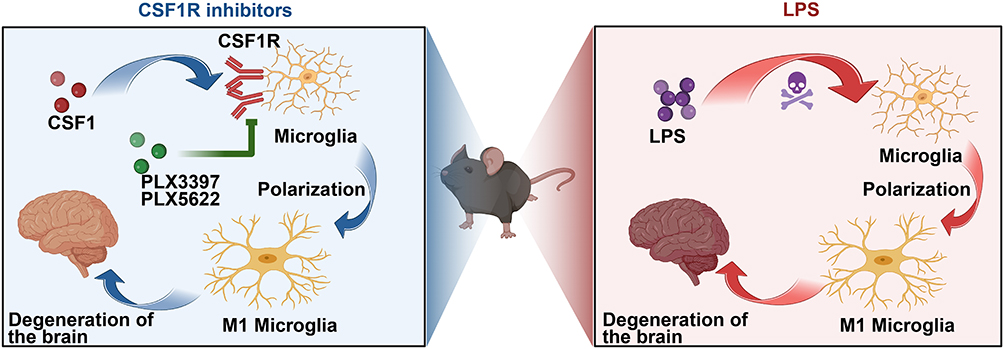 |
Figure 7 Microglia treated with CSF1R inhibitor and LPS mouse models showed different changes. Microglia in mice treated with the inhibitor significantly decreased, and the onset of degenerative disease was delayed, whereas microglia in mice treated with LPS showed proliferation and differentiation, accelerating the onset of degenerative disease. The flowchart was created using Biorender.com. |
Neurodegenerative diseases are effectively controlled after using inhibitors. By contrast, activators may aggravate neurodegenerative diseases, indicating that microglial cells are closely related to degenerative diseases. They can be used as a target for the treatment of degenerative diseases.
Discussion
The function of microglia is complex, and the states of microglia are different under various conditions, which is difficult to discuss uniformly. This review focuses on the disease state and explores the commonality of one aspect of its complex manifestations so as to provide constructive guidance for the pathogenesis and treatment strategies of other types of degenerative diseases in the future. Microglial cells critically affect CNS degenerative diseases, such as AHRL, retinal degenerative diseases, AD, and PNS degenerative diseases, such as decreased sensitivity to pain caused by aging. Microglial cells affect disease genesis and progression because of its dual role and other complex mechanisms. Additionally, microglial cells can also activate astrocytes through cascaded amplification, further stimulating neuronal degeneration and leading to the generation of degenerative diseases. For example, in AD and PD brains, microglial cells and astrocytes can acquire active inflammatory phenotypes, efficiently phagocytize accumulated cell debris and proteins, and produce different inflammatory factors and cytokines. The astrocyte–microglial cell interaction triggers reactive astrocytes. Astrocytes may have a common molecular language with microglia within diverse disorders, such as neuroinflammatory mechanisms and complement pathway mechanisms. As technology advances, astrocyte–microglial cell interaction may become a precise and efficient target for disease treatment. In most instances, microglia and astrocytes operate harmoniously and complementary across a broad spectrum of pathophysiological processes. These processes encompass synaptic phagocytosis and remodeling, the regulation of the blood-brain barrier, maintenance of homeostasis, and involvement in immune responses. Disturbance in astrocytoma–microglia interaction leads to the development of degenerative diseases.
In summary, the commonality of microglia as the target of degenerative diseases has not been thoroughly discussed. In this review of CNS and PNS degenerative diseases, we found that these diseases activate microglial cells through aging and other damage factors and regulate their polarized phenotypes (M1/M2) that play dual roles in regulating the genesis and progression of degenerative disorders. Moreover, microglial cells can also impact the occurrence of degenerative disorders, such as AD, PD, through complement pathway activation and exosome secretion.91 There is a close link between exosomes and the polarization of microglia, and exosomes such as microRNA play a regulatory role in the M1/M2 polarization of microglia.92 Microglial exosomes can be transported to and taken up by neurons, which may be beneficial or harmful in central nervous system diseases. Microglial cells have an important effect on phagocytic synapses, particularly in MS and AD. Moreover, they exert a dual effect on PNS neurodegenerative disorders, such as the decline of pain-sensing neurons and OA. Similarly, the crosstalk between microglia and astrocytes should be considered. Damage factors from aging and other factors activate microglia to produce inflammatory factors, such as TNF-α and complement factors, including C1q, to activate astrocytes further, and this pro-inflammatory circulation between glial cells also promotes the development of degenerative diseases.
Further Directions and Limitation
Another innovation of this paper is the incorporation of microglia agonists and inhibitors in the discussion to explore potential treatments for neurodegenerative diseases. The management of neurodegenerative diseases has always posed a challenging problem within the medical community, and our fundamental research aims to provide effective treatment options for the numerous patients worldwide suffering from these conditions. The establishment and application of animal models have significantly advanced the medical field for most researchers. Model tests have led to the development of numerous targeted drugs and treatments. Among neurodegenerative diseases, Parkinson’s disease animal model is the most well-known and mature.93–95 The use of this model has yielded unexpected progress in exploring treatment options for Parkinson’s disease, providing valuable reference experience.96–98 This prompts us to consider whether microglia inhibitors could effectively delay PD progression when applied on a mature PD model, which may also extend to other degenerative diseases.
Microglia activators like lipopolysaccharide can directly activate microglia into an M1 pro-inflammatory form while also enhancing interaction between microglia and astrocytes, thereby inducing neuroinflammation that leads to neurodegenerative diseases. Conversely, microglia inhibitors specifically counteract chronic activation of systemic microglia without causing organic lesions. Therefore, continuous administration of microglial inhibitors may also exert a positive inhibitory effect on degenerative diseases in elderly individuals. However, exploring how we can ensure that both inhibitors and activators maintain their normal physiological effects after acting upon microglia itself remains crucial. This suggests that further study should focus on refining pathways or surface receptors associated with microglia; for instance, investigating the use of C3aR inhibitors to inhibit the C3a receptor on the surface of microglia selectively could not only block chronic activation but also preserve most functions of these cells—an avenue worth exploring. Future studies should continue broadening the horizon and explore the relationship between glial cells and degenerative diseases to find new opportunities for treating degenerative diseases.
This review also has limitations; the state of microglia in different states can not be generalized. This article mainly discusses microglia in disease states, and the crosstalk between glial cells cannot be ignored. When we explore the connectivity and relationship between microglial cells and degenerative diseases, their interaction with other glial cells should be considered. This study focused on only a small portion.
Conclusion
Therefore, we clarified that the dual role of microglia is the key point of communication in CNS and PNS degenerative diseases, and exosome and complement pathways provide us with ideas, especially in ARHL. Current data show the importance of the complement pathway, particularly C3 and C3aR. In conclusion, it is hoped that this review of microglia in disease state can provide good ideas and suggestions for exploring unknown degenerative diseases. In the future, When studying neurodegenerative diseases with unknown mechanisms, focusing on the dual role of central microglia may receive unexpected gains, and we may be able to control the activation states of the microglial cell, exosome, and complement pathways, thereby reducing the occurrence of degenerative diseases and achieving therapeutic effects.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported in part by the National Natural Science Foundation of China (#82371153), the China Postdoctoral Science Foundation (#2023M731845), the Natural Science Foundation of Shandong Province (#ZR2021MH378 and #ZR2022QH073), the Shandong Society of Geriatric Science and Technology Project (#LKJGG2021Z020), and the Yantai Science and Technology Innovation Development Project (#2022YD009, #2023YD050 and #2020MSGY078).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sahu MR, Rani L, Subba R, Mondal AC. Cellular senescence in the aging brain: a promising target for neurodegenerative diseases. Mech Ageing Dev. 2022;204:111675. doi:10.1016/j.mad.2022.111675
2. Combes GF, Pellay FX, Radman M. Cause commune et mécanisme commun aux maladies du vieillissement ? [Common cause and mechanism for all pathologies of aging?]. Med Sci. 2020;36(12):1129–1134. doi:10.1051/medsci/2020221 French.
3. Esquerda-Canals G, Montoliu-Gaya L, Güell-Bosch J, Villegas S. Mouse Models of Alzheimer’s Disease. J Alzheimers Dis. 2017;57(4):1171–1183. doi:10.3233/JAD-170045
4. Hayes MT. Parkinson’s Disease and Parkinsonism. Am J Med. 2019;132(7):802–807. doi:10.1016/j.amjmed.2019.03.001
5. Patel R, McKinnon BJ. Hearing Loss in the Elderly. Clin Geriatr Med. 2018;34(2):163–174. doi:10.1016/j.cger.2018.01.001
6. Hindle JV. Ageing, neurodegeneration and Parkinson’s disease. Age Ageing. 2010;39(2):156–161. doi:10.1093/ageing/afp223
7. Fujiwara S, Urata K, Oto T, et al. Age-related Changes in Trigeminal Ganglion Macrophages Enhance Orofacial Ectopic Pain After Inferior Alveolar Nerve Injury. Vivo. 2023;37(1):132–142. doi:10.21873/invivo.13062
8. Karuppagounder V, Giridharan VV, Arumugam S. Modulation of Macrophage Polarization and HMGB1-TLR2/TLR4 Cascade Plays a Crucial Role for Cardiac Remodeling in Senescence-Accelerated Prone Mice. PLoS One. 2016;11(4):e0152922. doi:10.1371/journal.pone.0152922
9. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi:10.1146/annurev-immunol-032713-120240
10. Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi:10.1038/nn1472
11. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi:10.1146/annurev-immunol-051116-052358
12. Wright-Jin EC, Gutmann DH. Microglia as Dynamic Cellular Mediators of Brain Function. Trends Mol Med. 2019;25(11):967–979. doi:10.1016/j.molmed.2019.08.013
13. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–1369. doi:10.1038/s41593-018-0242-x
14. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649–665. doi:10.1111/bph.13139
15. Jorfi M, Maaser-Hecker A, Tanzi RE. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023;15(1):6. doi:10.1186/s13073-023-01155-w
16. Qian B, Yang Y, Tang N, et al. M1 macrophage-derived exosomes impair beta cell insulin secretion via miR-212-5p by targeting SIRT2 and inhibiting Akt/GSK-3β/β-catenin pathway in mice. Diabetologia. 2021;64(9):2037–2051. doi:10.1007/s00125-021-05489-1
17. Zhu B, Liu Y, Hwang S, et al. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol Neurodegener. 2022;17(1):58. doi:10.1186/s13024-022-00562-8
18. Stucky SR, Wolf KE, Kuo T. The economic effect of age-related hearing loss: national, state, and local estimates, 2002 and 2030. J Am Geriatr Soc. 2010;58:618–619.
19. Wu T, Zhou J, Qiu J, et al. Tumor necrosis factor-α mediated inflammation versus apoptosis in age-related hearing loss [J]. Front Aging Neurosci. 2022;14:956503.
20. Seicol BJ, Lin S, Xie R. Age-Related Hearing Loss Is Accompanied by Chronic Inflammation in the Cochlea and the Cochlear Nucleus. Front Aging Neurosci. 2022;14:846804. doi:10.3389/fnagi.2022.846804
21. Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi:10.1016/j.cell.2007.10.036
22. Mou W, Ma L, Zhu A, Cui H, Huang Y. Astrocyte-microglia interaction through C3/C3aR pathway modulates neuropathic pain in rats model of chronic constriction injury. Mol Pain. 2022;18:17448069221140532. doi:10.1177/17448069221140532
23. Pouw RB, Ricklin D. Tipping the balance: intricate roles of the complement system in disease and therapy. Semin Immunopathol. 2021;43(6):757–771. doi:10.1007/s00281-021-00892-7
24. Ziyang L. Research on Specific Pro-Regression Mediators in Delaying the Progression of Retinal Degenerative Diseases and Their Mechanisms[D]. Chinese People’s Liberation Army Army Medical University; 2019; doi:10.27001/d.cnki.gtjyu.2019.000109
25. Fan W, Huang W, Chen J, Li N, Mao L, Hou S. Retinal microglia: functions and diseases. Immunology. 2022;166(3):268–286. doi:10.1111/imm.13479
26. Gao H, Huang X, He J, Zou T, Chen X, Xu H. The roles of microglia in neural remodeling during retinal degeneration. Histol Histopathol. 2022;37(1):1–10. doi:10.14670/HH-18-384
27. Chen M, Luo C, Zhao J, Devarajan G, Xu H. Immune regulation in the aging retina. Prog Retin Eye Res. 2019;69:159–172. doi:10.1016/j.preteyeres.2018.10.003
28. Yulin Q, Qianyu J, Hejiang Y. CX3CR1 Research progress of microglia in retinal degenerative diseases. Int J Ophthalmol. 2021;21(8):1363.
29. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. doi:10.1111/ene.13439
30. Soria Lopez JA, González HM, Léger GC. Alzheimer’s disease. Handb Clin Neurol. 2019;167:231–255. doi:10.1016/B978-0-12-804766-8.00013-3
31. Zhang H, Zheng Y. β淀粉样蛋白级联假说相关的阿尔茨海默病发病机制及防治策略研究进展 [β Amyloid Hypothesis in Alzheimer’s Disease:Pathogenesis, Prevention, and Management]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2019;41(5):702–708. doi:10.3881/j.issn.1000-503X.10875 Chinese.
32. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6(6176). doi:10.1038/ncomms7176
33. Nguyen AT, Wang K, Hu G, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020;140(4):477–493. doi:10.1007/s00401-020-02200-3
34. Bemiller SM, McCray TJ, Allan K, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. 2017;12(74):216.
35. Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–1593. doi:10.1038/nn.4132
36. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–472. doi:10.1083/jcb.201709069
37. George S, Rey NL, Tyson T, et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener. 2019;14(1):34. doi:10.1186/s13024-019-0335-3
38. Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between α-synuclein and microglia in Parkinson’s disease: recent developments. Neuroscience. 2015;302:47–58. doi:10.1016/j.neuroscience.2015.02.008
39. Liu TW, Chen CM, Chang KH. Biomarker of Neuroinflammation in Parkinson’s Disease. Int J Mol Sci. 2022;23(8):4148. doi:10.3390/ijms23084148
40. Zhang YN, Fan JK, Gu L, Yang HM, Zhan SQ, Zhang H. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J Neuroinflammation. 2021;18(1):23. doi:10.1186/s12974-021-02079-1
41. Xu Y, Li Y, Wang C, et al. The reciprocal interactions between microglia and T cells in Parkinson’s disease: a double-edged sword. J Neuroinflammation. 2023;20(1):33. doi:10.1186/s12974-023-02723-y
42. Ho MS. Microglia in Parkinson’s Disease. Adv Exp Med Biol. 2019;1175:335–353. doi:10.1007/978-981-13-9913-8_13
43. El Andaloussi S, Lakhal S, Mäger I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65(3):391–397. doi:10.1016/j.addr.2012.08.008
44. Guo M, Wang J, Zhao Y, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143(5):1476–1497. doi:10.1093/brain/awaa090
45. Guo Y, Wei X, Yan H, et al. TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 2019;33(11):12164–12174. doi:10.1096/fj.201900992R
46. Zhang Y, Feng S, Nie K, et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem Biophys Res Commun. 2018;499(4):797–802. doi:10.1016/j.bbrc.2018.03.226
47. Boillée S, Ande V Elde C V, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59.
48. Kassa RM, Mariotti R, Bonaconsa M, Bertini G, Bentivoglio M. Gene, cell, and axon changes in the familial amyotrophic lateral sclerosis mouse sensorimotor cortex. J Neuropathol Exp Neurol. 2009;68:59–72.
49. Moisse K, Strong MJ. Innate immunity in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1083–1093.
50. Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4(4):389–398. doi:10.1007/s11481-009-9171-5
51. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D’Ambrosi N. The Dual Role of Microglia in ALS: mechanisms and Therapeutic Approaches. Front Aging Neurosci. 2017;9:242. doi:10.3389/fnagi.2017.00242
52. Brites D, Vaz AR. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci. 2014;8:117. doi:10.3389/fncel.2014.00117
53. Choi S, Guo L, Cordeiro MF. Retinal and Brain Microglia in Multiple Sclerosis and Neurodegeneration. Cells. 2021;10(6):1507. doi:10.3390/cells10061507
54. McNamara NB, Munro DAD, Bestard-Cuche N, et al. Microglia regulate central nervous system myelin growth and integrity. Nature. 2023;613(7942):120–129. doi:10.1038/s41586-022-05534-y
55. Guerrero BL, Sicotte NL. Microglia in Multiple Sclerosis: friend or Foe? Front Immunol. 2020;11:374. doi:10.3389/fimmu.2020.00374
56. Yong VW. Microglia in multiple sclerosis: protectors turn destroyers. Neuron. 2022;110(21):3534–3548. doi:10.1016/j.neuron.2022.06.023
57. Gillen KM, Mubarak M, Nguyen TD, Pitt D. Significance and In Vivo Detection of Iron-Laden Microglia in White Matter Multiple Sclerosis Lesions. Front Immunol. 2018;9:255. doi:10.3389/fimmu.2018.00255
58. Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014;128(2):191–213. doi:10.1007/s00401-014-1310-2
59. Rawji KS, Yong VW. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol. 2013;2013:948976. doi:10.1155/2013/948976
60. Voet S, Prinz M, van Loo G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol Med. 2019;25(2):112–123. doi:10.1016/j.molmed.2018.11.005
61. Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180–186. doi:10.1038/nature20411
62. Taguchi T, Ota H, Matsuda T, Murase S, Mizumura K. Cutaneous C-fiber nociceptor responses and nociceptive behaviors in aged Sprague–Dawley rats. Pain. 2010;151:771–782. doi:10.1016/j.pain.2010.09.011
63. Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and pro-inflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83.
64. Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7:816–822. doi:10.1016/j.jpain.2006.04.004
65. Pan TT, Pan F, Gao W, Hu SS, Wang D. Involvement of Macrophages and Spinal Microglia in Osteoarthritis Pain. Curr Rheumatol Rep. 2021;23(5):29. doi:10.1007/s11926-021-00997-w
66. Hahm SC, Lee JS, Yoon YW, Kim J. Analgesic Tolerance Development during Repetitive Electric Stimulations Is Associated with Changes in the Expression of Activated Microglia in Rats with Osteoarthritis. Biomedicines. 2020;8(12):575. doi:10.3390/biomedicines8120575
67. Sago T, Ono K, Harano N, et al. Distinct time courses of microglial and astrocytic hyperactivation and the glial contribution to pain hypersensitivity in a facial cancer model. Brain Res. 2012;1457:70–80. doi:10.1016/j.brainres.2012.03.039
68. Shinoda M, Kubo A, Hayashi Y, Iwata K. Peripheral and central mechanisms of persistent orofacial Pain. Front Neurosci. 2019;13:1227. doi:10.3389/fnins.2019.01227
69. Ikutame D, Urata K, Oto T, et al. Aging-Related Phenotypic Conversion of Medullary Microglia Enhances Intraoral Incisional Pain Sensitivity. Int J Mol Sci. 2020;21(21):7871. doi:10.3390/ijms21217871
70. Jin MM, Wang F, Qi D, et al. A Critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci. 2018;10:378. doi:10.3389/fnagi.2018.00378
71. Laffer B, Bauer D, Wasmuth S, et al. Loss of IL-10 Promotes Differentiation of Microglia to a M1 Phenotype. Front Cell Neurosci. 2019;13:430. doi:10.3389/fncel.2019.00430
72. Du X, Xu Y, Chen S, Fang M. Inhibited CSF1R Alleviates Ischemia Injury via Inhibition of Microglia M1 Polarization and NLRP3 Pathway. Neural Plast. 2020;2020:8825954. doi:10.1155/2020/8825954
73. Lin W, Xu D, Austin CD, et al. Function of CSF1 and IL34 in Macrophage Homeostasis, Inflammation, and Cancer. Front Immunol. 2019;10:2019. doi:10.3389/fimmu.2019.02019
74. Marzan DE, Brügger-Verdon V, West BL, Liddelow S, Samanta J, Salzer JL. Activated microglia drive demyelination via CSF1R signaling. Glia. 2021;69(6):1583–1604. doi:10.1002/glia.23980
75. Sosna J, Philipp S, Albay R 3rd, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener. 2018;13(1):11. doi:10.1186/s13024-018-0244-x
76. Kiani Shabestari S, Morabito S, Danhash EP, et al. Absence of microglia promotes diverse pathologies and early lethality in Alzheimer’s disease mice. Cell Rep. 2022;39(11):110961. doi:10.1016/j.celrep.2022.110961
77. Feng X, Valdearcos M, Uchida Y, Lutrin D, Maze M, Koliwad SK. Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI Insight. 2017;2(7):e91229. doi:10.1172/jci.insight.91229
78. Liu YJ, Spangenberg EE, Tang B, Holmes TC, Green KN, Xu X. Microglia Elimination Increases Neural Circuit Connectivity and Activity in Adult Mouse Cortex. J Neurosci. 2021;41(6):1274–1287. doi:10.1523/JNEUROSCI.2140-20.2020
79. Fujiwara T, Yakoub MA, Chandler A, et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol Cancer Ther. 2021;20(8):1388–1399. doi:10.1158/1535-7163.MCT-20-0591
80. Henry RJ, Ritzel RM, Barrett JP, et al. Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. J Neurosci. 2020;40(14):2960–2974. doi:10.1523/JNEUROSCI.2402-19.2020
81. Chokr SM, Milinkeviciute G, Jimenez GA, Abubakr H, Cramer KS. Long-term microglia depletion impairs synapse elimination and auditory brainstem function. Sci Rep. 2022;12(1):18521. doi:10.1038/s41598-022-23250-5
82. Van Zeller M, Sebastião AM, Valente CA. Microglia Depletion from Primary Glial Cultures Enables to Accurately Address the Immune Response of Astrocytes. Biomolecules. 2022;12(5):666. doi:10.3390/biom12050666
83. Qiu S, Palavicini JP, Wang J, et al. Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Mol Neurodegener. 2021;16(1):64. doi:10.1186/s13024-021-00488-7
84. Elmore MR, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi:10.1016/j.neuron.2014.02.040
85. Catorce MN, Gevorkian G. LPS-induced Murine Neuroinflammation Model: main Features and Suitability for Pre-clinical Assessment of Nutraceuticals. Curr Neuropharmacol. 2016;14(2):155–164. doi:10.2174/1570159x14666151204122017
86. Cheng J, Dong Y, Ma J, et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci Adv. 2021;7(35):eabe3600. doi:10.1126/sciadv.abe3600
87. Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi:10.1523/JNEUROSCI.4268-04.2005
88. Kenkhuis B, Somarakis A, Kleindouwel LRT, van Roon-Mom WMC, Höllt T, van der Weerd L. Co-expression patterns of microglia markers iba-1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol Dis. 2022;167:105684. doi:10.1016/j.nbd.2022.105684
89. Kondo S, Kohsaka S, Okabe S. Long-term changes of spine dynamics and microglia after transient peripheral immune response triggered by LPS in vivo. Mol Brain. 2011;4:27. doi:10.1186/1756-6606-4-27
90. Du Y, Brennan FH, Popovich PG, Zhou M. Microglia maintain the normal structure and function of the hippocampal astrocyte network. Glia. 2022;70(7):1359–1379. doi:10.1002/glia.24179
91. Guo M, Hao Y, Feng Y, et al. Microglial Exosomes in Neurodegenerative Disease. Front Mol Neurosci. 2021;14:630808. doi:10.3389/fnmol.2021.630808
92. Wan T, Huang Y, Gao X, Wu W, Guo W. Microglia Polarization: a Novel Target of Exosome for Stroke Treatment. Front Cell Dev Biol. 2022;10:842320. doi:10.3389/fcell.2022.842320
93. Rai SN, Singh P. Advancement in the modelling and therapeutics of Parkinson’s disease. J Chem Neuroanat. 2020;104:101752. doi:10.1016/j.jchemneu.2020.101752
94. Yadav SK, Rai SN, Singh SP. Mucuna pruriens reduces inducible nitric oxide synthase expression in Parkinsonian mice model. J Chem Neuroanat. 2017;80:1–10. doi:10.1016/j.jchemneu.2016.11.009
95. Rai SN, Singh P, Steinbusch HWM, Vamanu E, Ashraf G, Singh MP. The Role of Vitamins in Neurodegenerative Disease: an Update. Biomedicines. 2021;9(10):1284. doi:10.3390/biomedicines9101284
96. Rai SN, Dilnashin H, Birla H, et al. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox Res. 2019;35(3):775–795. doi:10.1007/s12640-019-0003-y
97. Ramakrishna K, Nalla LV, Naresh D, et al. WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: current Status and Future Perspective. Diseases. 2023;11(3):89. doi:10.3390/diseases11030089
98. Rai SN, Yadav SK, Singh D, Singh SP. Ursolic acid attenuates oxidative stress in nigrostriatal tissue and improves neurobehavioral activity in MPTP-induced Parkinsonian mouse model. J Chem Neuroanat. 2016;71:41–49. doi:10.1016/j.jchemneu.2015.12.002
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Dexmedetomidine Alleviates Neuropathic Pain via the TRPC6-p38 MAPK Pathway in the Dorsal Root Ganglia of Rats
Xu S, Yi Y, Wang Y, Wang P, Zhao Y, Feng W
Journal of Pain Research 2022, 15:2437-2448
Published Date: 19 August 2022
Selenium Nanoparticles-Enriched Lactobacillus casei ATCC 393 Prevents Cognitive Dysfunction in Mice Through Modulating Microbiota-Gut-Brain Axis

Qiao L, Chen Y, Song X, Dou X, Xu C
International Journal of Nanomedicine 2022, 17:4807-4827
Published Date: 13 October 2022
Neural Stem Cell-Derived Exosomes Improve Neurological Function in Rats with Cerebral Ischemia-Reperfusion Injury by Regulating Microglia-Mediated Inflammatory Response

Zhao X, Zhu J, Chen S, Liu R, Long T
Journal of Inflammation Research 2023, 16:3079-3092
Published Date: 24 July 2023
Advances in the Understanding of the Correlation Between Neuroinflammation and Microglia in Alzheimer’s Disease
Yan H, Wang W, Cui T, Shao Y, Li M, Fang L, Feng L
ImmunoTargets and Therapy 2024, 13:287-304
Published Date: 12 June 2024
Prevention and Treatment Strategies for Alzheimer’s Disease: Focusing on Microglia and Astrocytes in Neuroinflammation
Zhang S, Gao Z, Feng L, Li M
Journal of Inflammation Research 2024, 17:7235-7259
Published Date: 13 October 2024