Back to Journals » Journal of Inflammation Research » Volume 15
Exploring Long Non-Coding RNAs Associated with IP3/DAG Signaling Pathway as Potential Biomarkers Involved in Mast Cell Degranulation in Chronic Spontaneous Urticaria with 2-Year Follow-Up
Authors Liang Y ![]() , Kong Q, Luo H, Tan J, Zhu H
, Kong Q, Luo H, Tan J, Zhu H
Received 15 October 2021
Accepted for publication 6 January 2022
Published 14 January 2022 Volume 2022:15 Pages 267—283
DOI https://doi.org/10.2147/JIR.S343826
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Monika Sharma
Yudan Liang,1,2,* Qinghuo Kong,1,* Huiwen Luo,1 Jinhua Tan,1 Huizheng Zhu3
1Department of Acupuncture and Rehabilitation, The Affiliated Jiangmen Traditional Chinese Medicine Hospital of Jinan University, Jiangmen, Guangdong, People’s Republic of China; 2Integrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou, Guangdong, People’s Republic of China; 3Department of Traditional Chinese Medicine, The 2nd Clinical Medical College (Shenzhen People’s Hospital) of Jinan University, Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huizheng Zhu
Department of Traditional Chinese Medicine, The 2nd Clinical Medical College (Shenzhen People’s Hospital) of Jinan University, No. 1017, Dongmen North Road, Luohu District, Shenzhen, Guangdong, 518020, People’s Republic of China
Email [email protected]
Purpose: Chronic spontaneous urticaria (CSU) pathogenesis involves mast cell degranulation induced by the inositol 1,4,5-trisphosphate/diacylglycerol (IP3/DAG) pathway, but the condition lacks specific biomarkers. This study was performed to investigate long non-coding RNA (lncRNA) expression profiles, identify those associated with IP3/DAG pathway, and assess their diagnostic and prognostic value for CSU.
Methods: Ten samples were selected from CSU and control groups, and microarray was performed to screen differentially expressed (DE) lncRNAs and mRNAs. Bioinformatic and co-expression network analyses were used to identify lncRNAs associated with IP3/DAG pathway. Quantitative real-time polymerase chain reaction was used to validate lncRNA expression levels. Combined with disease characteristics and serum indices detected with enzyme-linked immunosorbent assays, Spearman analysis and logistic regression were applied to analyze lncRNA-associated disease risk. Receiver operating characteristic (ROC) curves and 2-year follow-up data were applied to evaluate lncRNA diagnostic and prognostic value.
Results: A total of 678 up- and 573 downregulated DE lncRNAs and 609 up- and 176 downregulated DE mRNAs were identified. Seven lncRNAs (upregulated T264761, T280622, ENST00000587970, T224062, ENST00000562459, and his-1_RNA_dna; downregulated ENST00000417930) were associated with the IP3/DAG pathway. D-dimer and histamine levels were significantly different between the two groups. Correlation analysis showed that his-1_RNA_dna positively correlated with the frequency of symptom appearance, while his-1_RNA_dna, ENST00000417930, T264761, and T280622 negatively correlated with the maximum wheal diameter. Regression analysis showed T264761 was associated with CSU risk. ROC analysis showed that the specificity of T264761 was 90%, with an area under the curve of 0.666. In follow-up, the rate of well-controlled disease in the low T264761 expression group was 82.61%.
Conclusion: This study established lncRNA and mRNA expression profiles in CSU and identified lncRNAs associated with IP3/DAG pathway, which is mechanistically involved in this disease. T264761 may be a novel biomarker for CSU, but further study is needed to confirm its specific mechanism.
Keywords: lncRNA, biomarker, urticaria, mRNA, microarray
Introduction
Chronic spontaneous urticaria (CSU) is a common dermatological condition characterized by sudden itchy wheals or angioedema lasting for up to 6 weeks and occurring twice a week or more.1 The symptoms are spontaneous, with no known specific triggers. CSU diagnosis is mainly based on clinical symptoms and medical history, the 7-day Urticaria Activity Score (UAS7), quality of life assessments, and other questionnaires that assist in evaluation of the disease and treatment effects. Some reviews suggest that there are statistical differences in the contents of D-dimer, C-reactive protein (CRP), and microRNAs between patients with CSU and healthy controls. These factors may reflect disease course and activity and treatment effects, but they are not specific to CSU and therefore cannot sensitively or accurately distinguish CSU from other diseases.2–4 Specific biomarkers of CSU that may facilitate early diagnosis, accurate prognosis, and individualized treatments remain elusive.
Urticaria is a mast cell-driven disease. The high-affinity receptor for immunoglobulin E (IgE) (FcεRI) is considered a key factor in mast cell degranulation, which induces characteristic skin wheals in CSU. Antigen-dependent mast cell activation is regulated by a complex series of intracellular signaling processes that are initiated following FcεRI aggregation.5 In downstream signaling processes, phospholipase Cγ1/2 hydrolyzes phosphatidylinositol 4,5-bisphosphate in the cell membrane to produce two second messenger molecules: inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG).6 IP3 induces calcium release from the endoplasmic reticulum, triggering extracellular calcium influx; DAG binds to protein kinase C, which is activated by cell membrane and cytoskeleton proteins.7 IP3 and DAG combine to trigger mast cell degranulation, increase microvascular permeability, and induce the release of inflammatory mediators such as histamine (HIS), leukotriene B4 (LTB4), prostaglandin D2 (PGD2), and mast cell tryptase (MCT).8 Additionally, the mitogen-activated protein kinase and phosphoinositide 3-kinase pathways are mainly involved in generating eicosanoids and cytokines.5 The IP3/DAG signaling pathway is thus a key pathway in mast cell degranulation. Currently, there are no known biomarkers for this signaling pathway.
Long non-coding RNAs (lncRNAs) are a group of functional RNAs which non or rarely in coding transcripts that longer than 200 nucleotides.9 LncRNAs regulate gene expression at epigenetic and transcriptional levels in the nucleus and at post-transcriptional and translational levels in the cytoplasm. LncRNAs are closely related to the physiology and pathology of allergic diseases including asthma, atopic dermatitis, and rhinitis, suggesting that they could act as biomarkers and therapeutic targets for allergic disorders, aid in the diagnosis and prognosis of disease, and identify novel therapeutic agents.4 Some lncRNAs were shown to be closely related to skin physiology and pathologies. For example, COL1A2-AS1 promotes the apoptosis of normal skin fibroblasts by inhibiting p-Smad3 and promoting β-catenin expression, WAKMAR1 regulates wound healing by promoting keratinocyte migration, and MEG3 affects the proliferation and apoptosis of psoriatic epidermal cells by targeting miR-21/caspase-8.10–12 Since CSU pathogenesis is closely related to allergic responses, differential lncRNA expression may therefore serve as a biomarker for this disease.
This study aimed to identify lncRNAs that are closely related to CSU physiology and pathology. LncRNA and mRNA expression profiles in blood samples from CSU patients were compared with those in healthy individuals using microarray technology; bioinformatic analyses, quantitative real-time polymerase chain reaction (qRT-PCR), receiver operating characteristic (ROC) curve analysis, logistic regression analysis, and a 2-year clinical follow-up were used to explore the value of key lncRNAs as biomarkers for CSU.
Materials and Methods
Recruitment and Inclusion/Exclusion Criteria
Patients were recruited from the affiliated Jiangmen Traditional Chinese Medicine Hospital of Jinan University between November 2018 and May 2019. All participants had a documented history of CSU. Patients suffering from the spontaneous appearance of wheals and/or angioedema for ≥6 weeks due to known or unknown causes, and who were aged 18 years or older at the time of recruitment were included in the study. Patients with blood system diseases, tumors or other serious diseases (severe cardiovascular, hepatic, or renal insufficiency), pregnant or lactating women, and those with current drug abuse, alcoholism, or mental illness were excluded. Healthy controls were included in the study if they had no obvious abnormalities on routine physical examination and laboratory imaging, no history of disease or drug treatment before blood collection, and were aged 18 years or older. All subjects signed written informed consent forms prior to participating in this study.
The present study was approved by the Ethics Committee of the affiliated Jiangmen Traditional Chinese Medicine Hospital of Jinan University (approval KY [2017]-c14) and was conducted in compliance with the Declaration of Helsinki. The study was registered in the Chinese Clinical Trial Registry (No. ChiCTR1800018653).
Blood Sample Collection
Two vacutainer tubes of peripheral blood were collected from all participants. One tube contained ethylene diamine tetraacetic acid (EDTA) and was used for lncRNA microarrays or qRT-PCR, the second standard tube was used for enzyme-linked immunosorbent assay (ELISA). Peripheral blood mononuclear cells (PBMCs) in the EDTA tube were isolated by density gradient centrifugation, and total RNA was isolated from the PBMCs using TRIzol reagent. Blood serum was extracted after centrifugation in the standard tube.
lncRNA Profiling
The Arraystar Human lncRNA microarray v4.0, which contains 40,173 lncRNAs and 20,730 coding transcripts, was used to profile the expression of lncRNAs and mRNAs. Five samples from CSU patients and 5 samples from healthy controls were selected to be detected by Shanghai Kangchen Bio-tech, Inc. (Shanghai, China). After the total RNA was isolated, the concentration and quality were assessed using absorbance spectrometry, measuring absorbance ratios of A260/A280 and A260/230 using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Supplementary Table 1 shows RNA quantification and quality assurance, and Supplementary Figure 1 shows an assessment of RNA integrity and gDNA contamination test by denaturing agarose gel electrophoresis. According to the manufacturer’s instructions and as in previously published research,13 RNA samples were labeled by Quick Amp Labeling Kit (Agilent p/n 5190–0442, Santa Clara, CA, USA), and purified by RNeasy Mini Kit (Qiagen p/n 74,104, Hilden, Germany). Microarray hybridization was performed using the Agilent Gene Expression Hybridization Kit (Agilent p/n 5188–5242). After washing and fixing, the hybridized arrays were scanned by Agilent Microarray Scanner (Agilent p/n G2565BA) and data were extracted using Agilent Feature Extract Software (Agilent, v10.5.1.1, Palo Alto, CA, USA). Data were deposited in the Gene Expression Omnibus database (GSE185516). Raw data were normalized using a quantile algorithm in Agilent GeneSpring GX v12.1. Differentially expressed (DE) lncRNAs and mRNAs were identified according to their fold change (FC) at a threshold ≥1.5-fold with significance p < 0.05.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses
The GO and KEGG databases were used to analyze potential biological functions and signaling pathways associated with DE mRNAs. GO enrichment analysis of target genes was performed by GOseq R package (http://www.geneontology.org) and included biological processes (BP), cellular components (CC), and molecular functions (MF). Signaling pathways related to target genes were explored using the KEGG database (http://www.geneontology.org) and performed using DAVID software. Target genes with p < 0.05 were considered significantly enriched.
Pearson Analysis of DE lncRNAs and mRNAs Associated with IP3/DAG Signaling Pathway
Pearson correlation analysis was carried out on selected DE mRNAs and lncRNAs identified in the KEGG analysis; those with a Pearson correlation coefficient (PCC) ≥ 0.8 with significance p < 0.5 and a false discovery rate (FDR) ≤ 1 were selected for further exploration of closely related DE lncRNAs.
Validation of lncRNAs Using qRT-PCR and lncRNA–mRNA Co-Expression Networks
LncRNAs for which the p value of significance is lower, FC is higher, raw intensity is greater than 200, and which were closely related to the IP3/DAG pathway were selected for qRT-PCR validation. Total RNA was extracted using TRIzol reagent, and cDNA was synthesized using EasyScript First-Strand cDNA Synthesis SuperMix (TransGen, Beijing, China). qRT-PCR was performed using SYBR Green (Yeasen, Shanghai, China). The specific primers were designed by LandM Biotech Inc. (Guangzhou, China). Cycle threshold (Ct) values were used to quantify the expression levels of lncRNAs using the 2−ΔΔCt method, and β-actin was used as a control to normalize values.
Selected lncRNAs and total mRNAs with PCC ≥ 0.9, p < 0.5, and FDR ≤1, were used to construct a co-expression network which was visualized using Cytoscape v2.8.3 (Institute of Systems Biology, Seattle, WA, USA). GO analysis was used to assess the putative biological functions of the co-expressed mRNAs, which likely reflect the biological function of the lncRNAs.
ELISA
The levels of HIS (ab213975, Abcam, Cambridge, UK), MCT (CSB-E09012h, CUSABIO, Wuhan, China), LTB4 (CSB-E08033h), PGD2 (CSB-E13898h), hs-CRP (CSB-E08617h), and D-dimer (CSB-E05175h) were assessed in peripheral blood serum using human ELISA kits according to the manufacturer’s instructions. The optical density (OD) value of samples was evaluated at the wavelength of 450 nm.
Statistical Analysis
All data are presented as mean ± standard deviation (SD), median (range), or n, as appropriate. Chi-square and Mann–Whitney U-tests were used to analyze qualitative and quantitative data, respectively. Spearman coefficient analysis was performed to assess correlations between lncRNA levels and clinical characteristics of CSU. Logistic regression was used to analyze the CSU risk associated with selected lncRNAs. ROC curves were generated to evaluate the prognostic value of selected lncRNAs. lncRNA expression values were sorted into high- and low-level groups, based on these ROC curves using Youden’s index correction.14 Data were analyzed using SPSS v19.0 (IBM Corp., Armonk, NY, USA) and graphs were drawn using GraphPad Prism v8.3.0 (San Diego, CA, USA). p values < 0.05 were regarded as statistically significant.
Results
Figure 1 is a flow diagram summarizing the study methods, participant inclusion and exclusion, and key results.
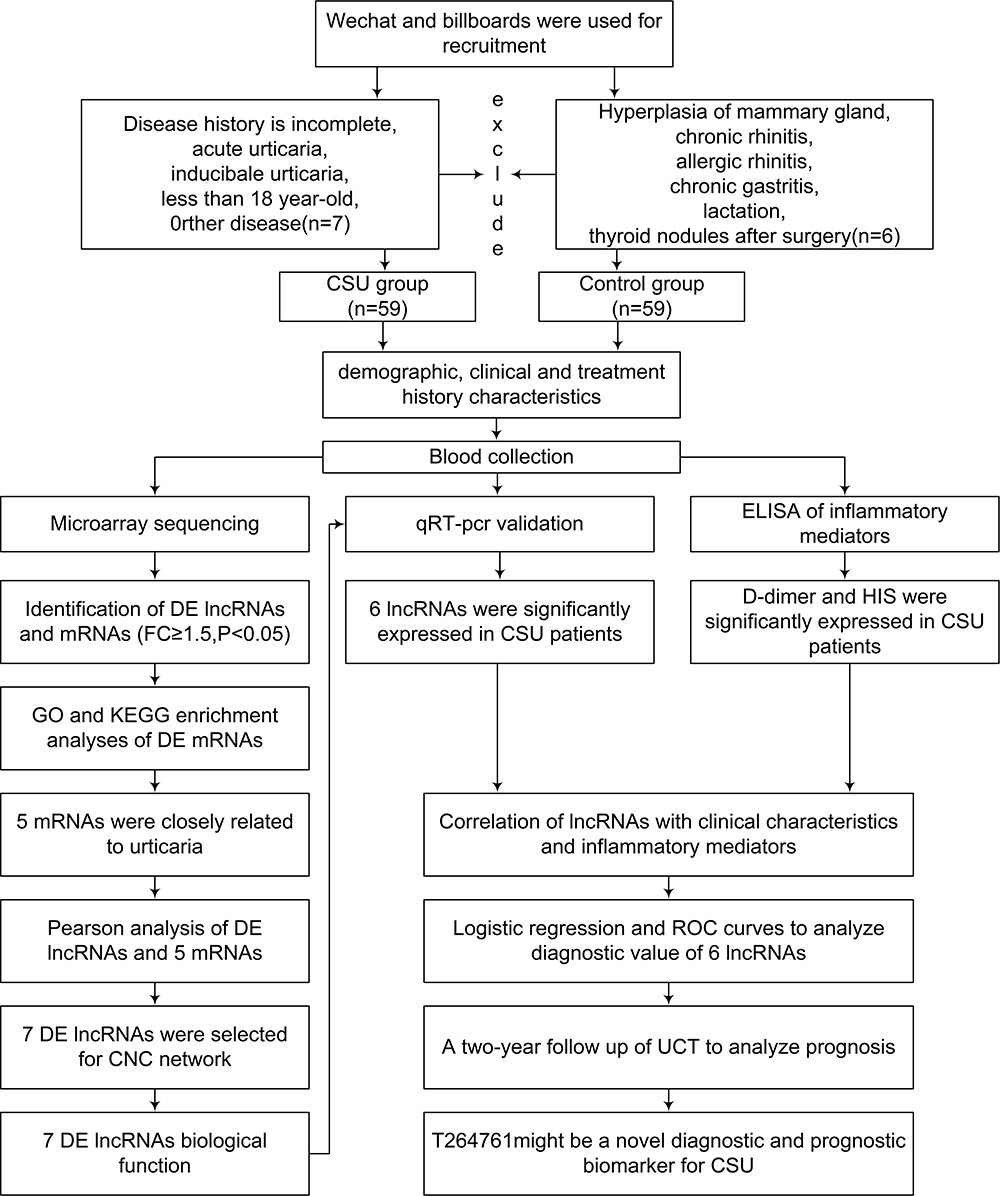 |
Figure 1 Flow diagram showing recruitment and inclusion/exclusion of participants and a summary of the study methods and key results. Abbreviations: CNC, coding and noncoding co-expression; CSU, chronic spontaneous urticaria; DE, differentially expressed; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; ROC, receiver operating characteristic. |
Patient Demographics
In total, 59 people with CSU and 59 healthy controls were recruited for this study; a summary of their demographic characteristics is shown in Table 1.
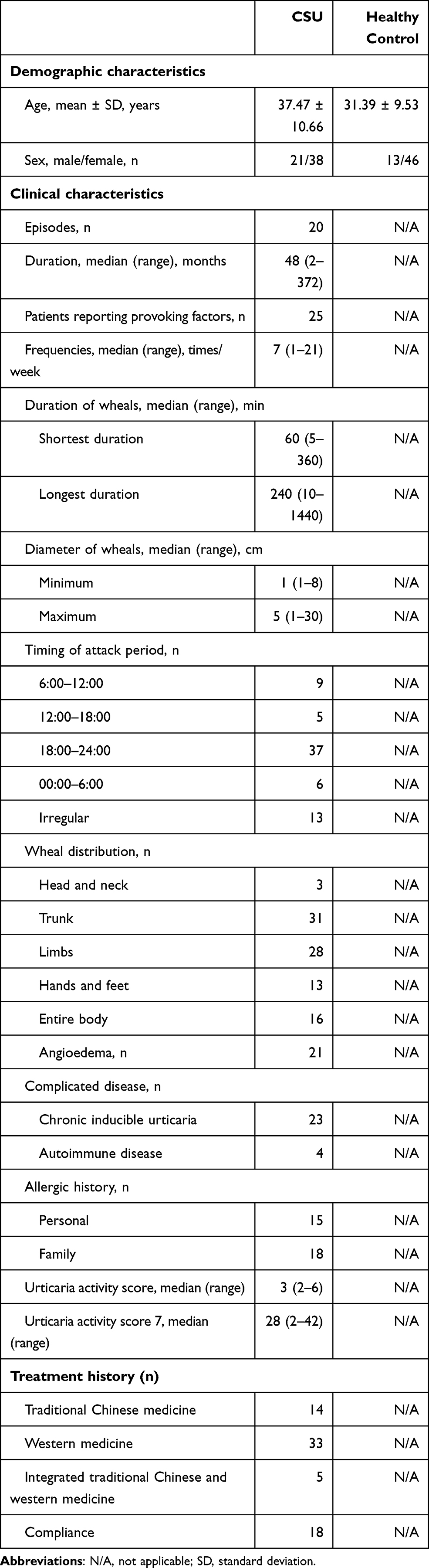 |
Table 1 Demographic, Clinical, and Treatment History Characteristics |
LncRNA and mRNA Expression Profiles
A total of 1482 lncRNAs were found to be upregulated and 1775 downregulated with FC ≥ 1.5 in CSU patients versus healthy control individuals (Figure 2A). Of these up- and downregulated lncRNAs, 678 and 573, respectively, were significantly differentially expressed (FC ≥ 1.5, p < 0.05). ENST00000562459 was upregulated to the greatest extent, with an FC of 6.4280631, while NR_122077 was downregulated to the greatest extent, with an FC of 5.1422487 (Figure 2B, Table 2). LncRNA expression profiles were distinguishable between CSU patients and healthy control individuals according to the hierarchical clustering (Figure 2C).
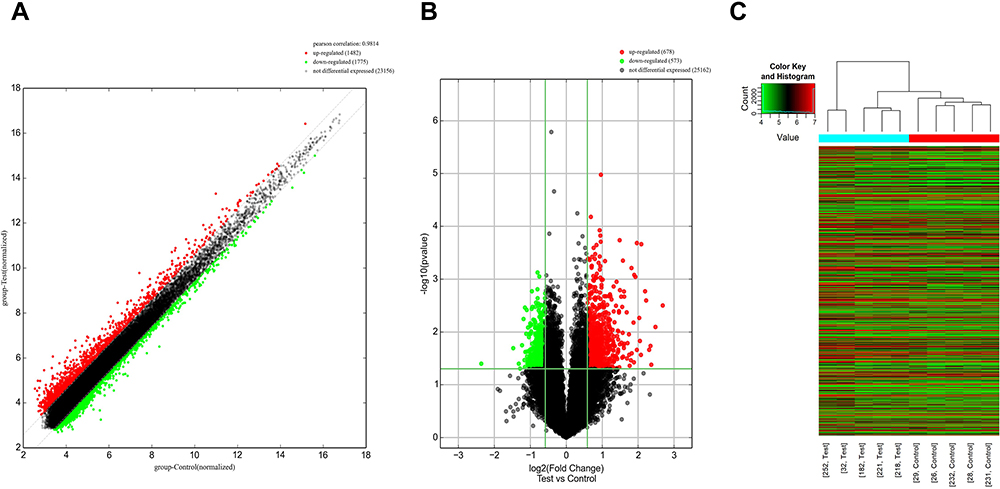 |
Figure 2 lncRNA expression profile. (A) Scatter plot. (B) Volcano plot of differentially expressed lncRNA. (C) Heatmap analysis. Red and green indicate up- and downregulated lncRNAs, respectively, and black indicates RNAs with no significant differential expression. The five left and right columns represent CSU patients and healthy controls, respectively. |
A total of 1072 mRNAs were upregulated and 549 downregulated with an FC ≥ 1.5 in CSU patients versus healthy controls (Figure 3A). Of these up- and downregulated mRNAs, 609 and 176, respectively, were significantly differentially expressed (FC ≥ 1.5, p < 0.05). NM_033130 was upregulated to the greatest extent with an FC of 4.2383847, while NM_002538 was downregulated to the greatest extent with an FC of 11.41218 (Figure 3B, Table 3). mRNA expression profiles were distinguishable between CSU patients and healthy controls according to the hierarchical clustering (Figure 3C).
 |
Figure 3 mRNA expression profile of mRNA. (A) Scatter plot of mRNA. (B) Volcano plot of differentially expressed mRNA. (C) Heatmap analysis of mRNA. Red and green indicate upregulated and downregulated mRNAs, respectively, and black indicates mRNAs with no significant differential expression. The five left and right columns represent CSU patients and healthy controls, respectively. |
We constructed a Circos graph to visualize the chromosomal distributions and classifications of the dysregulated lncRNAs and mRNAs (Figure 4). There were 114 lncRNAs in chromosome 2 and 77 mRNAs in chromosome 1.
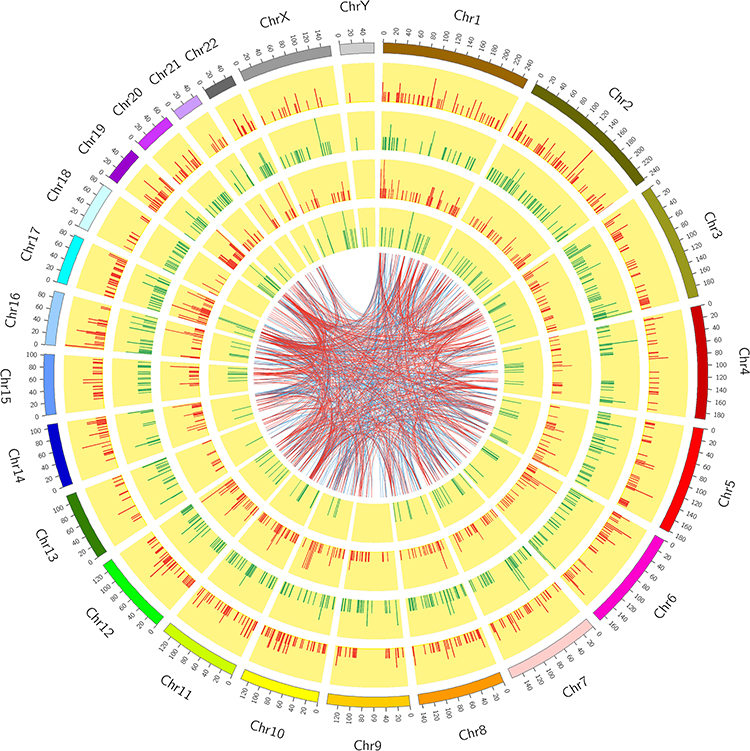 |
Figure 4 Circos plot showing differentially expressed lncRNAs and mRNAs on human chromosomes. The first circle is the human autosomal distribution map. The second and third circles show the distributions of differentially expressed genes among the chromosomes. Red and green lines show up- and downregulated RNAs, respectively. A higher column indicates greater differential expression of genes in the region. The fourth and fifth circles show the distributions of differentially expressed lncRNAs on chromosomes. The expression form is related to the expression of RNA. Internal connections indicate relationships between the top 500 co-expressed lncRNAs and mRNAs (PCC ≥ 0.8, p < 0.5). Red and blue show positive and negative correlations, respectively. |
GO and KEGG Pathway Analyses
Among upregulated target genes, GO analysis showed that vesicle-mediated transport had the highest enrichment score in the BP category (Figure 5A), cytoplasmic vesicle in the CC category (Figure 5B), and carbohydrate binding in the MF category (Figure 5C). Among downregulated target genes, establishment of epithelial cell apical/basal polarity had the highest enrichment score in the BP category (Figure 6A), the apical part of the cell in the CC category (Figure 6B), and intracellular chloride channel activity in the MF category (Figure 6C).
 |
Figure 5 GO enrichment analyses showing upregulated differentially expressed mRNAs. (A) Biological process (BP) analysis. (B) Cellular component (CC) analysis. (C) Molecular function (MF) analysis. |
 |
Figure 6 GO enrichment analyses showing downregulated differentially expressed mRNAs. (A) Biological process (BP) analysis. (B) Cellular component (CC) analysis. (C) Molecular function (MF) analysis. |
KEGG pathway analysis of all upregulated target genes showed enrichment in 19 pathways including lysosome, osteoclast differentiation and human cytomegalovirus infection, and vascular endothelial growth factor (VEGF) signaling pathways (Supplementary Figure 2, Table 4). Of these, the VEGF pathway was the most closely related to the IP3/DAG pathway (Figure 7), and granules secreted by mast cells contain cytokines including VEGF.15 VEGF increases microvascular permeability, which is partially consistent with the pathogenesis of urticaria, so we hypothesized that the DE mRNAs in this pathway might be closely related to disease occurrence. Downregulated target genes were enriched in four pathways including salmonella infection, tight junction, and cytokine-cytokine receptor pathways (Supplementary Figure 3, Supplementary Table 2).
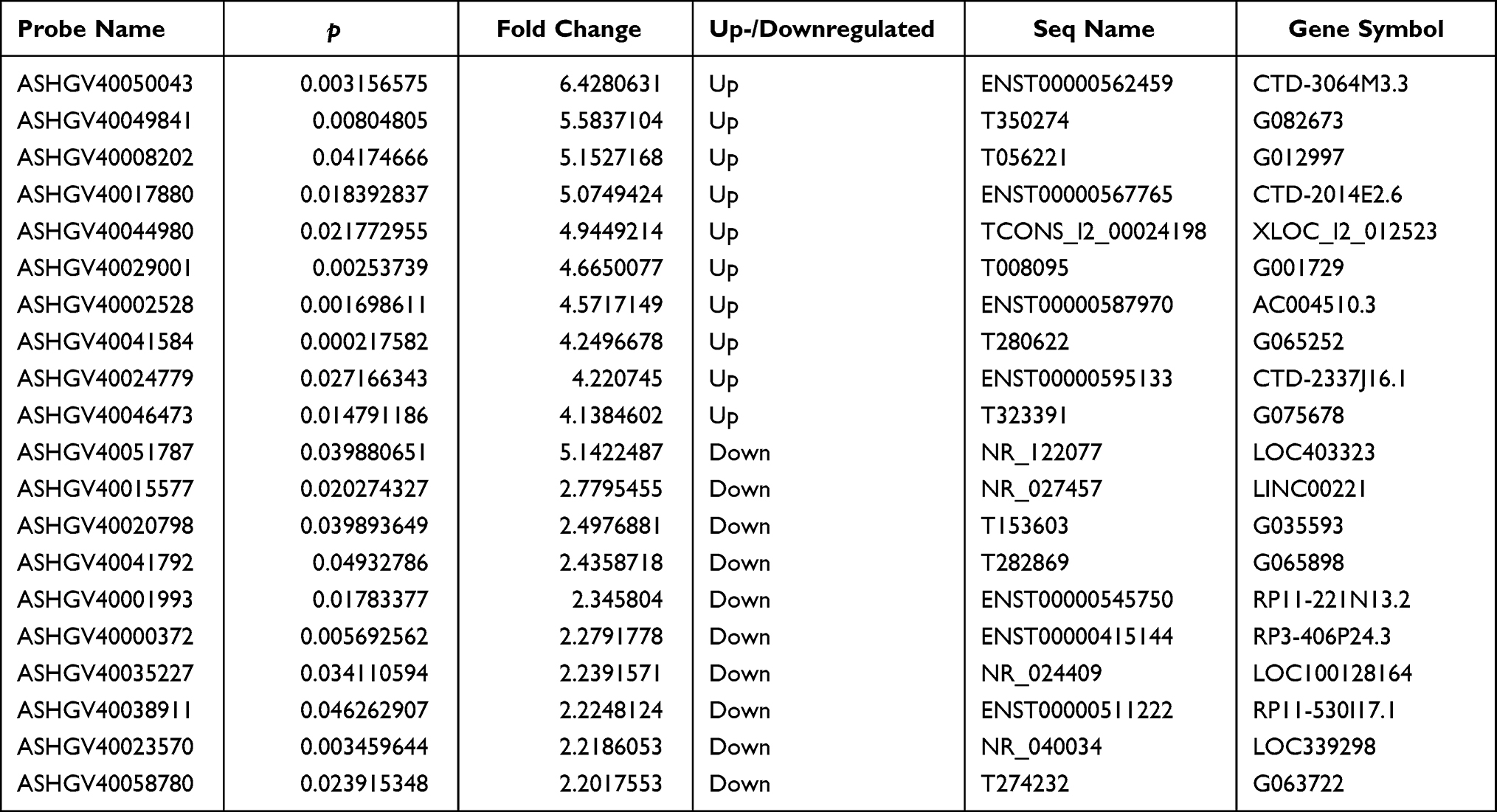 |
Table 2 The Top 10 Up- and Downregulated lncRNAs in the CSU Group Compared with the Control Group |
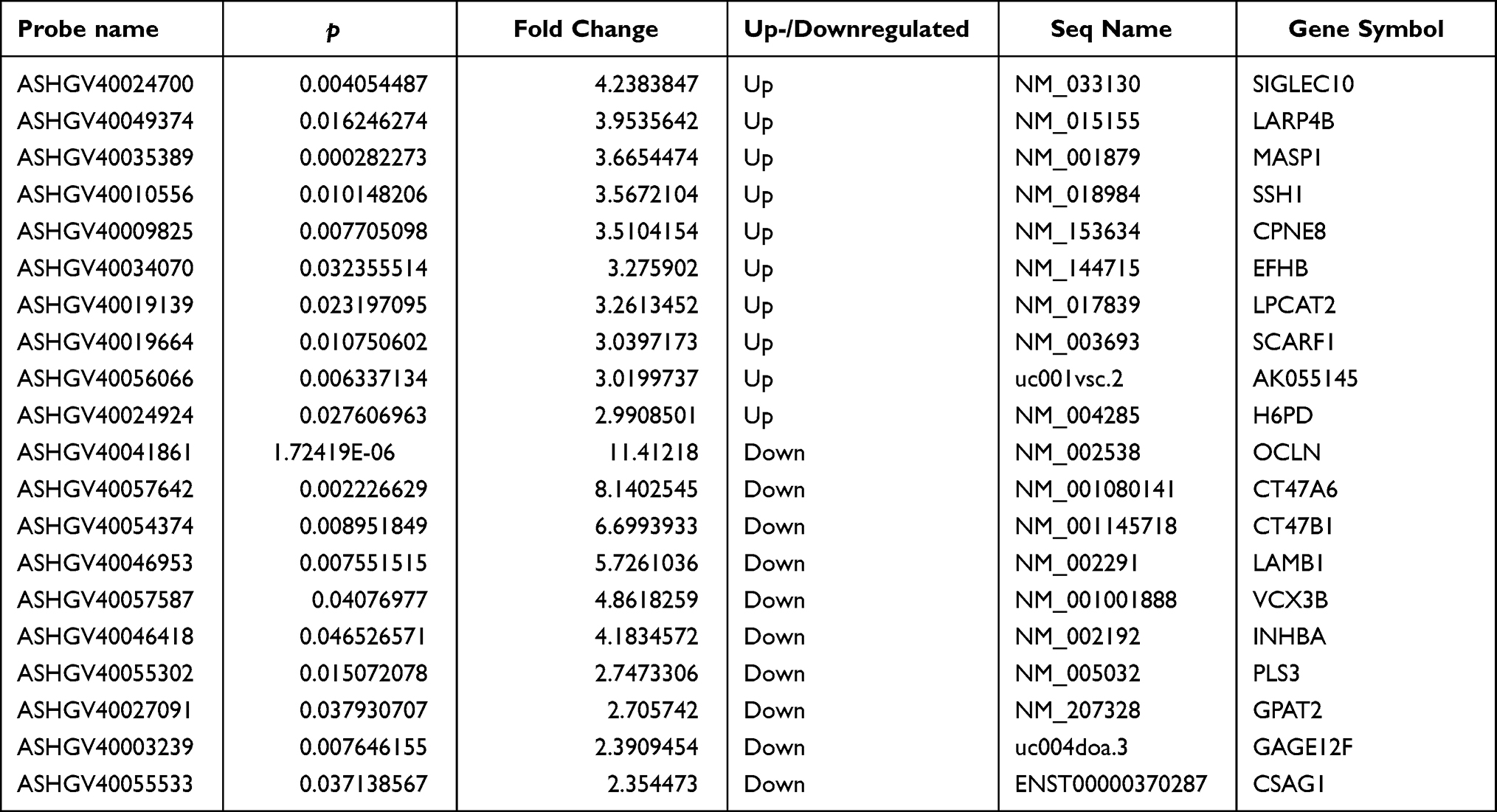 |
Table 3 The Top 10 Up- and Downregulated mRNAs in the CSU Group Compared with the Control Group |
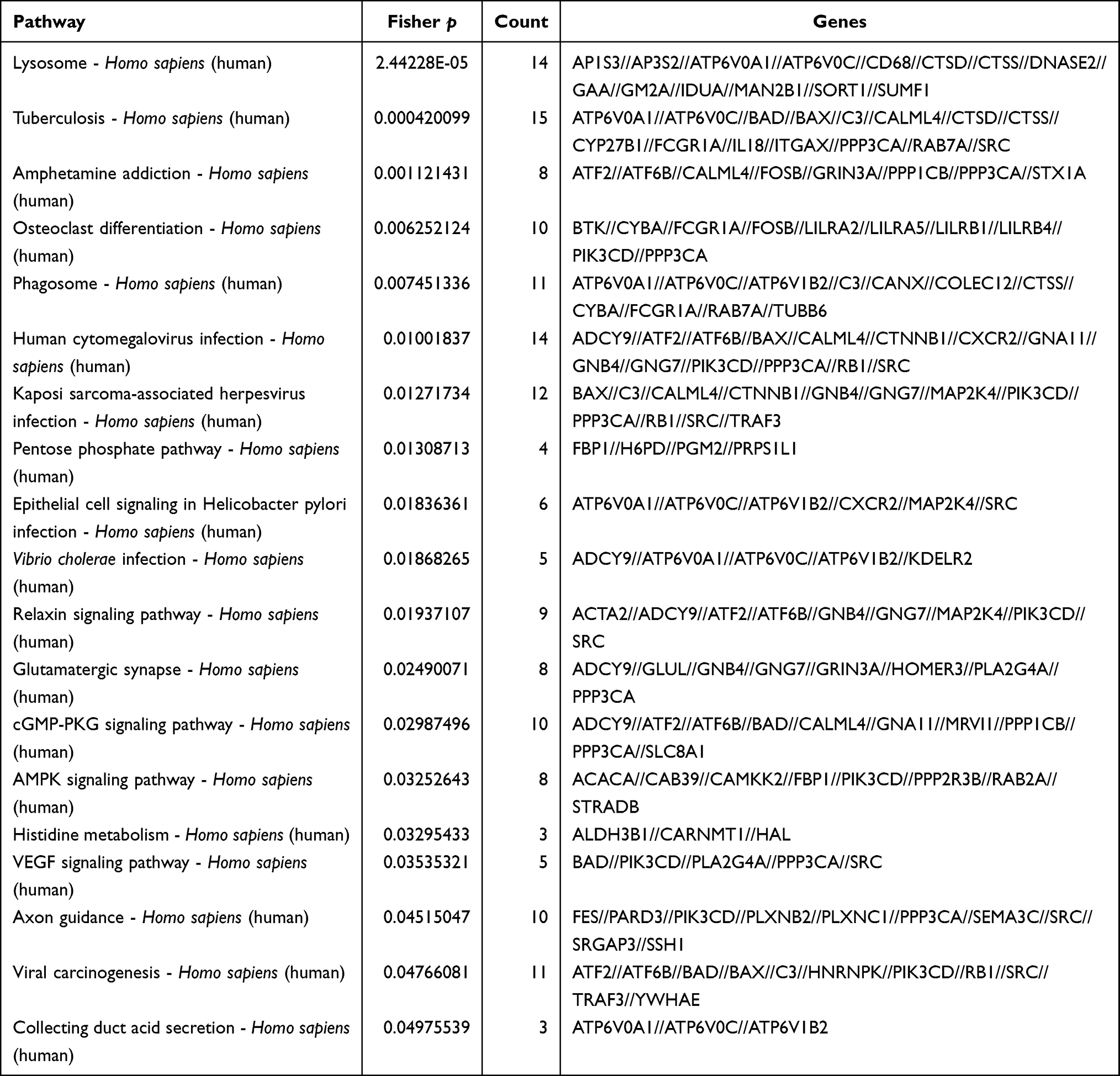 |
Table 4 Signaling Pathways Associated with Differentially Expressed Upregulated mRNAs |
 |
Figure 7 KEGG pathway annotations within the vascular endothelial growth factor (VEGF) signaling pathway. |
DE lncRNAs Related to the IP3/DAG Pathway
According to KEGG analysis, the VEGF pathway was the most closely related to IP3/DAG pathway. There were five DE mRNAs in the VEGF pathway in our analysis: BAD, PIK3CD, PLA2G4A, PPP3CA, and SRC. Pearson analysis of these five mRNAs and all DE lncRNAs revealed that there were 514 upregulated and 488 downregulated DE lncRNAs related to the 5 VEGF pathway mRNAs (PCC ≥ 0.8, p < 0.5, FDR ≤ 1). Supplementary Table 3 shows the most closely related lncRNAs based on PCC values.
Validation of lncRNAs Using qRT-PCR and lncRNA–mRNA Co-Expression Networks
In total, seven lncRNAs were selected for validation, of which six were upregulated (T264761, T280622, ENST00000587970, T224062, ENST00000562459 and his-1_RNA_dna) and one was downregulated (ENST00000417930).
These 7 lncRNAs and 959 mRNAs were used to construct a co-expression network (Figure 8). GO analysis revealed those co-expressed mRNAs were most enriched in the regulation of intracellular transport within the BP category, cell leading edge within the CC category, and receptor ligand activity within the MF category (Figure 9). These biological functions are closely related to the process of mast cell degranulation, indicating that the lncRNAs were indeed related to CSU development.
 |
Figure 8 Co-expression network of lncRNAs and mRNAs. Red and yellow nodes represent up- and downregulated lncRNAs, respectively, and blue nodes represent mRNAs. Dotted and solid lines indicate negative and positive correlations, respectively. |
 |
Figure 9 GO enrichment analyses of co-expressed mRNAs associated with seven biomarker candidate lncRNAs. |
Samples from 53 CSU patients and 20 healthy controls were selected for qRT-PCR validation. Supplementary Table 4 shows the specific primers sequences for the seven lncRNAs. The results demonstrated that T264761 (1.62-fold), T280622 (3.03-fold), ENST00000587970 (2.67-fold), ENST00000562459 (2.70-fold), and his-1_RNA_dna (1.38-fold) were significantly increased in CSU, while ENST00000417930 (2.06-fold) was significantly decreased in CSU (Figure 10, Supplementary Table 5). However, T224062 was not dysregulated in the CSU group.
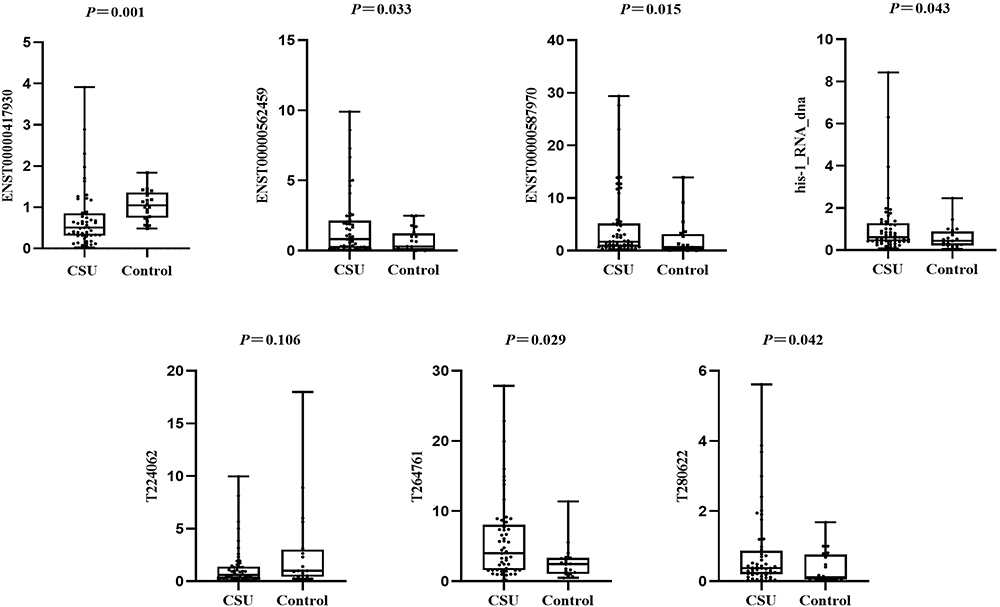 |
Figure 10 Expression levels of seven lncRNAs in the CSU (n = 53) and healthy control (n = 20) groups measured by qRT-PCR. Six of seven lncRNAs were confirmed to be significantly differentially expressed; T224062 was the exception. |
Correlation of lncRNAs with Clinical Characteristics and Inflammatory Mediators
Samples from 56 CSU patients and 13 healthy controls were selected for ELISA, which showed increased expression levels of D-dimer and HIS in the serum of CSU patients (Figure 11, Supplementary Table 6).
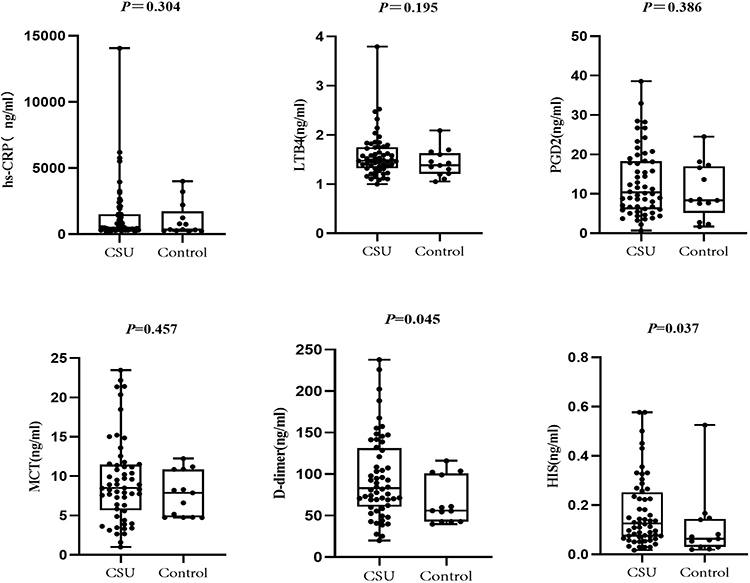 |
Figure 11 Serum concentrations of hs-CRP, LTB4, PGD2, MCT, D-dimer and HIS in the CSU (n = 56) and healthy control (n = 13) groups. Differences between groups were assessed using Mann–Whitney U-tests; experiments were repeated three times. Abbreviations: hs-CRP, high sensitivity C-reactive protein; LTB4, leukotriene B4; PGD2, prostaglandin D2; MCT, mast cell tryptase; HIS, histamine. |
Spearman coefficient analysis was used to assess the correlations of six lncRNAs (T264761, T280622, ENST00000587970, ENST00000562459, his-1_RNA_dna, and ENST00000417930) with clinical characteristics (age, disease duration, frequency of symptom occurrence, duration of wheals, size of wheals, and UAS7 score) and inflammatory mediators (D-dimer and HIS). A positive correlation was observed between his-1_RNA_dna and the frequency of symptom occurrence, and negative correlations were noted between four lncRNAs (T264761, T280622, his-1_RNA_dna, ENST00000417930) and the maximum size of wheals (Figure 12, Supplementary Table 7).
 |
Figure 12 Correlations between lncRNA levels and clinical characteristics in CSU patients. The expression levels of T264761, T280622, his-1_RNA_dna, and ENST00000417930 were associated with the maximum size of wheals. The expression level of his-1_RNA_dna was associated with the frequency of symptom occurrence. |
ROC Curves of Selected lncRNAs for CSU Risk Prediction
Logistic regression was used to analyze the CSU risk associated with selected lncRNAs. Six lncRNAs with significant differences in univariate analysis were included as independent variables, with CSU occurrence as the dependent variable (1 = occurrence, 0 = no occurrence). ENST00000417930 (odds ratio [OR] = 0.385; 95% confidence interval [CI] = 0.156–0.948) and T264761 (OR = 1.266, 95% CI = 1.023–1.567) were identified as risk factors for CSU (Table 5). Furthermore, at the cut-off value of 4.138, the area under the ROC curve for T264761 was 0.666, and the sensitivity and specificity values were 49.06% and 90%, respectively (Figure 13, Supplementary Table 8). This finding suggested that the T264761 expression level may be useful for differentiating CSU patients from healthy controls.
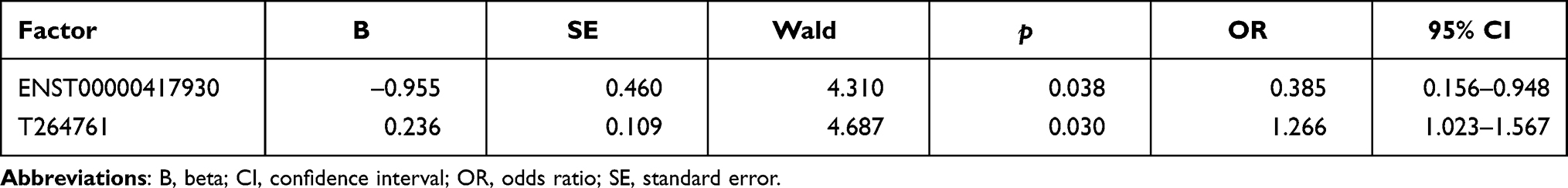 |
Table 5 Logistic Regression Analyses to Assess the Potential Value of lncRNAs in the Prediction of CSU Risk |
 |
Figure 13 The ROC curve for lncRNAs. |
Prognostic Value of T264761
The last follow-up data was collected in 4th September 2021. A total of 47 patients were included in this analysis. According to a previous study in China, a Urticaria Control Test (UCT) score ≥12 indicates well-controlled CSU.16 Based on a T264761 expression level cut-off value of 4.138, CSU patients were sorted into high and low expression groups.14 The rate of well-controlled disease in those with low T264761 expression was 82.61% compared with 54.17% in those with high T264671 expression. Table 6 shows that lower T264671 expression could predict higher disease control rates.
 |
Table 6 Correlations Between T264761 Levels and UCT Scores in CSU Group After 2-Year Follow-Up |
Discussion
In this study, we identified several DE lncRNAs and mRNAs in CSU patients compared with healthy controls using microarray sequencing. Some were found to be closely related to IP3/DAG signaling pathway, a key pathway in the induction of mast cell degranulation, which is involved in CSU. Importantly, we identified a potential biomarker for this disease, where no known biomarkers existed previously: lncRNA T264761 was found to have diagnostic and prognostic value for CSU in our study.
Upregulated lncRNAs and mRNAs outnumbered those that were downregulated in CSU samples compared with healthy controls, indicating the activation of multiple biological processes or signaling pathways under pathological conditions. In our study, we identified DE lncRNAs using a FC threshold of 1.5, compared to 2 in other studies.13,17,18 A lower threshold in this study may be related to the characteristic CSU symptoms of sudden itchy wheals or angioedema. Most of the CSU patients were in remission, and were therefore symptomatically similar to healthy control individuals except for those who experienced sudden attacks, so the overall differential expression may have been relatively low.
DE mRNAs were analyzed using GO term and KEGG pathway enrichment analyses. GO analysis showed that most upregulated mRNAs were associated with vesicle-mediated transport within the BP category, cytoplasmic vesicle within the CC category and carbohydrate binding within the MF category. This pattern is consistent with the process of mast cell degranulation, which involves the secretion of mediators via a vesicle secretion system.19,20 Mast cells also require carbohydrates for granule generation and maintenance.21 Our KEGG analysis showed that the VEGF signaling pathway was the most closely related to the occurrence of urticaria. VEGF is a mediator of vascular permeability that could be involved in inducing and maintaining urticaria symptoms; the IP3/DAG pathway is known to be involved in urticaria pathology and is included in the pathway diagram in Figure 7.22 We therefore identified five DE mRNAs in the VEGF pathway that were used to identify seven lncRNAs closely related to the IP3/DAG pathway (T264761, T280622, ENST00000587970, T224062, ENST00000562459, his-1_RNA_dna, and ENST00000417930). Co-expression network and GO analyses confirmed that these lncRNAs were closely related to the development of CSU.
Co-expressed mRNAs were found to be enriched in the regulation of intracellular transport within the BP category, cell leading edge within the CC category, and receptor ligand activity within the MF category. Many high-affinity receptors for IgE are expressed on the mast cell surface, and the intracellular transportation and release of pro-inflammatory mediators following antigen-induced aggregation causes allergic reactions.5,23 Our qRT-PCR results suggested that T224062 was not significantly dysregulated in CSU patients, which contradicted our microarray sequence data; however, this may be due to the relatively small sample size.
Given that our bioinformatics analysis revealed putative associations between several lncRNAs and CSU, we then assessed their correlations with clinical characteristics of the disease to determine their usefulness as disease indicators. Our ELISA results revealed that D-dimer and HIS expression were increased in CSU. Previous studies showed a positive correlation between D-dimer levels and the severity of CSU activity.24,25 However, we found no correlation between the seven lncRNAs we identified in our bioinformatics analysis and D-dimer or HIS levels. Some of the lncRNAs were correlated with the frequency of symptom occurrence and the maximum size of wheals. It suggested that lncRNAs did not seem to be promising biomarkers for disease activity. Logistic regression showed that high levels of T264761 could indicate a greater risk of CSU. The ROC curve analysis suggested that T264761 may differentiate CSU patients from healthy control individuals with high specificity; however, the sensitivity was not high. A combination of medical history and symptom evaluation could improve its diagnostic value. We performed a 2-year follow-up to assess the prognostic value of T264761 in CSU patients based on the UCT score. This assessment was the first valid and reliable tool to assess disease control in patients with CSU.26 This test was rarely used in CSU clinical research in China before 2020; however, Yu et al translated the UCT into Chinese and assessed the reliability, validity, sensitivity, and screening accuracy of the new scale, enabling its application in China.16 In our follow-up, lower T264761 expression (≤4.138) could predict a higher disease control rate. Notably, in five women, CSU resolved after pregnancy during the follow-up period; further studies of lncRNA levels in pregnant and postnatal women with CSU could help identify the underlying mechanism.
Many recently developed techniques have enabled the identification of potentially useful CSU biomarkers, including proteomic analysis, transcriptome analysis, microRNA or gene sequencing, and gut microbiome analysis.27–31 However, this study is the first to assess the diagnostic and prognostic value of lncRNAs as CSU biomarkers using microarrays, bioinformatics analyses, and co-expression network construction. T264761 was found to be related to the IP3/DAG signal pathway, which is known to induce mast cell degranulation that is involved in CSU. This lncRNA was also associated with the maximum size of the wheals that are symptomatic of this disease. High levels of T264761 may indicate an increased CSU risk with high specificity. This lncRNA may also predict longer-term disease control status. This study provides novel insights into CSU biomarkers and guide further basic and clinical research.
Our results should be considered in the context of some limitations. First, the samples were only from CSU patient and healthy controls; in future studies we will include more samples including those from subjects acute urticaria and other similar allergic diseases as controls to further investigate the diagnostic value of lncRNAs. Secondly, the frequency and duration of follow-up were relatively short, because urticaria has a self-healing tendency without treatment, so the findings should be interpreted with caution. Thirdly, the results of this study are mostly based on bioinformatics predictions, and further cell culture and animal experiments are needed to clarify the specific roles of lncRNAs in CSU. Additionally, it is worth mentioning that although some CSU patients included were complicated with chronic idiopathic urticaria, the results are still plausible. This is consistent with clinical practice and other relevant CSU clinical literatures.28,32
Conclusions
Our study established lncRNA and mRNA expression profiles in CSU using microarrays. Some lncRNAs and mRNAs were differentially expressed in CSU patients compared with healthy controls; these were shown to be involved in diverse biological pathways related to mast cell degranulation, which is associated with CSU, and the IP3/DAG signal pathway that is involved in producing the characteristic wheals and itchiness associated with CSU. The lncRNA T264761 may be a clinically useful novel diagnostic biomarker for CSU, with lower levels predicting a better UCT score and thus indicating less severe disease. Further investigations are required to clarify the specific function of T264761 in CSU pathogenesis.
Data Sharing Statement
Supporting data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
This study was jointly funded by grants from the National Natural Science Foundation of China (81804176,81904074), the China Postdoctoral Science Foundation (2017M622919, 2018M631049), the Natural Science Foundation of Guangdong Province of China (2020A1515011421), and the Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (20191369, 20201380).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Powell RJ, Leech SC, Till S, Huber PA, Nasser SM, Clark AT. BSACI guideline for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2015;45(3):547–565. doi:10.1111/cea.12494
2. Puxeddu I, Petrelli F, Angelotti F, Croia C, Migliorini P. Biomarkers in chronic spontaneous urticaria: current targets and clinical implications. J Asthma Allergy. 2019;12:285–295. doi:10.2147/jaa.S184986
3. Kolkhir P, André F, Church MK, Maurer M, Metz M. Potential blood biomarkers in chronic spontaneous urticaria. Clin Exp Allergy. 2017;47(1):19–36. doi:10.1111/cea.12870
4. Ghafouri-Fard S, Shoorei H, Taheri M, Sanak M. Emerging role of non-coding RNAs in allergic disorders. Biomed Pharmacother. 2020;130:110615. doi:10.1016/j.biopha.2020.110615
5. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. 2006;6(3):218–230. doi:10.1038/nri1782
6. Kim YJ, Sekiya F, Poulin B, Bae YS, Rhee SG. Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol Cell Biol. 2004;24(22):9986–9999. doi:10.1128/mcb.24.22.9986-9999.2004
7. Gong L, Li J, Tang Y, et al. The antinociception of oxytocin on colonic hypersensitivity in rats was mediated by inhibition of mast cell degranulation via Ca(2+)-NOS pathway. Sci Rep. 2016;6:31452. doi:10.1038/srep31452
8. Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G. The role of human mast cell-derived cytokines in eosinophil biology. J Interferon Cytokine Res. 2004;24(5):271–281. doi:10.1089/107999004323065057
9. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. doi:10.1038/nrg2521
10. Li J, Gao Y, Li Q, Chen L, Chen Y, Li J. LncRNA COL1A2-AS1 promotes skin fibroblast apoptosis by repressing p-Smad3 and promoting β-catenin expression. Exp Dermatol. 2021;30(8):1090–1098. doi:10.1111/exd.14269
11. Li D, Kular L, Vij M, et al. Human skin long noncoding RNA WAKMAR1 regulates wound healing by enhancing keratinocyte migration. Proc Natl Acad Sci U S A. 2019;116(19):9443–9452. doi:10.1073/pnas.1814097116
12. Jia HY, Zhang K, Lu WJ, Xu GW, Zhang JF, Tang ZL. LncRNA MEG3 influences the proliferation and apoptosis of psoriasis epidermal cells by targeting miR-21/caspase-8. BMC Mol Cell Biol. 2019;20(1):46. doi:10.1186/s12860-019-0229-9
13. Lin F, Gong X, Yu P, et al. Distinct circulating expression profiles of long noncoding RNAs in heart failure patients with ischemic and nonischemic dilated cardiomyopathy. Front Genet. 2019;10:1116. doi:10.3389/fgene.2019.01116
14. Ruopp MD, Perkins NJ, Whitcomb BW, Schisterman EF. Youden index and optimal cut-point estimated from observations affected by a lower limit of detection. Biom J. 2008;50(3):419–430. doi:10.1002/bimj.200710415
15. Grützkau A, Krüger-Krasagakes S, Baumeister H, et al. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206. Mol Biol Cell. 1998;9(4):875–884. doi:10.1091/mbc.9.4.875
16. Yu M, Chen Y, Liu B, Song X, Zhao Z. The Chinese version of the urticaria control test and validation of its reliability and validity. Chin J Dermatol. 2020;53(7):533–538. doi:10.35541/cjd.20191190
17. Feng Y, Shen Y, Chen H, et al. Expression profile analysis of long non-coding RNA in acute myeloid leukemia by microarray and bioinformatics. Cancer Sci. 2018;109(2):340–353. doi:10.1111/cas.13465
18. Shi H, Cao N, Pu Y, Xie L, Zheng L, Yu C. Long non-coding RNA expression profile in minor salivary gland of primary Sjögren’s syndrome. Arthritis Res Ther. 2016;18(1):109. doi:10.1186/s13075-016-1005-2
19. Serrano-López EM, López-Martínez D, Gómez-Fernández JC, Egea-Jiménez AL, Corbalán-García S. PKCε controls the fusion of secretory vesicles in mast cells in a phosphatidic acid-dependent mode. Int J Biol Macromol. 2021;185:377–389. doi:10.1016/j.ijbiomac.2021.06.019
20. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10(6):440–452. doi:10.1038/nri2782
21. Plum T, Wang X, Rettel M, Krijgsveld J, Feyerabend TB, Rodewald HR. Human mast cell proteome reveals unique lineage, putative functions, and structural basis for cell ablation. Immunity. 2020;52(2):404–416.e405. doi:10.1016/j.immuni.2020.01.012
22. Zhao JW, Ping JD, Wang YF, et al. Vitamin D suppress the production of vascular endothelial growth factor in mast cell by inhibiting PI3K/Akt/p38 MAPK/HIF-1α pathway in chronic spontaneous urticaria. Clin Immunol. 2020;215:108444. doi:10.1016/j.clim.2020.108444
23. Amin K. The role of mast cells in allergic inflammation. Respir Med. 2012;106(1):9–14. doi:10.1016/j.rmed.2011.09.007
24. Triwongwaranat D, Kulthanan K, Chularojanamontri L, Pinkaew S. Correlation between plasma D-dimer levels and the severity of patients with chronic urticaria. Asia Pac Allergy. 2013;3(2):100–105. doi:10.5415/apallergy.2013.3.2.100
25. Deza G, Ricketti PA, Giménez-Arnau AM, Casale TB. Emerging biomarkers and therapeutic pipelines for chronic spontaneous urticaria. J Allergy Clin Immunol Pract. 2018;6(4):1108–1117. doi:10.1016/j.jaip.2018.02.024
26. Weller K, Groffik A, Church MK, et al. Development and validation of the urticaria control test: a patient-reported outcome instrument for assessing urticaria control. J Allergy Clin Immunol. 2014;133(5):
27. Kim JH, Lee HY, Ban GY, Shin YS, Park HS, Ye YM. Serum clusterin as a prognostic marker of chronic spontaneous urticaria. Medicine. 2016;95(19):e3688. doi:10.1097/md.0000000000003688
28. Giménez-Arnau A, Curto-Barredo L, Nonell L, et al. Transcriptome analysis of severely active chronic spontaneous urticaria shows an overall immunological skin involvement. Allergy. 2017;72(11):1778–1790. doi:10.1111/all.13183
29. Lin CE, Kaptein JS, Sheikh J. Differential expression of microRNAs and their possible roles in patients with chronic idiopathic urticaria and active hives. Allergy Rhinol. 2017;8(2):67–80. doi:10.2500/ar.2017.8.0199
30. Patel OP, Giorno RC, Dibbern DA, Andrews KY, Durairaj S, Dreskin SC. Gene expression profiles in chronic idiopathic (spontaneous) urticaria. Allergy Rhinol. 2015;6(2):101–110. doi:10.2500/ar.2015.6.0124
31. Wang D, Guo S, He H, Gong L, Cui H. Gut microbiome and serum metabolome analyses identify unsaturated fatty acids and butanoate metabolism induced by gut microbiota in patients with chronic spontaneous urticaria. Front Cell Infect Microbiol. 2020;10:24. doi:10.3389/fcimb.2020.00024
32. Grieco T, Dies L, Sernicola A, et al. Potential clinical and serological predictors of chronic spontaneous urticaria relapse in patients under omalizumab treatment. Immunotherapy. 2020;12(16):1173–1181. doi:10.2217/imt-2020-0088
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.