Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 7
Exploring targets and therapies for amyotrophic lateral sclerosis: current insights into dietary interventions
Authors Ngo ST ![]() , Mi JD, Henderson RD, McCombe PA, Steyn FJ
, Mi JD, Henderson RD, McCombe PA, Steyn FJ
Received 1 May 2017
Accepted for publication 24 May 2017
Published 25 July 2017 Volume 2017:7 Pages 95—108
DOI https://doi.org/10.2147/DNND.S120607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Shyuan T Ngo,1–5 Jia D Mi,1 Robert D Henderson,3,4 Pamela A McCombe,3,4 Frederik J Steyn3–5
1School of Biomedical Sciences, 2Queensland Brain Institute, 3Centre for Clinical Research, The University of Queensland, 4Department of Neurology, Royal Brisbane & Women’s Hospital, 5Wesley Medical Research, The Wesley Hospital, Auchenflower, Brisbane, QLD, Australia
Abstract: A growing number of preclinical and human studies demonstrate a disease-modifying effect of nutritional state in amyotrophic lateral sclerosis (ALS). The management of optimal nutrition in ALS is complicated, as physiological, physical, and psychological effects of the disease need to be considered and addressed accordingly. In this regard, multidisciplinary care teams play an integral role in providing dietary guidance to ALS patients and their carers. However, with an increasing research focus on the use of dietary intervention strategies to manage disease symptoms and improve prognosis in ALS, many ALS patients are now seeking or are actively engaged in using complementary and alternative therapies that are dietary in nature. In this article, we review the aspects of appetite control, energy balance, and the physiological effects of ALS relative to their impact on overall nutrition. We then provide current insights into dietary interventions for ALS, considering the mechanisms of action of some of the common dietary interventions used in ALS, discussing their validity in the context of clinical trials.
Keywords: ALS, treatment, alternative off-label therapies, antioxidants, clinical trials, nutrition
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by the loss of motor neurons in the cerebral cortex, brainstem, and spinal cord. Death of these neurons results in progressive paralysis and disability, culminating in respiratory failure and death within 2–5 years following diagnosis.1,2 In the absence of an effective treatment for ALS, there is increased emphasis on interventions to improve the quality and extend the duration of life.
Engagement in ALS multidisciplinary care increases the quality of life while extending survival.3 As part of this process, patients receive dietary guidance preferably by a dietician. While addressing dysphagia, nutritional management also seeks to address changes in appetite that can occur as part of the disease, possibly due to physiological and psychological effects of ALS. Worsening of nutritional state, as reflected by weight loss, is associated with worsening motor symptoms and survival.4,5 This highlights the critical need for patients to meet energy requirements. In this review, we summarize the merits and mechanisms of action of dietary interventions proposed for ALS. We discuss the variable impact of ALS on overall energy balance, noting the effects on appetite control and energy expenditure. We also discuss dietary interventions within the context of risk factors known to impact energy balance and dietary needs in ALS.
Meeting energy needs and maintaining energy homeostasis in ALS
Undernutrition is observed in ALS;6 however, there is significant nutritional heterogeneity among individuals. This is of clinical importance as nutritional state may negatively impact survival,7 whereas maintenance of body weight is an effective strategy to extend survival.8 Moreover, recent discoveries of the compounding effects of coexisting neurodegenerative conditions (including behavioral variants of frontotemportal dementia [bvFTD]) on nutritional status and appetite control in ALS patients highlight additional complexities and heterogeneity of dietary control.9 This heterogeneity indicates a need for well-directed treatments to improve nutrition in ALS while calling for a greater understanding of all factors that might impair energy homeostasis in ALS.
In ALS, a number of studies provide evidence for impairments in control of energy homeostasis (the balance of energy supply and use), which may compromise the capacity to meet energy needs. In particular, patients can experience reduced appetite, leading to reduced intake of food, and thus a failure to meet their energy requirements.
Complications of appetite control and ALS
Hunger represents our need to eat, a sensation that is generally guided by intrinsic mechanisms that promote behaviors that seek food. Appetite, by contrast, is a far more complex behavior, defined as our desire and capacity to consume energy. Many intrinsic and external factors may contribute to appetite, including emotions and/or our physical capacity to access food.10 Considerable changes in appetite and eating behavior are observed in ALS.11 Impaired appetite control in ALS could directly contribute to calorie deficit or excess, compounding weight loss, or weight gain. In ALS, appetite can be affected by physical difficulties, including difficulties in swallowing (dysphagia) or progressive disability that restricts access to food (including paralysis resulting in loss of use of hands), or dysfunction of endogenous processes that regulate hunger, satiety, and mood. Mechanisms of impaired appetite control discussed in this review are summarized in Figure 1.
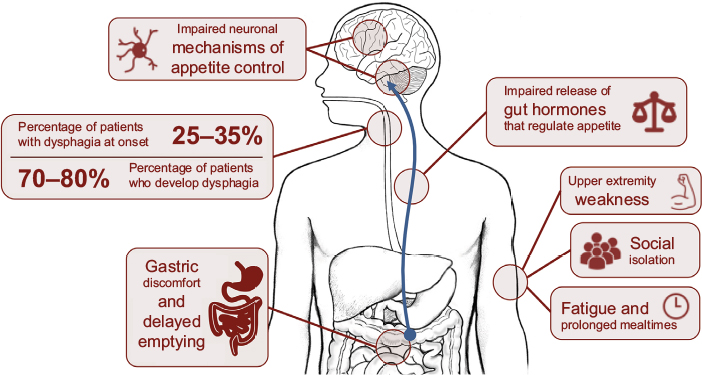 | Figure 1 Mechanisms of impaired appetite control in amyotrophic lateral sclerosis (ALS). Notes: Appetite control in ALS may be compromised by progressive worsening of disability or dysfunction of mechanisms that control hunger and satiety. Disability may result in dysphagia, upper extremity weakness resulting in a reduced capacity to access food or self-feed, social isolation during mealtimes, and/or fatigue and prolonged mealtimes resulting in reduced calorie or liquid consumption. Evidence suggests dysfunction of endogenous processes that control appetite regulation in ALS, including gastric discomfort and delayed gastric emptying, reduced release of gastric hormones that control appetite, or dysfunction of neuronal processes that control appetite. |
Progressive loss of cranial nerves contributes to difficulties in regulating the complex swallowing process.12 This results in poor control of the food bolus during swallowing. When observed alongside declining respiratory status, weakening oral control can lead to fear of choking, a reduction in calorie and fluid consumption, and possibly malnutrition.13 In ALS, 25%–35% of patients experience bulbar onset of weakness with dysphagia, and 70%–80% of patients develop dysphagia throughout the course of the disease.14,15 Initial management of dysphagia involves modification of food and fluid consistency (blending food, adding thickeners to liquids) and education of patients and carers in swallowing techniques.16 Swallowing difficulties can be managed through enteral feeding.
Disabilities other than dysphagia can increase the risk of calorie deficit in ALS; upper extremity weakness can impair the capacity to prepare or manipulate food. This can be circumvented through assistance from a caregiver; however, prolonged mealtimes and fatigue increase the risk of developing dehydration and suboptimal nutrition.17 Disability can also result in social isolation and embarrassment, further compromising a patient’s nutritional status. Social aspects of eating can be affected by dysphagia or time-consuming and exhausting practices associated with eating. Introduction of adapted eating utensils can help a patient to maintain autonomy.18 Sialorrhoea (drooling or excessive salivation) can further burden patients during mealtimes, especially within social settings,16,19 lessening the desire to eat, and the enjoyment of meals. Treatment options range from anticholinergic drugs and botulinum toxin injections to reduce saliva production, through to radiological and surgical interventions.19 Gastric emptying and colonic transit times can also be delayed in ALS.20 While contributing to discomfort associated with eating, impairments in gut function or delayed gastric emptying can also impact the release of gut hormones21 associated with appetite control,22 resulting in altered perception of hunger and satiety.
Appetite is controlled by peripheral and central mechanisms that sense and respond to changing energy balance. Energy homeostasis is achieved when circulating signals inform the brain of available fat stores and the brain then responds by stimulating corrective adjustments to food intake to maintain these stores. Various brain centers receive information on energy availability, calorie provision, and energy needs while also responding to behaviors and rewards (pleasure) associated with eating.23 Dysfunction of any component within this network or changes in the way our brain responds to food could result in impairments in appetite control, leading to impairments in maintenance of body weight.
There is evidence of impairment of appetite-regulating pathways in ALS.24 Hypothalamic neurons that promote satiety are less responsive in mouse models of ALS and in ALS patients than in controls.25 Dysfunction of appetite control is further highlighted by TDP43 pathology in the lateral hypothalamus of some ALS patients,26 with the accumulation of TDP43 thought to impair the function of, or contribute to the death of neurons that control appetite. TDP43 proteinopathy is also observed in areas of the brain associated with appetite motivation,26 and thus impairments in appetite control in ALS may extend beyond hypothalamic control of energy homeostasis.
Clinical implications of altered appetite regulation in ALS, particularly with respect to weight management and prognosis, are still under investigation. Understanding of these pathways could lead to suggestions for therapy, and so it is important that we understand the changing energy needs of ALS patients.
Challenges to metabolic balance and ALS
Hypermetabolism in ALS5,27,28 has been suggested to contribute energy deficit, weight loss,29 and reduced survival.27 However, weight loss in ALS occurs regardless of the metabolic status.30 Therefore, hypermetabolism in ALS may only contribute to energy deficit and weight loss, should appetite be impaired. Given that hypermetabolism is linked to shorter survival,27 attempts to define the cause of hypermetabolism and to manage hypermetabolism in ALS remains of interest.
Dietary interventions in ALS
There is evidence that ALS patients are adopting self-prescribed dietary strategies. Over 600 dietary strategies and/or supplements are reported to be used by ~9500 ALS patients currently registered with Patients Like Me, a web-based community where members share details about their treatments.31 This is in line with findings that ~80% of ALS patients use high-dose vitamins, minerals, and other nutraceuticals.32 Common dietary strategies include avoidance of certain dietary components, adoption of very specific dietary regimes, or use of dietary supplements. Of these strategies, the majority have not been tested in clinical trials, or have produced no positive effects when tested. Below, we discuss dietary interventions for which there is some evidence of benefit. Outcomes are summarized in Table 1.
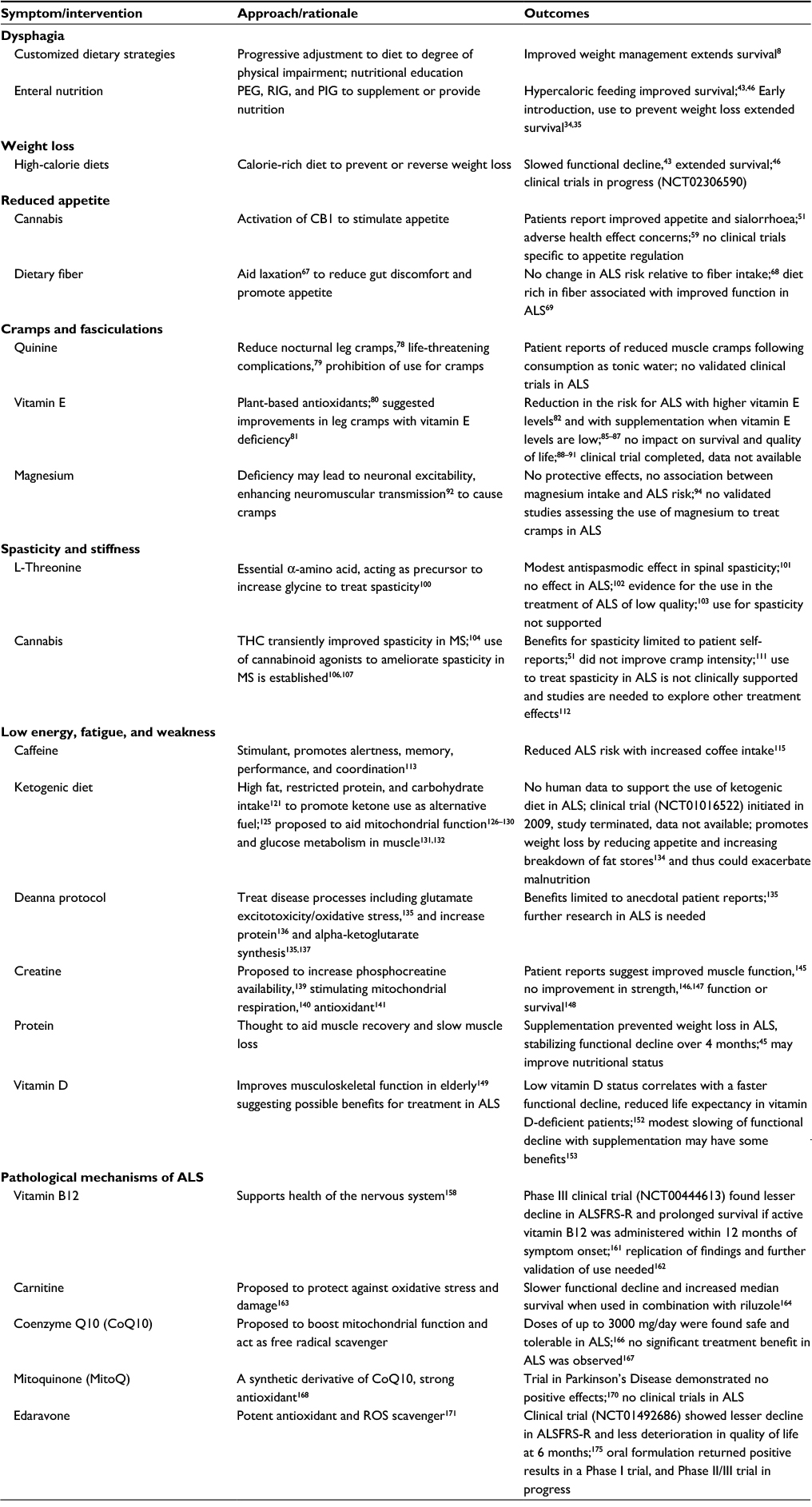 | Table 1 Dietary interventions proposed to improve capacity to meet energy needs or treat complications associated with ALS Abbreviations: PEG, endoscopic gastrostomy; RIG, radiologically inserted gastrostomy; PIG, per-oral image-guided gastrostomy; CBI, cannabinoid receptors; MS, multiple sclerosis; ROS, reactive oxygen species; ALS, amyotrophic lateral sclerosis; ALSFRS-R, ALS Functional Rating Scale-Revised. |
Dietary interventions that address dysphagia in ALS
Customizing dietary strategies to suit ALS
Management of dysphagia needs to be progressively adjusted to accommodate the changing degree of impairment throughout the course of ALS. While dysphagia may not be observed early in disease course, early education regarding nutrition should be considered. At the onset of upper extremity and/or bulbar dysfunction, greater care should be taken to maintain stable body weight, and it is recommended that assessments of dietary efficiency occur at 3-month intervals. Weight and respiratory capacity should be assessed, and patients are encouraged to consult with speech pathologists for advice on improving swallowing techniques.17 Patients may consider more effective dietary strategies to reduce the risk of choking or aspiration including altering the texture and consistency of food,15,16,33 consuming smaller portions more frequently, consuming calorie-rich foods, and increasing mastication time.17 With progressive bulbar dysfunction, the introduction of enteral nutrition should be offered. While not an attractive option to all patients,17 this does improve survival.34,35
It is important that caregivers are actively engaged. Those who take on a caregiver role need to adapt to cater for the specific issue of ALS. Due to physical disabilities in ALS, carers may be responsible for the majority of meal preparation36 and, thus, should be aware of dietary needs specific to ALS. Recently, the Motor Neurone Disease Association released information, recipes, and advice to assist patients and caregivers in sustaining optimal dietary intake while also providing advice on how to maintain enjoyment for food in social settings.37
Enteral nutrition
As bulbar symptoms progress, or when patients lose >10% of their baseline body weight, enteral feeding is often indicated.38 A recent prospective study assessing the use of gastrostomy in ALS suggests that enteral feeding should be considered when patients lose 5% of their baseline body weight, and when their nutritional intake does not match their energy requirements.39,40
Enteral feeding in ALS can be delivered via nasogastric tubing (NGT) or gastrostomy. Although feeding through NGT is a minor and noninvasive procedure, migration of the NGT, oropharyngeal secretion, nasopharyngeal discomfort, pain, and ulceration limits its long-term use in ALS.16,17 Gastrostomy is commonly used for patients who require long-term enteral nutrition,41 and methods used in ALS include percutaneous endoscopic gastrostomy (PEG), radiologically inserted gastrostomy (RIG), and per-oral image-guided gastrostomy (PIG).42 For more information on the use of gastrostomy in ALS, we encourage the reader to consult recent reviews.41,42
Recently, a double-blind, Phase II randomized clinical trial reported benefits of hypercaloric feeding via enteral nutrition. High-calorie supplementation improved survival outcome compared to an isocaloric intervention.43 Importantly, it has also been found that placement of PEG in patients with dysphagia34 and early introduction and efficient use of PEG to prevent weight loss is able to extend survival in ALS.35 Hence, refinements in the use of PEG might further improve disease outcome for those with ALS.
Dietary interventions that address weight loss in ALS
High-calorie diets
Use of high-calorie supplementation in ALS has stemmed from the finding that calorie-rich diets prevent or reverse weight loss in ALS5,44 by offsetting malnutrition and increasing energy requirements. In ALS, a limited number of studies have assessed caloric supplementation by modifying protein,45 carbohydrate, or fat43,46 content in dietary regimens. While such studies report improvements or stabilization of body weight45 or ALS Functional Rating Scale-Revised (ALSFRS-R),43 only the prospective interventional study by Wills et al was able to demonstrate improvements in survival following high-calorie/high-carbohydrate, or high-calorie/high-fat supplementation.43 Congruent with this, Dorst et al recently reported that high-calorie intake via PEG was associated with longer survival in ALS.46 While these data are promising, small study sample size and limited experimental design necessitate that larger, double-blinded randomized clinical trials are conducted to clarify dietary strategies that improve prognosis in ALS. In this regard, a double-blinded randomized clinical trial to assess the efficacy, safety, and tolerability of high-lipid and -calorie supplementation in 200 ALS participants (NCT02306590) is currently underway. With the primary endpoint of the trial being survival time from randomization until death or date of study completion, data from this trial will shed some light on the utility of high-calorie supplementation as a treatment approach for ALS.
Dietary interventions that address reduced appetite and digestive health in ALS
Cannabis
Cannabis plants contain over 500 distinct chemical functional molecules, of which delta-9-tetrahydrocannabinol (THC) is considered the most biologically active.47 Cannabinoids (CBs) such as THC modulate their effects via CB receptors expressed throughout the brain (predominantly CB1 receptors) and immune system (predominantly CB2 receptors).48 The reader is encouraged to consult recent reviews on the use of cannabis and pharmacological targeting of the endocannabinoid system in treating disease.49,50 We limit the discussion to advances in the use of cannabis and cannabis-derived factors in treating symptoms associated with ALS, including appetite and spasticity.
ALS patients report improvements in appetite and sialorrhoea following the use of cannabis,51 suggesting that CBs may negate some effects of ALS on appetite control.52 Studies in mice show that CBs act via the CB1 receptor to promote food intake. Specifically, CBs promote the activity of hypothalamic pro-opiomelanocorti-(POMC)-expressing neurons (a neuronal population better known to promote satiety53), stimulating β-endorphin release.54 In turn, β-endorphins promote food intake, acting on additional brain circuitry.55 These actions of CBs on POMC-expressing neurons are thought to occur via selective modification of mitochondrial activity.54 Since mitochondrial dysfunction56–58 and aberrant hypothalamic control of appetite regulation occurs in ALS,25,26 use of CBs may not improve appetite control across all ALS patients. There are potential adverse health effects of cannabis,59 so there is a need for alternative compounds to promote hunger in ALS. Ghrelin is a potent endogenous orexigenic factor that stimulates appetite to increase calorie consumption.60 Given the reduced circulating levels of ghrelin in ALS,21,61 treatment with ghrelin may improve appetite. Moreover, as ghrelin has been shown to protect spinal cord motoneurons from chronic glutamate excitotoxicity while inhibiting microglial activation,62,63 benefits of ghrelin treatment may extend beyond improvements in appetite control.64 To our knowledge, no clinical trials have assessed the use of cannabis, CBs, or orexigenic factors such as ghrelin in promoting appetite in ALS. Of interest, a patent for the use of an analog that targets the ghrelin receptor as treatment in ALS has been filed (Patent Cooperation Treaty number JP2013/078743).
Dietary fiber
Fiber lowers the concentrations of inflammatory markers and can reduce the risk of mortality in some inflammatory diseases.65,66 Moreover, dietary fiber is known to promote gut health, thereby improving laxation.67 Improvements in gut function following increased ingestion of fiber may also aid the production and release of key gut hormones (including ghrelin), thereby enhancing appetite control.67 Comparing outcomes from five large cohort studies, no change in ALS risk relative to dietary fiber intake was observed.68 Of interest, a diet rich in antioxidant nutrients, carotenoids and fiber, and vegetables is associated with improved function in ALS patients.69 Whether improved function was due to increased fiber intake was not specifically addressed. It remains unclear whether dietary fiber led to improved gut health, reduced colonic transit times to improve laxation, or improved appetite control in ALS.
Dietary interventions directed toward physical disability in ALS
Dietary modification for cramps and fasciculations
Muscle cramping and fasciculations are widely reported in ALS,70 with symptoms presenting prior to the onset of weakness and/or muscle wasting.71,72 Fasciculations occur as a consequence of abnormal excitation of the terminal branches of motor axons,73 whereas cramps may occur as a consequence of mechanical excitation of the motor nerve terminal,73 hyperexcitability or bistability of spinal motor neurons,74 or through impairments in inhibitory mechanisms of interneuron activity.75 Muscle cramping in ALS can be persistent and can cause debilitating pain. Various drugs have been proposed to treat cramps in ALS, including antispasmodics such as Baclofen or Zanaflex.76 To manage cramping, patients report the use of quinine and increased consumption of dietary supplements including vitamin E and magnesium.
Quinine
Quinine is used for the treatment of malaria.77 While showing some benefits in managing nocturnal leg cramps,78 the occurrence of life-threatening complications79 has resulted in a ruling by the FDA to prohibit the use of quinine for the treatment of cramps. Promotion of quinine for treatment of leg cramps has been prohibited in the USA since February 1995, with the marketing of quinine-containing preparations banned by the FDA since December 2006 (FDA, Docket No. 2006N-0476). ALS patients report reduced muscle cramps following the consumption of quinine as tonic water, with amounts of quinine ingested per liter of tonic water estimated to be ~25-fold lower than the proposed daily dosage for the treatment of malaria.77
Vitamin E
Vitamin E is a group of lipid-soluble antioxidants commonly found in plant products.80 Investigation of the effects of vitamin E on dermatological conditions led to the suggestion of treatment benefits on nocturnal leg cramping in individuals with possible vitamin E deficiency.81 While vitamin E deficiency in ALS is not universal,82–84 a potential reduction in risk for ALS is suggested in those with higher vitamin E levels,82 or those with low baseline vitamin E levels who are receiving vitamin E supplementation.85–87 Despite these positive associations, oral administration of vitamin E does not impact ALS survival or the quality of life.88–91 A randomized crossover trial to assess the impact of vitamin E on the number, duration, and severity of cramps was initiated in 2006 (NCT00372879). While this trial is now complete, the results have not been made public.
Magnesium
Magnesium deficiency can lead to neuronal excitability, enhancing neuromuscular transmission.92 Accordingly, magnesium supplementation has been proposed for the treatment of cramps. A recent systematic review of randomized controlled trials with meta-analysis found no evidence for effective management of cramps using magnesium. A small effect in reducing nocturnal cramps was observed in pregnant women only.93 Comparing the results from five large cohort studies, recent observations found no association between magnesium intake and ALS risk, or protective effects of magnesium on ALS,94 although modest effects had been suggested previously.95 We could not identify any studies assessing the specific use of magnesium to manage cramps in ALS.
Dietary modification for spasticity and stiffness
Spasticity, an increase in tonic stretch reflexes due to hyperexcitability, is observed in a number of neurological disorders as a result of loss of upper motor neurons.96 Symptoms include increased tone, spasms, and/or clonus, and spasticity can lead to severe disability, pain, or incapacitation.97 Pharmacological treatments include Baclofen, alpha-2 agonists such as clonidine, or use of anticonvulsants such as benzodiazepines.96 Given the possible side effects of pharmacological treatment,98 a multidisciplinary approach combining medication and physical therapy is recommended.96 Use of nutraceuticals including L-threonine and CBs can be considered for treatment of spasticity.
L-Threonine
L-Threonine is an essential α-amino acid, acting as a glycine precursor to increase glycine levels in the central nervous system.99 Potential use of glycine to treat spasticity has led to the assessment of L-threonine as a possible treatment for spasticity in ALS.100 A double-blind, placebo-controlled, crossover study of oral L-threonine to treat spinal spasticity found a modest antispasmodic effect.101 Similar treatment effects were not observed in ALS, with a pilot observation100 and a double-blind, placebo-controlled, crossover study of L-threonine showing no improvements in ALS.102 These observations were unexpected, given suggested positive treatment effects following short-term L-threonine treatment of ALS.102 A recent Cochrane review found evidence for the effective use of L-threonine in the treatment of ALS to be of low quality,103 and thus the use of L-threonine for treatment of spasticity in ALS remains unsupported.
Cannabis
Clinical use of cannabis and CBs in treating spasticity originates from observations in multiple sclerosis (MS), where synthetic THC treatment transiently improved spasticity scores.104 This observation is in line with studies that implicate the endocanabinoid system in the pathophysiology of MS.105 The use of CB agonists to ameliorate spasticity in MS is established.106,107 Evidence supporting the use of cannabis or cannabis-derived compounds in treating ALS is mostly limited to animal studies, and in particular observations of delayed disease progression following treatment of SOD1G93A mice with a CB1/CB2 agonist.108 Moreover, the upregulation in CB2 receptor expression109 and potentiation of CB1 receptors controlling glutamatergic and GABAergic transmission in ALS mice110 suggest the involvement of the endocannabinoid system in the pathophysiology of ALS. Unlike MS, clinical evidence showing positive treatment effects of CB use in ALS is mostly limited to patient self-reports of subjective improvement in spasticity.51 These patient reports are contradicted by a small randomized double-blind crossover trial, where twice-daily treatment with THC did not improve cramp intensity.111 Given the lack of evidence, use of THC in treating spasticity in ALS is not clinically supported, and more studies are needed to explore other treatment effects.112
Dietary modification for low energy, fatigue, and weakness
Caffeine
Caffeine is a commonly consumed stimulant, acting within the central nervous system via various mechanisms to promote alertness, memory, performance, and coordination.113 Chronic consumption of caffeine in the SOD1G93A mouse model negatively influenced motor performance and reduced survival duration,114 prompting fears that caffeine might worsen disease survival. However, subsequent studies in humans found no association between caffeine and risk for developing ALS.115 This contradicts findings from a case–control study, observing a reduction in ALS risk relative to increased coffee consumption.116 Recent reviews discuss the possible mechanisms of action and perceived benefits of caffeine in ALS and other neurodegenerative diseases.117–119
Ketogenic diet
Introduced in 1921 for the treatment of epilepsy,120 the ketogenic diet is a regimen that encompasses high dietary intake of fat alongside the restriction of protein and carbohydrates.121 The ketogenic diet has been trialed as a nonpharmacological therapeutic approach across a number of neurological conditions.122–124 While its neuroprotective effects are proposed to range from mitigating inflammation to alleviating neuronal excitotoxicity, data remain contentious.123 In ALS, the most pertinent mechanism by which the ketogenic diet could exert neuroprotection is through the provision of fat, which is converted into ketone bodies for use as a fuel substrate for the generation of cellular energy.125 In light of defective mitochondrial function in skeletal muscle126–130 of ALS patients, and mouse data highlighting muscle weakness in the presence of defective skeletal muscle glucose metabolism,131,132 the ketogenic diet offers an avenue through which skeletal muscle can bypass mitochondrial dysfunction and the need for glucose to sustain function. Improved motor performance in SOD1G93A mice fed a ketogenic diet implies that this may be the case. However, given that improved motor performance was observed alongside the attenuation of motor neuron death,133 it remains unknown whether the ketogenic diet has direct effects on muscle to mitigate weakness. To date, there is limited mouse data, and no human data to support the adoption of a ketogenic diet in ALS. While a clinical trial to assess the safety and tolerability of the ketogenic diet in ALS (NCT01016522) was initiated in 2009, the study was terminated, and no data have been disseminated. Critically, since the ketogenic diet promotes weight loss by reducing appetite and increasing the breakdown of endogenous fat stores,134 this could exacerbate malnutrition and weight loss in ALS. Further research is needed to clarify the clinical efficacy of the ketogenic diet in ALS.
Deanna protocol
The Deanna protocol is a commercially available dietary supplement cocktail. Components of the Deanna protocol are thought to target pathophysiological processes of ALS including glutamate excitotoxicity and oxidative stress.135 In addition, the key components of the Deanna protocol are L-arginine, which improves blood flow to promote protein synthesis,136 and alpha-ketoglutarate, a fuel substrate that can be fed directly into mitochondrial pathways for the generation of cellular energy.135,137 Thus, it is plausible that L-arginine and alpha-ketoglutarate could have positive effects in ALS by promoting skeletal muscle growth and bypassing defects in skeletal muscle glucose metabolism131 to improve energy production to sustain muscle function. Congruent with this, a 2014 study in SOD1G93A mice found that supplementation with arginine-alpha-ketoglutarate led to improved motor function.137 However, this mouse data has not been independently replicated, and there are no published clinical trials on the use of the Deanna protocol in ALS. Given that the current clinical value of the Deanna protocol rests solely on anecdotal patient reports135, further research into the adoption of the Deanna protocol in ALS is needed.
Creatine
Creatine is a naturally occurring guanidine derivative that is synthesized in the liver and found predominantly in muscle and the brain.138 Dietary supplementation with creatine is thought to increase energy output during anaerobic activity, increasing phosphocreatine availability139 and stimulating mitochondrial respiration.140 Creatine is also thought to act as an antioxidant, stabilizing mitochondrial membranes.141 Study outcomes in SOD1G93A mice are mixed. Initial studies found that creatine was able to extend survival142 while reducing glutamate excitotoxicity;143 however, improvements in muscle function were not observed.144 Creatine use by ALS patients was reported to lead to improvements in muscle function,145 with reduced muscle fatigue and improved endurance. Comprehensive studies, however, showed no positive outcomes on strength.146,147 A recent Cochrane review of trials in ALS found no improvement in ALSRFS-R scores following the use of creatine, suggesting no functional benefits of creatine use. The same review found no significant benefit of the use of creatine in ALS survival, noting studies are needed to assess the potential benefits of creatine when taken earlier in the disease course.148
Protein
Few studies have assessed the impact of protein supplementation in ALS, despite the belief that protein supplementation could aid muscle recovery and slow loss of muscle mass. Supplementation with milk whey protein in a small cohort of ALS patients prevented weight loss, contributing to a modest but significant rise in BMI. Maintenance of body weight was associated with a stable ALSFRS-R score over the 4-month assessment period, whereas ALSRFS-R scores declined in individuals not supplemented with protein.45 These results suggest that whey protein supplementation can improve the nutritional status of patients; specific effects of increased protein supply versus calorie supplementation remain unclear.
Vitamin D
Vitamin D is a secosteroid (fat-soluble sub-class of steroid-derived hormones) involved in a range of biological processes including calcium regulation. While some foods contain vitamin D, it is primarily produced endogenously following direct sun exposure to the skin.149 Vitamin D supplementation alone, or in combination with calcium in aged individuals, improves musculoskeletal function,149 suggesting possible benefits for treatment in ALS. Supplementation of vitamin D in SOD1G93A mice show mixed results, with studies documenting improvements in functional capacity but not survival. Treatment was found to be toxic in female mice.150,151 In patients, low vitamin D status correlates with a faster functional decline, with reduced life expectancy observed in those with vitamin D deficiency.152 Assessment of ALS patients found vitamin D levels <30 ng/mL in ~80% of patients, and vitamin D levels <20 ng/mL in ~40% of patients. Noting a modest slowing of functional decline following vitamin D supplementation, the authors concluded that vitamin D supplementation was safe and may have some benefits.153 Additional prospective control trials are required to study the effect of vitamin D on the progression of disability in ALS patients.
Dietary interventions that address pathological mechanisms in ALS
Antioxidants
A balance between oxidation/reduction states (redox homeostasis) allows for the generation of reactive oxygen species (eg, free radicals) and reactive nitrogen species that play essential roles in regulating cellular pathways that maintain function and survival. Naturally occurring antioxidant defense systems in cells protect against cell damage by preventing the oxidation of molecules and by scavenging free radicals. When the balance between free radical production and antioxidant defenses becomes unfavorable, oxidative stress ensues.154 With oxidative stress considered to be one of the primary pathogenic mechanisms in ALS,155 it is not surprising that dietary supplements with antioxidant effects are commonly used. Common antioxidants include vitamins, carnitine, and coenzyme Q10 (CoQ10). Despite the lack of bona fide clinical trial data to demonstrate positive effects in ALS for the majority of antioxidants,156,157 high tolerance and safety combined with ease of availability do not discourage their use by patients.
Vitamin B12
Vitamin B12 is a water-soluble vitamin that plays an essential role supporting the health of the nervous system.158 Humans are unable to synthesize vitamin B12, and hence must obtain it through dietary sources.159 Methylcobalamin, an active form of vitamin B12, has been trialed at high doses in ALS. In a double-blind controlled trial, Kaji et al found that compared to low-dose methylcobalamin, high-dose methylcobalamin was able to prevent the deterioration in the amplitude of the compound muscle action potential after 4 weeks of treatment.160 A randomized double-blind, Phase III clinical trial (NCT00444613) conducted by the same group found that patients receiving high-dose methylcobalamin (intramuscularly) had a lesser decline in ALSFRS-R and prolonged survival if the compound was administered within 12 months of symptom onset.161 Based on current clinical data, ALSUntangled have provided recommendations to encourage the replication of findings and the validation of the use of vitamin B12 in ALS.162
Carnitine
Carnitine is an essential nutrient that plays an integral role in fatty acid metabolism. L-carnitine, the biologically active form of carnitine, can be synthesized endogenously and obtained through dietary sources. As an antioxidant, L-carnitine is proposed to protect against oxidative stress and damage.163 A randomized double-blind placebo-controlled trial assessing the effects on disability and mortality of acetyl-L-carnitine in combination with riluzole showed that there was a slower decline in ALSFRS-R and an increase in median survival.164
Coenzyme Q10 (CoQ10)
CoQ10, also known as ubiquinone, is a lipid-soluble antioxidant. Serving as a diffusible electron carrier in the mitochondrial electron transport chain, CoQ10 plays a vital role in cellular energy production.165 Hence, CoQ10 could exert beneficial effects in ALS by boosting mitochondrial function, and by scavenging free radicals to protect against oxidative stress. In an open-label dose-escalation trial, doses of up to 3000 mg/day of CoQ10 were found to be safe and tolerable in ALS.166 However, a subsequent Phase II double-blind randomized controlled trial assessing daily CoQ10 at 2700 mg/day demonstrated no significant benefit of CoQ10 over placebo.167
Mitoquinone (MitoQ)
A synthetic derivative of CoQ10, MitoQ is a strong antioxidant that was designed to accumulate in and protect against oxidative damage in mitochondria.168 Administration of MitoQ to symptomatic SOD1G93A mice slowed the decline in mitochondrial function in skeletal muscle and spinal cord, improved hindlimb strength, and extended survival.169 These data suggest that specific targeting of antioxidants to mitochondria could be beneficial in ALS. A previous trial of MitoQ in Parkinson’s disease demonstrated no positive effects,170 and there have been no clinical trials of MitoQ in ALS.
Edaravone
Currently being developed by Mitsubishi Tanabe Pharma Corporation (Japan), edaravone (MCI-186, Radicut®) is a potent antioxidant that scavenges reactive oxygen species.171 Preclinical studies demonstrate that edaravone improves motor function, slows symptom progression, and attenuates motor neuron degeneration in rodent models of ALS.172,173 A confirmatory double-blind, parallel-group, placebo-controlled study of edaravone in ALS patients demonstrated a smaller reduction in ALSFRS-R in patients receiving edaravone over a 24-week treatment period.174 More recently, Tanaka et al presented results from a 24-week, Phase III, double-blind, parallel-group study of edavarone in a defined cohort of patients (NCT01492686), revealing a lesser decline in ALSFRS-R at 6 months and less deterioration in quality of life.175 Radicut® (The Japanese Pharmacopoeia Edaravone injection) is currently approved for use as a treatment for ALS in Japan and South Korea. Radicava™ (intravenous) received FDA approval for use in ALS in the USA in May 2017. A Phase I trial of an oral formulation of edaravone (TW001) in the Netherlands has returned positive results, and a Phase II/III trial is in progress. For a more detailed overview of Edaravone, we encourage the reader to consult a recent review.176
Concluding remarks
In the absence of effective treatments for ALS, patients are increasingly adopting dietary regimens and using dietary supplements with the view to alleviate symptoms and to improve quality of life and disease outcome. While data suggest that the adoption of high-calorie or high-protein diets may have some benefit, larger clinical trials are needed to validate the effectiveness of these approaches in ALS. Although the majority of dietary supplements reviewed here have not been clinically validated, preliminary data from some trials suggest the potential usefulness of supplements that target specific pathogenic mechanisms in ALS. In these instances, further studies are needed.
Acknowledgments
STN acknowledges the support of a Scott Sullivan MND Research Fellowship funded by The MND and Me Foundation, The Royal Brisbane & Women’s Hospital Foundation, and the Queensland Brain Institute. The authors would like to acknowledge the valuable funding/salary support of Wesley Medical Research (Project number 2016-32) and The Motor Neurone Disease Research Institute of Australia.
Disclosure
The authors report no conflicts of interest in this work.
References
Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369(9578):2031–2041. | ||
Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):153–174. | ||
Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003;74(9):1258–1261. | ||
Jawaid A, Murthy SB, Wilson AM, et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotrophic Lateral Sclerosis. 2010;11(6):542–548. | ||
Marin B, Desport JC, Kajeu P, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2011;82(6):628–634. | ||
Genton L, Viatte V, Janssens JP, Heritier AC, Pichard C. Nutritional state, energy intakes and energy expenditure of amyotrophic lateral sclerosis (ALS) patients. Clin Nutr. 2011;30(5):553–559. | ||
Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44(1):20–24. | ||
Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioural impairment (an evidence-based review). Neurology. 2009;73:1227–1233. | ||
Ahmed RM, Caga J, Devenney E, et al. Cognition and eating behavior in amyotrophic lateral sclerosis: effect on survival. J Neurol. 2016;263(8):1593–1603. | ||
Castonguay TW, Applegate EA, Upton DE, Stern JS. Hunger and appetite: old concepts/new distinctions. Nutr Rev. 1983;41(4):101–110. | ||
Holm T, Maier A, Wicks P, et al. Severe loss of appetite in amyotrophic lateral sclerosis patients: online self-assessment study. Interact J Med Res. 2013;2(1):e8. | ||
Robbins J. Swallowing in ALS and motor neuron disorders. Neurol Clin. 1987;5(2):213–229. | ||
Neudert C, Oliver D, Wasner M, Borasio GD. The course of the terminal phase in patients with amyotrophic lateral sclerosis. J Neurol. 2001;248:612–616. | ||
Muscaritoli M, Kushta I, Molfino A, Inghilleri M, Sabatelli M, Rossi Fanelli F. Nutritional and metabolic support in patients with amyotrophic lateral sclerosis. Nutrition. 2012;28(10):959–966. | ||
Korner S, Hendricks M, Kollewe K, et al. Weight loss, dysphagia and supplement intake in patients with amyotrophic lateral sclerosis (ALS): impact on quality of life and therapeutic options. BMC Neurol. 2013;13(1):84–84. | ||
Heffernan C, Jenkinson C, Holmes T, et al. Nutritional management in MND/ALS patients: an evidence based review. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(2):72–83. | ||
Silani V, Kasarskis EJ, Yanagisawa N. Nutritional management in amyotrophic lateral sclerosis: a worldwide perspective. J Neurol. 1998;245(Suppl 2):S13–S19; discussion S29. | ||
Greenwood DI. Nutrition management of amyotrophic lateral sclerosis. Nutr Clin Pract. 2013;28(3):392–399. | ||
Andersen PM, Borasio GD, Dengler R, et al. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. An evidence-based review with good practice points. EALSC Working Group. Amyotroph Lateral Scler. 2007;8(4):195–213. | ||
Toepfer CFAKRLM. Gastrointestinal dysfunction in amyotrophic lateral sclerosis. Amyotrophic Lateral Scler Other Motor Neuron Disord. 2009;1(1):15–19. | ||
Ngo ST, Steyn FJ, Huang L, et al. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2015;357(1–2):22–27. | ||
Chaudhri O, Small C, Bloom S. Gastrointestinal hormones regulating appetite. Philos Trans R Soc Lond B Biol Sci. 2006;361(1471):1187–1209. | ||
Andrews ZB, Abizaid A. Neuroendocrine mechanisms that connect feeding behavior and stress. Front Neurosci. 2014;8:312. | ||
Ahmed RM, Irish M, Piguet O, et al. Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol. 2016;15(3):332–342. | ||
Vercruysse P, Sinniger J, El Oussini H, et al. Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain. 2016;139(Pt 4):1106–1122. | ||
Cykowski MD, Takei H, Schulz PE, Appel SH, Powell SZ. TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2014; 2:171. | ||
Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr. 1996;63(1):130–137. | ||
Desport JC, Preux PM, Magy L, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis.(Brief Article). Am J Clin Nutr. 2001;74(3):328. | ||
Ioannides ZA, Ngo ST, Henderson RD, McCombe PA, Steyn FJ. Altered metabolic homeostasis in amyotrophic lateral sclerosis: mechanisms of energy imbalance and contribution to disease progression. Neurodegener Dis. 2016;16(5–6):382–397. | ||
Bouteloup C, Desport JC, Clavelou P, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. 2009;256(8):1236–1242. | ||
Frost J, Okun S, Vaughan T, Heywood J, Wicks P. Patient-reported outcomes as a source of evidence in off-label prescribing: analysis of data from PatientsLikeMe. J Med Internet Res. 2011;13(1):e6. | ||
Bradley WG, Anderson F, Gowda N, Miller RG, Group ACS. Changes in the management of ALS since the publication of the AAN ALS practice parameter 1999. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(4):240–244. | ||
Salvioni CC, Stanich P, Almeida CS, Oliveira AS. Nutritional care in motor neurone disease/ amyotrophic lateral sclerosis. Arq Neuropsiquiatr. 2014;72(2):157–163. | ||
Spataro R, Ficano L, Piccoli F, La Bella V. Percutaneous endoscopic gastrostomy in amyotrophic lateral sclerosis: effect on survival. J Neurol Sci. 2011;304(1–2):44–48. | ||
Fasano A, Fini N, Ferraro D, et al. Percutaneous endoscopic gastrostomy, body weight loss and survival in amyotrophic lateral sclerosis: a population-based registry study. Amyotroph Lateral Scler Frontotemporal Degener. 2017:1–10. | ||
Mockford C, Jenkinson C, Fitzpatrick R. A review: carers, MND and service provision. Amyotroph Lateral Scler. 2006;7(3):132–141. | ||
MNDA. Eating and drinking with motor neurone disease (MND). 2017. Available from: https://www.mndassociation.org/wp-content/uploads/Eating-and-drinking-with-MND-final-web-PDF-2017.pdf. Accessed June 16, 2017. | ||
Rio A, Cawadias E. Nutritional advice and treatment by dietitians to patients with amyotrophic lateral sclerosis/motor neurone disease: a survey of current practice in England, Wales, Northern Ireland and Canada. J Hum Nutr Diet. 2007;20(1):3–13. | ||
Kasarskis EJ, Mendiondo MS, Matthews DE, et al. Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. Am J Clin Nutr. 2014;99(4):792–803. | ||
ProGas Study G. Gastrostomy in patients with amyotrophic lateral sclerosis (ProGas): a prospective cohort study. Lancet Neurol. 2015;14(7):702–709. | ||
Rahnemai-Azar AA, Rahnemaiazar AA, Naghshizadian R, Kurtz A, Farkas DT. Percutaneous endoscopic gastrostomy: indications, technique, complications and management. World J Gastroenterol. 2014;20(24):7739–7751. | ||
Stavroulakis T, Walsh T, Shaw PJ, McDermott CJ, Progas S. Gastrostomy use in motor neurone disease (MND): a review, meta-analysis and survey of current practice. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(2):96–104. | ||
Wills A-M, Hubbard J, Macklin EA, et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2014;383(9934):2065–2072. | ||
Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. 2007;9(1):17–20. | ||
Silva LBdC, Mourão LF, Silva AA, et al. Effect of nutritional supplementation with milk whey proteins in amyotrophic lateral sclerosis patients. Arquivos de Neuro-Psiquiatria. 2010;68:263–268. | ||
Dorst J, Cypionka J, Ludolph AC. High-caloric food supplements in the treatment of amyotrophic lateral sclerosis: a prospective interventional study. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7–8):533–536. | ||
Thomas BF, ElSohly MA. Chapter 1 – The botany of Cannabis sativa L. In: The Analytical Chemistry of Cannabis. Elsevier; 2016:1–26. | ||
Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3(9):771–784. | ||
Grotenhermen F, Muller-Vahl K. The therapeutic potential of cannabis and cannabinoids. Dtsch Arztebl Int. 2012;109(29–30):495–501. | ||
Alexander SP. Therapeutic potential of cannabis-related drugs. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:157–166. | ||
Amtmann D, Weydt P, Johnson KL, Jensen MP, Carter GT. Survey of cannabis use in patients with amyotrophic lateral sclerosis. Am J Hosp Palliat Care. 2004;21(2):95–104. | ||
Carter GT, Abood ME, Aggarwal SK, Weiss MD. Cannabis and amyotrophic lateral sclerosis: hypothetical and practical applications, and a call for clinical trials. Am J Hosp Palliat Care. 2010;27(5):347–356. | ||
Cowley MA, Smart JL, Rubinstein M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. | ||
Koch M, Varela L, Kim JG, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519(7541):45–50. | ||
Kalra SP, Horvath TL. Neuroendocrine interactions between galanin, opioids, and neuropeptide Y in the control of reproduction and appetite. Ann N Y Acad Sci. 1998;863:236–240. | ||
Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;5(2):77–87. | ||
Cozzolino M, Carri MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012;97(2):54–66. | ||
Muyderman H, Chen T. Mitochondrial dysfunction in amyotrophic lateral sclerosis – a valid pharmacological target? Br J Pharmacol. 2014;171(8):2191–2205. | ||
Volkow ND, Baler RD, Compton WM, Weiss SR. Adverse health effects of marijuana use. N Engl J Med. 2014;370(23):2219–2227. | ||
Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86(12):5992. | ||
Czell D, Baldinger R, Schneider U, Neuwirth C, Weber M. The role of the SenseWear device and ghrelin for metabolism in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(3–4):295–296. | ||
Lim E, Lee S, Li E, Kim Y, Park S. Ghrelin protects spinal cord motoneurons against chronic glutamate-induced excitotoxicity via ERK1/2 and phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3beta pathways. Exp Neurol. 2011;230(1):114–122. | ||
Lee S, Kim Y, Li E, Park S. Ghrelin protects spinal cord motoneurons against chronic glutamate excitotoxicity by inhibiting microglial activation. Korean J Physiol Pharmacol. 2012;16(1):43–48. | ||
Colldén G, Tschop MH, Muller TD. Therapeutic potential of targeting the ghrelin pathway. Int J Mol Sci. 2017;18(4):E798. | ||
Xu H, Huang X, Riserus U, et al. Dietary fiber, kidney function, inflammation, and mortality risk. Clin J Am Soc Nephrol. 2014;9(12):2104–2110. | ||
Young RP, Hopkins RJ. High dietary fiber lowers systemic inflammation: potential utility in COPD and lung cancer. Am J Med. 2014;127(8):e13. | ||
Slavin J. Fiber and prebiotics: mechanisms and health benefits. Nutrients. 2013;5(4):1417–1435. | ||
Fondell E, O’Reilly EJ, Fitzgerald KC, et al. Dietary fiber and amyotrophic lateral sclerosis: results from 5 large cohort studies. Am J Epidemiol. 2014;179(12):1442–1449. | ||
Nieves JW, Gennings C, Factor-Litvak P, et al. Association between dietary intake and function in amyotrophic lateral sclerosis. JAMA Neurol. 2016;73(12):1425–1432. | ||
Hanisch F, Skudlarek A, Berndt J, Kornhuber ME. Characteristics of pain in amyotrophic lateral sclerosis. Brain Behav. 2015;5(3):e00296. | ||
Layzer RB. Diagnostic implications of clinical fasciculation and cramps. Adv Neurol. 1982;36:23–29. | ||
Gubbay SS, Kahana E, Zilber N, Cooper G, Pintov S, Leibowitz Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J Neurol. 1985;232(5):295–300. | ||
Layzer RB. The origin of muscle fasciculations and cramps. Muscle Nerve. 1994;17(11):1243–1249. | ||
Baldissera F, Cavallari P, Dworzak F. Motor neuron ‘bistability’. A pathogenetic mechanism for cramps and myokymia. Brain. 1994;117 ( Pt 5):929–939. | ||
Obi T, Mizoguchi K, Matsuoka H, Takatsu M, Nishimura Y. Muscle cramp as the result of impaired GABA function–an electrophysiological and pharmacological observation. Muscle Nerve. 1993;16(11):1228–1231. | ||
Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227–1233. | ||
Achan J, Talisuna AO, Erhart A, et al. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J. 2011;10:144. | ||
Man-Son-Hing M, Wells G, Lau A. Quinine for nocturnal leg cramps: a meta-analysis including unpublished data. J Gen Intern Med. 1998;13(9):600–606. | ||
Dyson EH, Proudfoot AT, Prescott LF, Heyworth R. Death and blindness due to overdose of quinine. Br Med J (Clin Res Ed). 1985;291(6487):31–33. | ||
Tucker JM, Townsend DM. Alpha-tocopherol: roles in prevention and therapy of human disease. Biomed Pharmacother. 2005;59(7):380–387. | ||
Ayres S, Jr., Mihan R. Leg cramps (systremma) and “restless legs” syndrome. Response to vitamin E (tocopherol). Calif Med. 1969;111(2):87–91. | ||
Michal Freedman D, Kuncl RW, Weinstein SJ, Malila N, Virtamo J, Albanes D. Vitamin E serum levels and controlled supplementation and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(4):246–251. | ||
de Bustos F, Jimenez-Jimenez FJ, Molina JA, et al. Cerebrospinal fluid levels of alpha-tocopherol in amyotrophic lateral sclerosis. J Neural Transm (Vienna). 1998;105(6–7):703–708. | ||
Iwasaki Y, Ikeda K, Kinoshita M. Vitamin A and E levels are normal in amyotrophic lateral sclerosis. J Neurol Sci. 1995;132(2):193–194. | ||
Wang H, O’Reilly EJ, Weisskopf MG, et al. Vitamin E intake and risk of amyotrophic lateral sclerosis: a pooled analysis of data from 5 prospective cohort studies. Am J Epidemiol. 2011;173(6):595–602. | ||
Veldink JH, Kalmijn S, Groeneveld GJ, et al. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007;78(4):367–371. | ||
Ascherio A, Weisskopf MG, O’Reilly E J, et al. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann Neurol. 2005;57(1):104–110. | ||
Kwiecinski H, Janik P, Jamrozik Z, Opuchlik A. [The effect of selegiline and vitamin E in the treatment of ALS: an open randomized clinical trials]. Neurol Neurochir Pol. 2001;35(1 Suppl):101–106. | ||
Desnuelle C, Dib M, Garrel C, Favier A. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):9–18. | ||
Galbussera A, Tremolizzo L, Brighina L, et al. Vitamin E intake and quality of life in amyotrophic lateral sclerosis patients: a follow-up case series study. Neurol Sci. 2006;27(3):190–193. | ||
Graf M, Ecker D, Horowski R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. J Neural Transm (Vienna). 2005;112(5):649–660. | ||
Durlach J, Bac P, Bara M, Guiet-Bara A. Physiopathology of symptomatic and latent forms of central nervous hyperexcitability due to magnesium deficiency: a current general scheme. Magnes Res. 2000;13(4):293–302. | ||
Sebo P, Cerutti B, Haller DM. Effect of magnesium therapy on nocturnal leg cramps: a systematic review of randomized controlled trials with meta-analysis using simulations. Fam Pract. 2014;31(1):7–19. | ||
Fondell E, O’Reilly EJ, Fitzgerald KC, et al. Magnesium intake and risk of amyotrophic lateral sclerosis: results from five large cohort studies. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5–6):356–361. | ||
Longnecker MP, Kamel F, Umbach DM, et al. Dietary intake of calcium, magnesium and antioxidants in relation to risk of amyotrophic lateral sclerosis. Neuroepidemiology. 2000;19(4):210–216. | ||
Chang E, Ghosh N, Yanni D, Lee S, Alexandru D, Mozaffar T. A review of spasticity treatments: pharmacological and interventional approaches. Crit Rev Phys Rehabil Med. 2013;25(1–2):11–22. | ||
Kheder A, Nair KP. Spasticity: pathophysiology, evaluation and management. Pract Neurol. 2012;12(5):289–298. | ||
Simon O, Yelnik AP. Managing spasticity with drugs. Eur J Phys Rehabil Med. 2010;46(3):401–410. | ||
Maher TJ, Wurtman RJ. L-threonine administration increases glycine concentrations in the rat central nervous system. Life Sci. 1980;26(16):1283–1286. | ||
Testa D, Caraceni T, Fetoni V, Girotti F. Chronic treatment with L-threonine in amyotrophic lateral sclerosis: a pilot study. Clin Neurol Neurosurg. 1992;94(1):7–9. | ||
Lee A, Patterson V. A double-blind study of L-threonine in patients with spinal spasticity. Acta Neurol Scand. 1993;88(5):334–338. | ||
Blin O, Pouget J, Aubrespy G, Guelton C, Crevat A, Serratrice G. A double-blind placebo-controlled trial of L-threonine in amyotrophic lateral sclerosis. J Neurol. 1992;239(2):79–81. | ||
Ng L, Khan F, Young CA, Galea M. Symptomatic treatments for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2017;1:CD011776. | ||
Petro DJ, Ellenberger C, Jr. Treatment of human spasticity with delta 9-tetrahydrocannabinol. J Clin Pharmacol. 1981;21(8–9 Suppl):413S–416S. | ||
Pryce G, Baker D. Endocannabinoids in multiple sclerosis and amyotrophic lateral sclerosis. Handb Exp Pharmacol. 2015;231:213–231. | ||
Flachenecker P, Henze T, Zettl UK. Nabiximols (THC/CBD oromucosal spray, Sativex(R)) in clinical practice--results of a multicenter, non-interventional study (MOVE 2) in patients with multiple sclerosis spasticity. Eur Neurol. 2014;71(5–6):271–279. | ||
Fernandez O. Advances in the management of MS spasticity: recent observational studies. Eur Neurol. 2014;72(Suppl 1):12–14. | ||
Bilsland LG, Dick JR, Pryce G, et al. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J. 2006;20(7):1003–1005. | ||
Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101(1):87–98. | ||
Rossi S, De Chiara V, Musella A, et al. Abnormal sensitivity of cannabinoid CB1 receptors in the striatum of mice with experimental amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(1–2):83–90. | ||
Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135–1140. | ||
Group AL. ALSUntangled No. 16: cannabis. Amyotroph Lateral Scler. 2012;13(4):400–404. | ||
Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992;17(2):139–170. | ||
Potenza RL, Armida M, Ferrante A, et al. Effects of chronic caffeine intake in a mouse model of amyotrophic lateral sclerosis. J Neurosci Res. 2013;91(4):585–592. | ||
Fondell E, O’Reilly EI, Fitzgerald KC, et al. Intakes of caffeine, coffee and tea and risk of amyotrophic lateral sclerosis: results from five cohort studies. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5–6):366–371. | ||
Beghi E, Pupillo E, Messina P, et al. Coffee and amyotrophic lateral sclerosis: a possible preventive role. Am J Epidemiol. 2011;174(9):1002–1008. | ||
Onatibia-Astibia A, Franco R, Martinez-Pinilla E. Health benefits of methylxanthines in neurodegenerative diseases. Mol Nutr Food Res. 2017. | ||
Yenisetti SC, Manjunath MJ, Muralidhara C. Neuropharmacological properties of Withania somnifera – Indian Ginseng: an overview on experimental evidence with emphasis on clinical trials and patents. Recent Pat CNS Drug Discov. 2016;10(2):204–215. | ||
Kolahdouzan M, Hamadeh MJ. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci Ther. 2017;23(4):272–290. | ||
A DB. The ketogenic diet in epilepsy. Can Med Assoc J. 1931;24(1):106–107. | ||
Peterman MG. The ketogenic diet in epilepsy. JAMA. 1925;84(26):1979–1983. | ||
Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59. | ||
Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006;17(5–6):431–439. | ||
Paoli A, Bianco A, Damiani E, Bosco G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int. 2014;2014:474296. | ||
Vidali S, Aminzadeh S, Lambert B, et al. Mitochondria: The ketogenic diet–A metabolism-based therapy. Int J Biochem Cell Biol. 2015;63:55–59. | ||
Echaniz-Laguna A, Zoll J, Ribera F, et al. Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann Neurol. 2002;52(5):623–627. | ||
Wiedemann FR, Winkler K, Kuznetsov AV, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;156(1):65–72. | ||
Vielhaber S, Winkler K, Kirches E, et al. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci. 1999;169(1–2):133–139. | ||
Siciliano G, Pastorini E, Pasquali L, Manca ML, Iudice A, Murri L. Impaired oxidative metabolism in exercising muscle from ALS patients. J Neurol Sci. 2001;191(1–2):61–65. | ||
Crugnola V, Lamperti C, Lucchini V, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. 2010;67(7):849–854. | ||
Palamiuc L, Schlagowski A, Ngo ST, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526–546. | ||
Pradat PF, Bruneteau G, Gordon PH, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(1–2):166–171. | ||
Zhao Z, Lange DJ, Voustianiouk A, et al. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29. | ||
Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur J Clin Nutr. 2013;67(8):789–796. | ||
ALS Untangled Group, Fournier C, Bedlack B, et al. ALS Untangled No. 20: the Deanna protocol. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(4):319–323. | ||
Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–2012. | ||
Ari C, Poff AM, Held HE, et al. Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS One. 2014;9(7):e103526. | ||
Walker JB. Creatine: biosynthesis, regulation, and function. Adv Enzymol Relat Areas Mol Biol. 1979;50:177–242. | ||
Casey A, Greenhaff PL. Does dietary creatine supplementation play a role in skeletal muscle metabolism and performance? Am J Clin Nutr. 2000;72(2 Suppl):607S–617S. | ||
O’Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. 1996;1276(2):161–170. | ||
Strong MJ, Pattee GL. Creatine and coenzyme Q10 in the treatment of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(Suppl 4):17–20. | ||
Klivenyi P, Ferrante RJ, Matthews RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5(3):347–350. | ||
Andreassen OA, Jenkins BG, Dedeoglu A, et al. Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J Neurochem. 2001;77(2):383–390. | ||
Derave W, Van Den Bosch L, Lemmens G, Eijnde BO, Robberecht W, Hespel P. Skeletal muscle properties in a transgenic mouse model for amyotrophic lateral sclerosis: effects of creatine treatment. Neurobiol Dis. 2003;13(3):264–272. | ||
Mazzini L, Balzarini C, Colombo R, et al. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: preliminary results. J Neurol Sci. 2001;191(1–2):139–144. | ||
Rosenfeld J, King RM, Jackson CE, et al. Creatine monohydrate in ALS: Effects on strength, fatigue, respiratory status and ALSFRS. Amyotrophic Lateral Sclerosis. 2008;9(5):266–272. | ||
Drory VE, Gross D. No effect of creatine on respiratory distress in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(1):43–46. | ||
Pastula DM, Moore DH, Bedlack RS. Creatine for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2012;12:CD005225. | ||
Kulie T, Groff A, Redmer J, Hounshell J, Schrager S. Vitamin D: an evidence-based review. J Am Board Fam Med. 2009;22(6):698–706. | ||
Gianforcaro A, Solomon JA, Hamadeh MJ. Vitamin D(3) at 50× AI attenuates the decline in paw grip endurance, but not disease outcomes, in the G93A mouse model of ALS, and is toxic in females. PLoS One. 2013;8(2):e30243. | ||
Gianforcaro A, Hamadeh MJ. Dietary vitamin D3 supplementation at 10x the adequate intake improves functional capacity in the G93A transgenic mouse model of ALS, a pilot study. CNS Neurosci Ther. 2012;18(7):547–557. | ||
Camu W, Tremblier B, Plassot C, et al. Vitamin D confers protection to motoneurons and is a prognostic factor of amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35(5):1198–1205. | ||
Karam C, Barrett MJ, Imperato T, MacGowan DJ, Scelsa S. Vitamin D deficiency and its supplementation in patients with amyotrophic lateral sclerosis. J Clin Neurosci. 2013;20(11):1550–1553. | ||
McCord JM. The evolution of free radicals and oxidative stress. Am J Med. 2000;108(8):652–659. | ||
D’Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol Med. 2013;65:509–527. | ||
Orrell RW, Lane RJ, Ross M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph Lateral Scler. 2008;9(4):195–211. | ||
Orrell RW, Lane RJ, Ross M. Antioxidant treatment for amyotrophic lateral sclerosis / motor neuron disease. Cochrane Database Syst Rev. 2007(1):CD002829. | ||
Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006;5(11):949–960. | ||
Watanabe F. Vitamin B12 sources and bioavailability. Exp Biol Med (Maywood). 2007;232(10):1266–1274. | ||
Kaji R, Kodama M, Imamura A, et al. Effect of ultrahigh-dose methylcobalamin on compound muscle action potentials in amyotrophic lateral sclerosis: a double-blind controlled study. Muscle Nerve. 1998;21(12):1775–1778. | ||
Kaji R, Kuzuhara S, Iwasaki Y, et al. Ultra-high dose methylcobalamin (E0302) prolongs survival of ALS: report of 7 years’ randomised double-blind, phase 3 clinical trial (ClinicalTrials.gov NCT00444613). Paper presented at: AAN Annual Meeting; 2015. | ||
ALS-Untangled. ALSUntangled No. 30: methylcobalamin. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7–8):536–539. | ||
Gulcin I. Antioxidant and antiradical activities of L-carnitine. Life Sci. 2006;78(8):803–811. | ||
Beghi E, Pupillo E, Bonito V, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5–6):397–405. | ||
Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660(1–2):171–199. | ||
Ferrante KL, Shefner J, Zhang H, et al. Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology. 2005;65(11):1834–1836. | ||
Kaufmann P, Thompson JL, Levy G, et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann Neurol. 2009;66(2):235–244. | ||
Kelso GF, Porteous CM, Coulter CV, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276(7):4588–4596. | ||
Miquel E, Cassina A, Martinez-Palma L, et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med. 2014;70:204–213. | ||
Snow BJ, Rolfe FL, Lockhart MM, et al; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25(11):1670–1674. | ||
Watanabe T, Tahara M, Todo S. The novel antioxidant edaravone: from bench to bedside. Cardiovasc Ther. 2008;26(2):101–114. | ||
Aoki M, Warita H, Mizuno H, Suzuki N, Yuki S, Itoyama Y. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011;1382:321–325. | ||
Ito H, Wate R, Zhang J, et al. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp Neurol. 2008;213(2):448–455. | ||
Abe K, Itoyama Y, Sobue G, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):610–617. | ||
Tanaka M, Sakata T, Palumbo J, Akimoto M. A 24-week, phase III, double-blind, parallel-group study of edaravone (MCI-186) for treatment of amyotrophic lateral sclerosis (ALS). Paper presented at: AAN Annual Meeting; 2016; Vancouver, Canada. | ||
Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front Aging Neurosci. 2017;9:68. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.