Back to Journals » OncoTargets and Therapy » Volume 13
Exploring DNA Methylation Profiles Altered in Cryptogenic Hepatocellular Carcinomas by High-Throughput Targeted DNA Methylation Sequencing: A Preliminary Study for Cryptogenic Hepatocellular Carcinoma
Authors Wang X, Cheng Y, Yan LL, An R, Wang XY, Wang HY
Received 9 July 2020
Accepted for publication 4 September 2020
Published 6 October 2020 Volume 2020:13 Pages 9901—9916
DOI https://doi.org/10.2147/OTT.S267812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Xin Wang,1,* Ya Cheng,1,* Liang-liang Yan,2 Ran An,1 Xing-yu Wang,1 Heng-yi Wang1
1Department of Emergency Surgery, The First Affiliated Hospital of Anhui Medical University, Hefei 230032, People’s Republic of China; 2Department of Rheumatology and Immunology, The First Affiliated Hospital of Anhui Medical University, Hefei 230032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Heng-yi Wang Tel +86-156-0551-1180
Email [email protected]
Background: Hepatocellular carcinoma (HCC) includes cryptogenic hepatocellular carcinomas (CR-HCC) that lack a defined cause. Specific DNA methylation patterns and comparisons of the aberrant alterations in DNA methylation between CR-HCC and adjacent peritumor tissues (APTs) have not yet been reported.
Methods: The SureSelectXT Methyl-Seq Target Enrichment System was used to sequence targeted DNA methylation in three paired CR-HCC tissues and APTs. Gene Ontology (GO) enrichment and KEGG pathway analysis were performed to investigate the DNA methylation mechanism of CR-HCC. The mRNA expression levels of HOXB-AS3, HOXB6, HOXB3, USP18, MAP3K6, TIRAP, TNNI2, SHC3, CTTN, and TFAP2A, selected from the identified signaling pathways, were evaluated by quantitative real-time PCR (qPCR).
Results: A total of 1728 differentially methylated regions (DMRs) were identified in tumor tissues compared with non-tumor tissues, of which 868 DMRs were hypermethylated and 860 were hypomethylated. The DMRs were mapped within 2091 DMR-associated genes (DMGs). The mRNA expression of HOXB-AS3, HOXB3, and MAP3K6 was downregulated in CR-HCC tissues compared to the APTs. However, the mRNA expression of TIRAP, SHC3, and CTTN was upregulated in the CR-HCC tissues. Differences between the mRNA expression of HOXB6, USP18, TNNI2, and TFAP2A in the CR-HCC and APTS tissues were not statistically significant. GO analysis showed that the molecular functions of “binding”, “protein binding”, and “cytoskeletal protein binding” were the main categories for the hypermethylated DMGs. The hypomethylated DMGs were mostly enriched in the molecular functions “binding”, “protein binding”, “calcium ion binding”, among others. KEGG pathway analysis showed that the hypermethylated DMGs were enriched in several pathways such as “estrogen signaling pathway”, while hypomethylated DMGs were enriched in several pathways such as “proteoglycans in cancer”, suggesting that epigenetic modifications play important roles in the cryptogenic hepatocarcinogenesis.
Conclusion: These results provide useful information for future work to characterize the functions of epigenetic mechanisms on CR-HCC.
Keywords: cryptogenic hepatocellular carcinomas, DNA methylation, DMR
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer deaths worldwide.1 Improvement in HCC treatment and prognosis is, therefore, urgent, but is challenging. To our knowledge, various factors, including infection with hepatitis B (HBV) or C viruses (HCV) and alcoholic and non-alcoholic steatohepatitis (NASH),2–4 are primarily responsible for HCC. HCC patients with active HBV or HCV infection have worse prognoses for survival time than uninfected patients, namely non-B non-C HCC (nBnC-HCC).5,6 Although there have been many recent studies on the development of HCC the mechanisms promoting HCC initiation and development remain largely unknown. Thus, it is important to understand the carcinogenic mechanism of HCC and to explore new therapeutic targets for the disease.
We defined cryptic hepatocellular carcinoma (CR-HCC) as HCC patients without any background liver etiology after excluding all other testable liver disease etiologies, including occult HBV infection, NASH, primary biliary cirrhosis, hemochromatosis, Wilson’s disease, alcoholic liver damage, α1 antitrypsin deficiency, severe steatosis, and autoimmune hepatitis. Because there are no guidelines for early detection of CR-HCC, these tumors to be greater in size and at advanced stages when diagnosed compared with hepatitis virus-related HCCs.7 It has been reported that the survival time of advanced CR-HCC is even shorter than that of hepatitis B/C-HCC.8 Therefore, there is an urgent need for a surveillance program for the early detection of CR-HCC. Although studies have identified a few of the tumor characteristics and the postoperative prognosis of CR-HCC patients, the main mechanism underlying CR-HCC is still unclear because of the rarity of the disease.
Epigenetic mechanisms can play important roles as interfaces between the genome and environmental influences, including lifestyle and other risk factors.9,10 Some investigations have suggested that epigenetic multi-regulation causes different epigenetic changes related to liver disease and may account for the progression from liver disease to HCC. A number of studies have investigated novel HCC therapies from the perspective of epigenetic alterations, targeting epigenetic regulation.11,12 Accumulating evidence on genome-wide DNA methylation provides strong support for the significance of differences in methylation of CpG sites between HCC tissues and adjacent peritumor tissues (APTs).13,14 We have consistently observed aberrant DNA methylation in virus-associated HCC.15,16 Some studies have found abnormal DNA methylation in the carcinogenic progression of non-alcoholic fatty liver disease and NASH to HCC.17 One of the key molecular events in the development of NAFLD-derived HCC was found to be hypermethylation in the promoter region of the glycine N-methyltransferase (GNMT) gene resulting in reduced expression.18
In this study, we focused on cryptogenic HCC (CR-HCC), one of the HCCs of unknown etiology, investigating differences in DNA methylation between CR-HCC tissues and APTs with the aim of increasing our understanding of the role of epigenetic mechanisms in the carcinogenesis of CR-HCC.
Methods
Sample Collection and Preparation
Cryptogenic HCC patients were identified from patients undergoing hepatectomy in the First Affiliated Hospital of Anhui Medical University from January 2017 to May 2018. Patients with HBV or HCV infections or alcoholism-related HCC, or with chronic liver disease of known etiology, such as autoimmune hepatitis, severe steatosis, and α1 antitrypsin deficiency, were excluded A total of three CR-HCC patients were identified.
Specimens from these three patients were collected during the operation. The APTs were collected from a location at least 3 cm away from the tumor boundaries, and all specimens were stored at −80°C in liquid nitrogen. All the collections followed the same protocol. All of the cancerous tissues were diagnosed as primary hepatocellular carcinoma by two independent and experienced pathologists.
Library Preparation and Data Analysis
Agarose gels (1%) were used to monitor the genomic DNA degradation and contamination. The Nano Photometer® spectrophotometer (IMPLEN, CA, USA) was used to check DNA purity. The Qubit® DNA Assay Kit in the Qubit® 2.0 Fluorimeter (Life Technologies, CA, USA) was used to measure DNA concentrations. According to manufacturer’s protocols, the SureSelectXT Methyl-Seq Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA) and EZ-DNA Methylation-Gold Kit (Zymo Research, CA, USA) were used to build whole-genome bisulfite DNA libraries. SureSelectXT Human Methyl-Seq is the first comprehensive target enrichment system to enable researchers to focus on the regions where methylation is known to impact gene regulation: CpG islands, CpG island shores, promoters, and differentially methylated regions (DMRs). After generating clusters, we sequenced the library preparations on the Illumina Hiseq 2000/2500 platform (San Diego, CA, USA) to obtain 125/150bp paired-end reads. The Illumina CASAVA pipeline was used to perform image analysis and base calling. Lastly, we collected the 125/150bp paired-end reads.
Differentially Methylated Sites (DMS) and Differentially Methylated Regions (DMR)
The BiSeq software (version 1.6.0; Hebestreit K, 2013) was used to identify differentially methylated sites (DMSs) and differentially methylated regions (DMRs). Only the target regions were analyzed. The genomic regions termed CpG clusters are covered by CpG sites with a high spatial density. The definition of a DMR is the adjacent rejected CpG region in a CpG cluster. The status of all CpG sites that are hyper- or hypo-methylated is determined by the DMRs. The CpG site is hypomethylated when the difference in methylation switches from positive to negative. Finally, we identified DMRs with more than two CpG sites where the difference of methylation level was greater than 0.1. For experiments without replicates, DMSs and DMRs were identified using the methylKit software (version 1.0.0; Akalin A, 2012). For DMR identification, methylKit tiles the genome with windows of 1000bp in length and a step size of 1000bp and summarizes the methylation information on the tiles. MethylKit uses Fisher’s exact test to calculate the P-values for the differential methylation which are then adjusted to Q-values using the SLIM method (Wang, 2011). The differentially methylated regions/bases are selected from the calculated Q-values and percent methylation difference cutoffs. The threshold value was a Q-value <0.05 and a percent methylation difference larger than 10%.
GO, KEGG Enrichment Analysis of DMR-Related Genes
GO enrichment analysis was performed with the GOseq function in R after correction for the gene length bias. DMR-related genes were considered significantly enriched when the corrected P-value was less than 0.05 in GO terms. In this analysis, KOBAS software (Mao X, 2005) was used to analyze the statistical enrichment of DMG with the assistance of the KEGG database, which helps to understand the high-level functions and utilities of biological systems from the molecular level, particularly from large-scale molecular datasets generated by genome sequencing and other high-throughput experimental techniques (http://www.genome.jp/kegg/)(Kanehisa M, 2008).
Quantitative Real‐Time Polymerase Chain Reaction (qPCR)
Total RNA from the CR-HCC tissues and APTs was extracted using the TRIzol reagent according to the manual (Invitrogen, USA) and the cDNA was reverse‐transcribed using the HiScript IIQ RT SuperMix for qPCR kit (Nanjing Nuoweizan Biological Technology Co., Ltd., China) according to the manufacturer’s instructions. The real-time qPCR was performed using the AceQTM qPCR SYBR Green Master Mix Kit (Nanjing Nuoweizan Biological Technology Co., Ltd., China). GAPDH served as an internal control and the relative mRNA expressions were calculated using the equation 2−∆∆Ct. All reactions were performed at least three times. The PCR primer sequences are listed in Table 1.
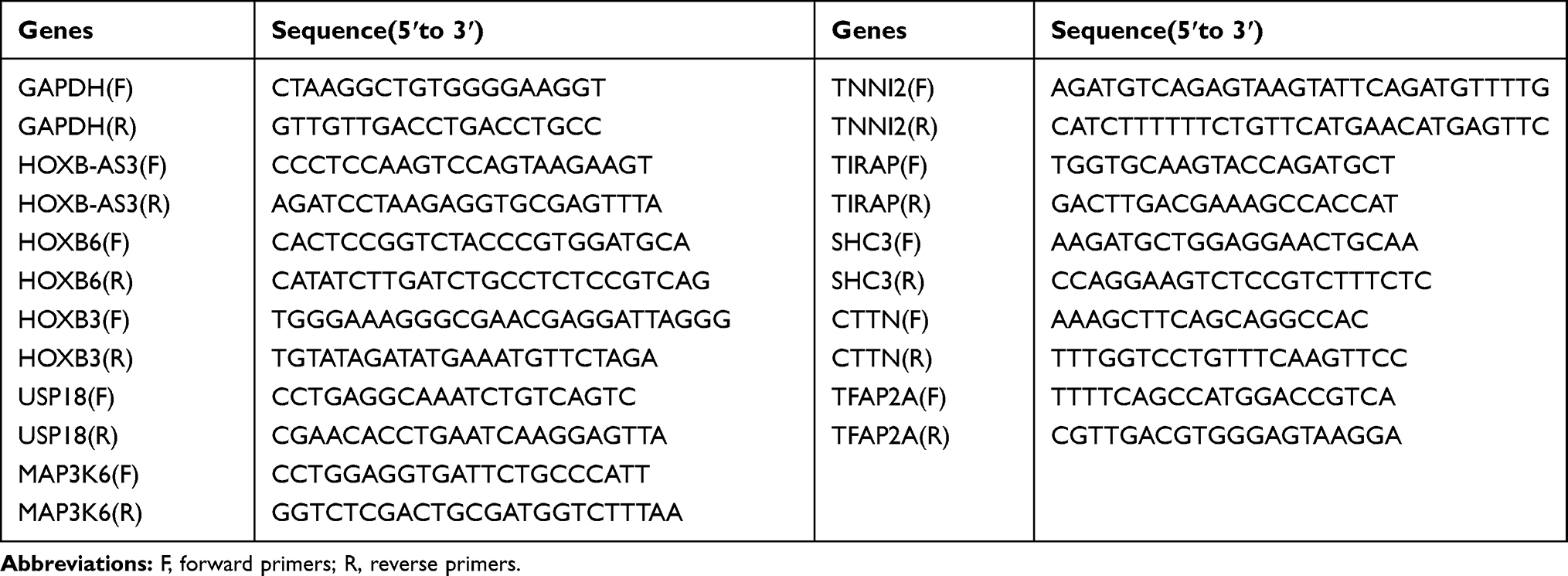 |
Table 1 Nucleotide Sequences of Primers for Real-Time qPCR |
Ethics Statement
All protocols in this study were approved by the Ethics Committee of the First Affiliated Hospital of Anhui Medical University. Written informed consent was gained from all patients in this study at the time of surgery.
Statistical Methods
Statistical analyses were performed with GraphPad Prism v. 8 (GraphPad Software 160 Inc., San Diego, CA, USA). The unpaired Student’s t-test was used to evaluate statistical significance. Results with P-values (P) < 0.05 were considered statistically significant.
Results
DNA Methylation Changes Between CR-HCC Tissues and APTs
To describe the methylome in CR-HCC and unravel the epigenetic basis of CR-HCC, we firstly examined if any individual sites exhibited differential DNA methylation in CR-HCC tissues compared with adjacent peritumor tissues. We showed substantial differences in DNA methylation between the CR-HCC tissues and the APTs and comprehensively identified a large subset of CpG sites/DMRs/genes that correlated with CR-HCC. A total of 38,554 DMSs were analyzed in CR-HCC tissues and APTs by using the human SureSelect XT arrays (Agilent Technologies, Santa Clara, CA, USA). Compared with the APTs, 21,853 DMSs in the CR-HCC tissues showed higher CpG methylation levels while 16,701 DMSs showed lower CpG methylation levels.
A DMR is a region in the genome that shows differences in methylation levels between samples. It is both more accurate and biologically relevant to use DMRs rather than CpG sites. The use of DMRs can also decrease potential artifacts due to random methylation in single CpG sites. The criterion used for DMR inclusion was a change in DNA methylation at a locus-specific level with an α≥10% methylation change with a p-value<0.05. Compared with the APTs, 1728 DMRs were identified in tumor tissues by using the Differential DNA Methylation Database (MethDiff) methods, of which 868 DMRs were hypermethylated and 860 DMRs were hypomethylated in the CR-HCC tissues. For characterization of the DMRs, we identified differences in the distribution of DMRs related to gene structures via the percentage of DMRs located within the promoter, exon, 5′ untranslated region (5′ UTR), and 3′untranslated region (3′ UTR). Similarly, we calculated the percentages of DMRs within a CpG island, CpG island shore, and CpG island shelf to determine differences in the distribution related to CpG islands. Our analyses showed prominent distribution of DMRs within promoter regions (1626), CpG islands (1821), CpG island shores (1792), CpG island shelves (982), and, more rarely, within 5′ UTRs (145) and 3′ UTRs (127)(Figure 1).
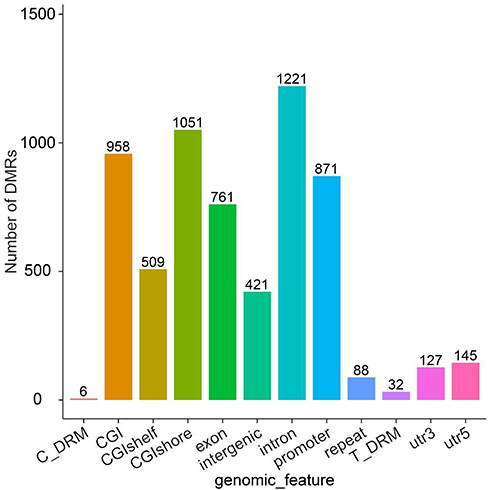 |
Figure 1 The distribution DMRs in the various genomic elements. The horizontal coordinate represents different functional areas, and the vertical coordinate represents the proportion of DMR in different functional areas. |
DMR Analysis During CR-HCC Tissues and APTs
After identifying the DMRs between the CR-HCC and APT groups, we used the physical position in the genome and the annotation information of the species to annotate the genes associated with the DMRs. A total of 2091 genes were identified from the DMR datasets, calculated by the MethDiff method. These1129 genes were related to DMRs with high methylation levels in the CR-HCC group and included 242 genes with methylation located in the gene promoter regions. A total of 962 genes were related to DMRs with low methylation levels in the CR-HCC group, of which 202 genes were differentially methylated the gene promoter. The data show that it is significantly more common for DMRs to be hypermethylated rather than hypomethylated in the CR-HCC tissues. Tables 2 and 3 show the top 30 hyper- or hypomethylated DMRs, ranked by statistical significance. After reviewing the GenBank database and the relevant literature, we selected the hypermethylated genes HOXB-AS3, HOXB6, HOXB3, USP18, and MAP3K6 and the hypomethylated genes TIRAP, TNNI2, SHC3, CTTN, and TFAP2A for further analysis of their expression by real-time qPCR. The results showed that the expression of HOXB-AS3, HOXB3, and MAP3K6 was downregulated in the CR-HCC tissues compared to the APTs, while the mRNA expression levels of TIRAP, SHC3, and CTTN were upregulated. The mRNA expression levels of HOXB6, USP18, TNNI2, and TFAP2A were not significantly different (Figure 2).
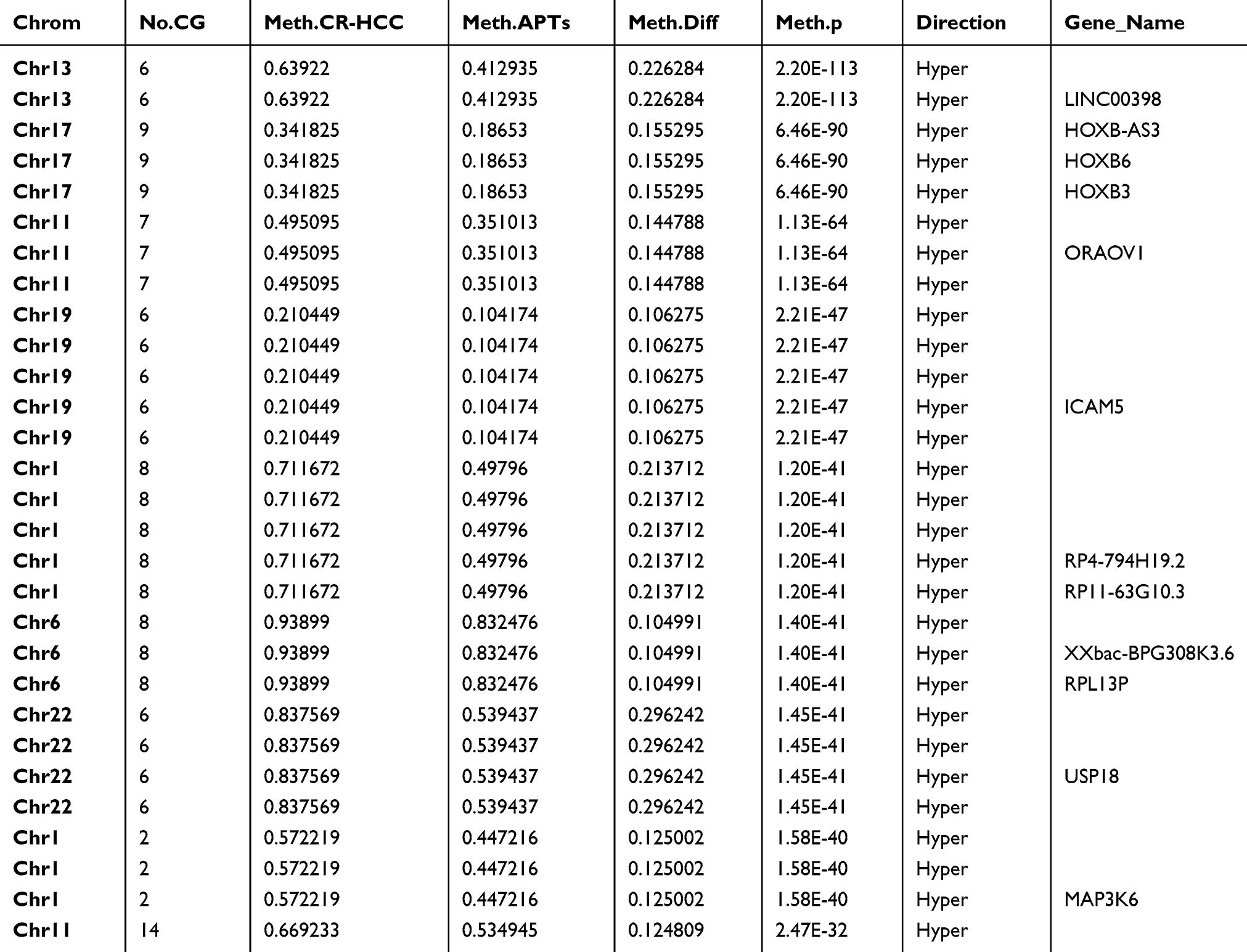 |
Table 2 Top30 Significant HyperMethylated DMRs in CR-HCC Tumor Tissues Compared with Adjacent Non-Tumor Tissues |
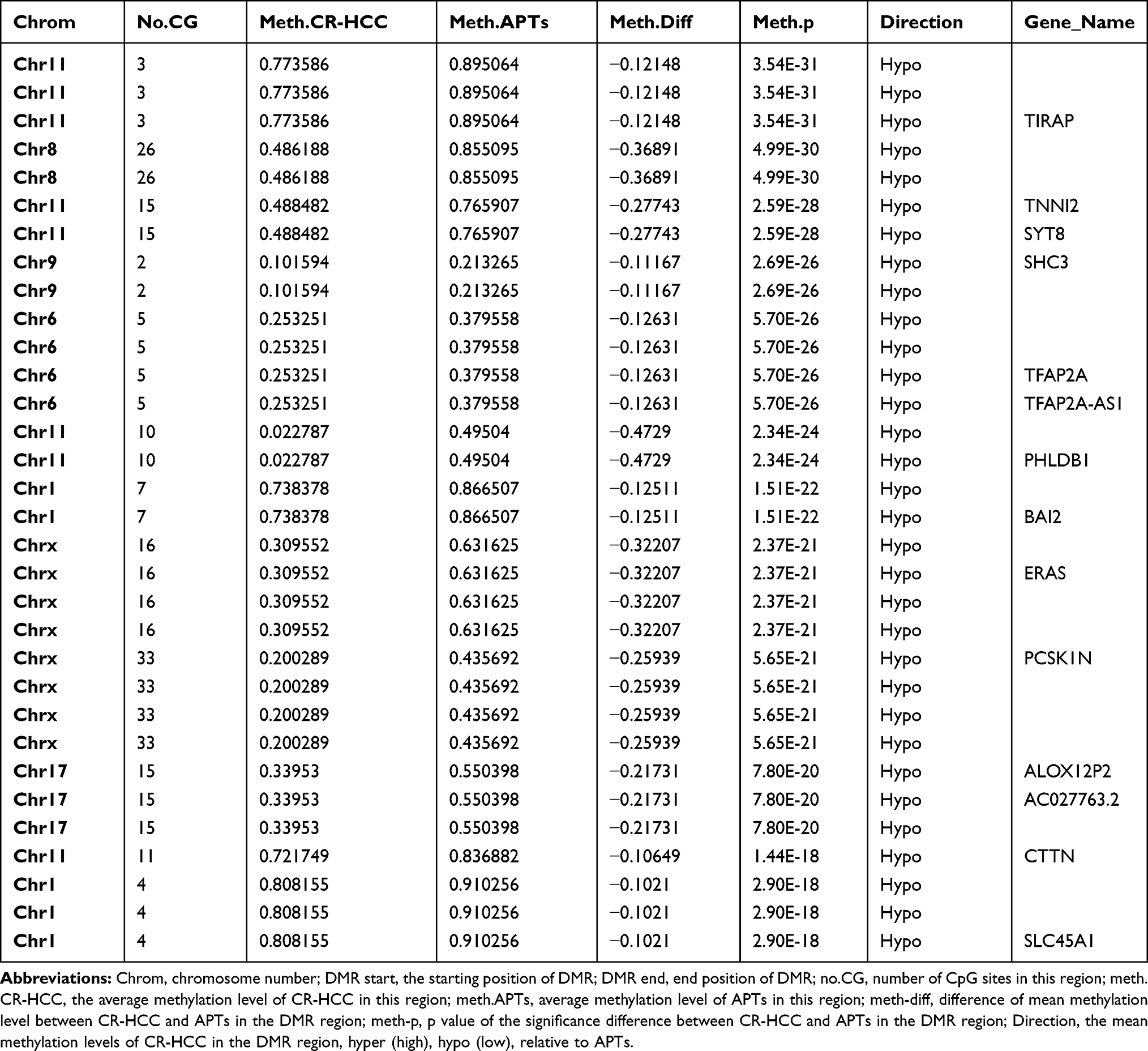 |
Table 3 Top 30 Significant hypoMethylated DMRs in CR-HCC Tumor Tissues Compared with Adjacent Non-Tumor Tissues |
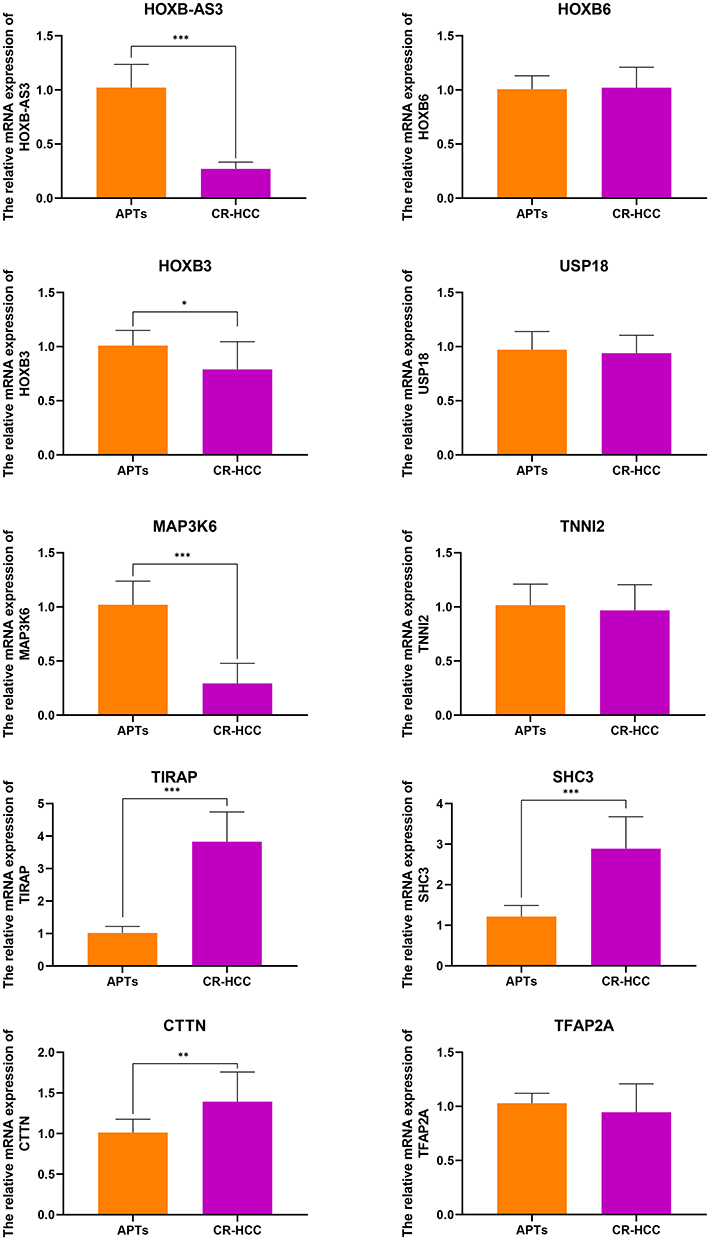 |
Figure 2 The relative mRNA expressions of HOXB-AS3, HOXB6, HOXB3, USP18, MAP3K6, TNNI2, TIRAP, SHC3, CTTN, and TFAP2A were detected in CR-HCC tissues compared with APTs (control). The mRNA levels of these genes were analyzed using real-time qPCR and normalized to the GAPDH. (*P<0.05, **P <0 0.01, ***P<0.001). |
Bioinformatic Analysis of Methylation Data
The hypergeometric distribution of these genes in relation to specific GO classifications was calculated for selected DMR genes. The number and functional classifications of the DMGs in relation to the GO categories, including the major enriched categories of molecular function, biological processes, and cellular components were displayed as a histogram. Further analysis of the enriched GO terms for both the hypermethylated and hypomethylated DMGs suggested that the molecular functions of “binding”, “protein binding”, and “cytoskeletal protein binding” were the main categories for the hypermethylated DMGs. The hypomethylated DMGs were mostly enriched in the molecular functions “binding”, “protein binding”, “calcium ion binding”, among others (Figure 3A and B). These molecular functions of the GO enrichment analysis in DMGs are shown in the Directed Acyclic Graph (DAG) graphical display (Figure 4A and B).
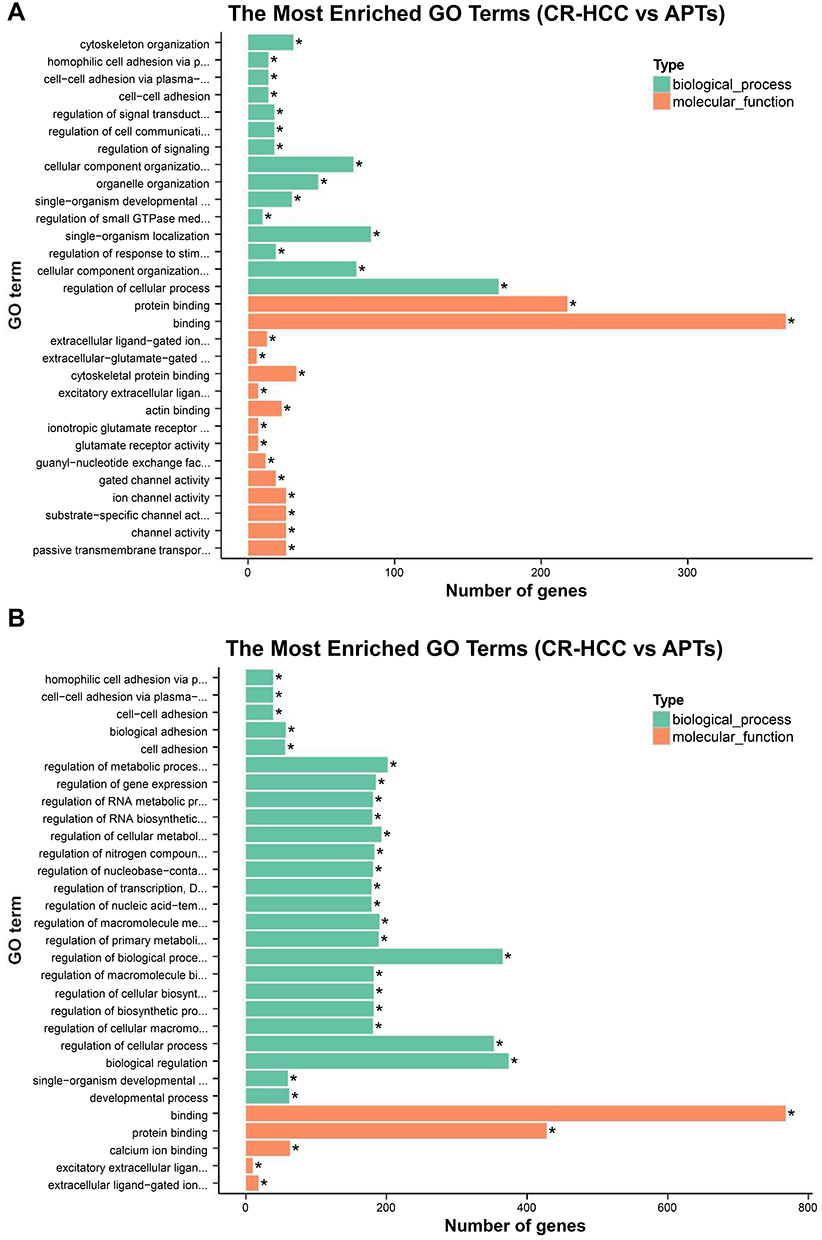 |
Figure 3 (A) The significant GO categories of hypermethylated genes in CR-HCC tissues compared with APTs; (P<0.05). (B) The significant GO categories of hypomethylated genes in CR-HCC tissues compared with APTs (P<0.05). “*” means the GO term is significantly enriched. |
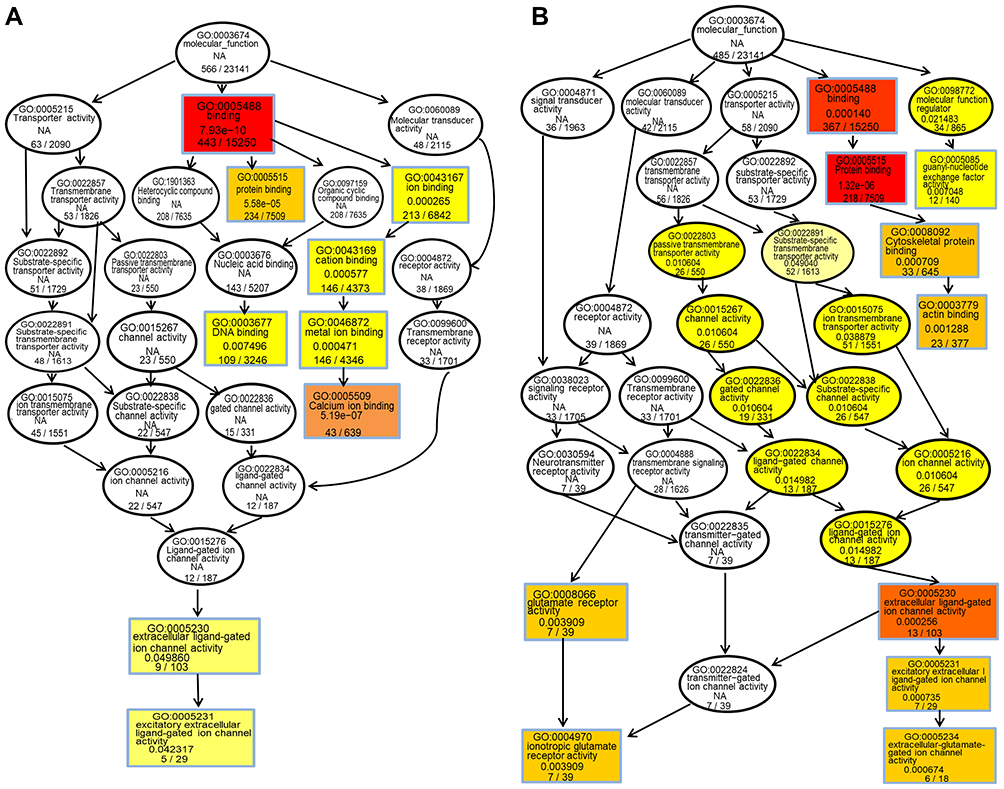 |
Figure 4 (A) Molecular function DAG of hypermethylated DMG. (B) Molecular function DAG of hypomethylated DMG. S each node represents A GO term, and the box represents the Top10 GO term with the degree of enrichment. The darker the color, the higher the degree of enrichment. The name of the TERM and the p-values of the enrichment analysis are displayed on each node. |
KEGG pathway analysis suggested that the significantly differentially hypermethylated genes were enriched in several pathways including the “Estrogen signaling pathway” (P=5.91E-03)(including the genes ADCY4, ADCY2, ADCY8, ADCY7, EGFR, AK72, FOS, GPER1, KCNJ6, KCNJ9, NRAS, PIK3R2, PLCB2, and MAP2K2), the “Notch signaling pathway” (P=9.04E-03) (including the genes RBPJL, CTBP1, DTX1, HES5, JAG2, NOTCH4, NUMBL, NCOR2, and MAML1), and ‘Signaling pathways regulating pluripotency of stem cells’ (P=1.26E-02)(including the genes DLX5, APC2, AKT2, FGFR3, JAK2, JAK3, ZFHX3, NRAS, MAPK11, REST, MAP2K2, IK3R2, BMPR11A, WNT10A, FZD1, WNT5B, and HAND1), the “Rap1 signaling pathway” (P=1.41E-02) (including the genesADCY4, EPHA2, RASGRP2, EFNA5, RAPGEF3, ADCY8, RGS14, EGFR, ADCY2, ADCY7, AKT2, FGFR3, PFN3, NRNAS, RALGDS, FIK3R2, RALB, RALA, MAP2K2, MAPK11, PLCB2, and CDH1) while differentially hypomethylated genes were enriched in several pathways such as “Proteoglycans in cancer” (P=5.04E-03) (including the genes FLNC, AKT1, CTTN, ANK1, GPC1, GFR, IGF2, HOXD10, MAPK11, MAP2K2, PIK3CD, PLCE1, CCND1, TGFB1, FZD3, WNT5A, ACTG1, WNT7B, KDR, CAMK2B, WNT5B, WNT10A, and HPSE2) and “Calcium signaling pathway” (P=7.79E-03) (including the genes ADORA2A, ADORA2B, CRIN2A, CRIN2D, GNAS, GNAL, GRIN2C, GRIN1, GRM5, HTR5A, NTSR1, PPP3CC, PLCE1, CALML5, P2RX1, RYR1, CACNA1A, CAMK2B, EGFR, CACNA1H, and ORAI3). Table 4(A–B) shows all the significant KEGG pathways (p<0.05). Both hypermethylated and hypomethylated genes with hypermethylation and hypomethylation were mostly related to specific pathways including the “Rap1 signaling pathway” and ‘Signaling pathways regulating pluripotency of stem cells’.
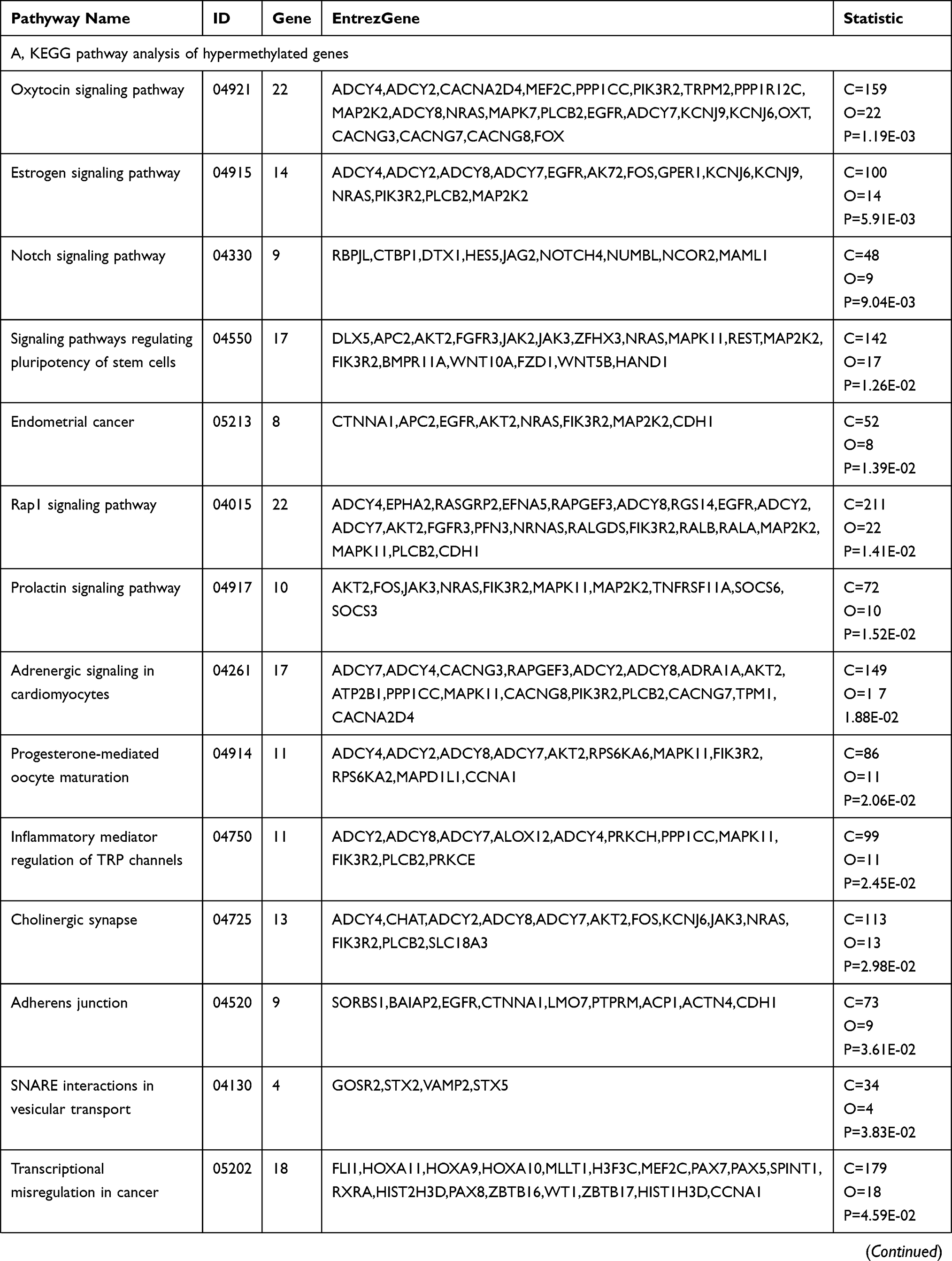 | 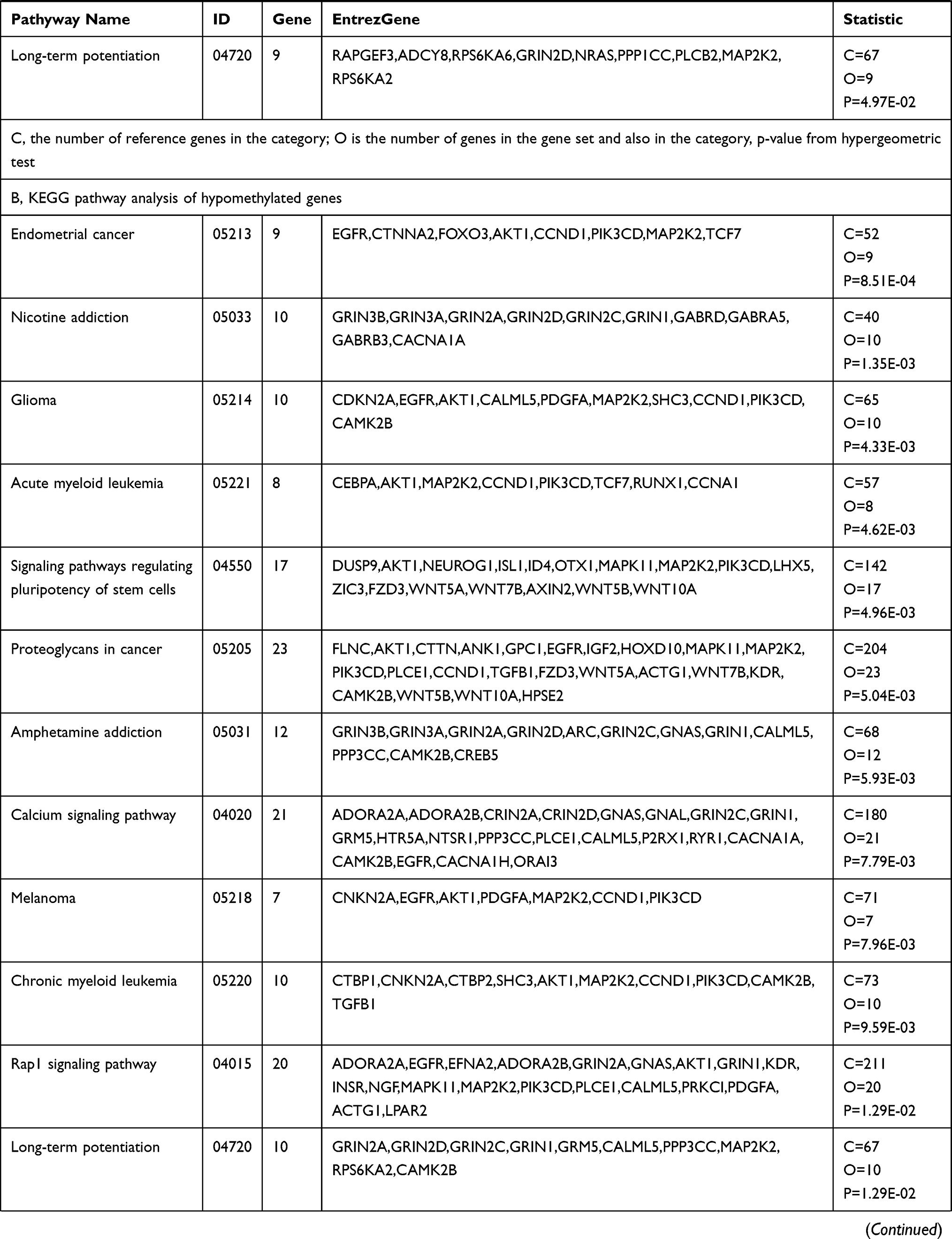 | 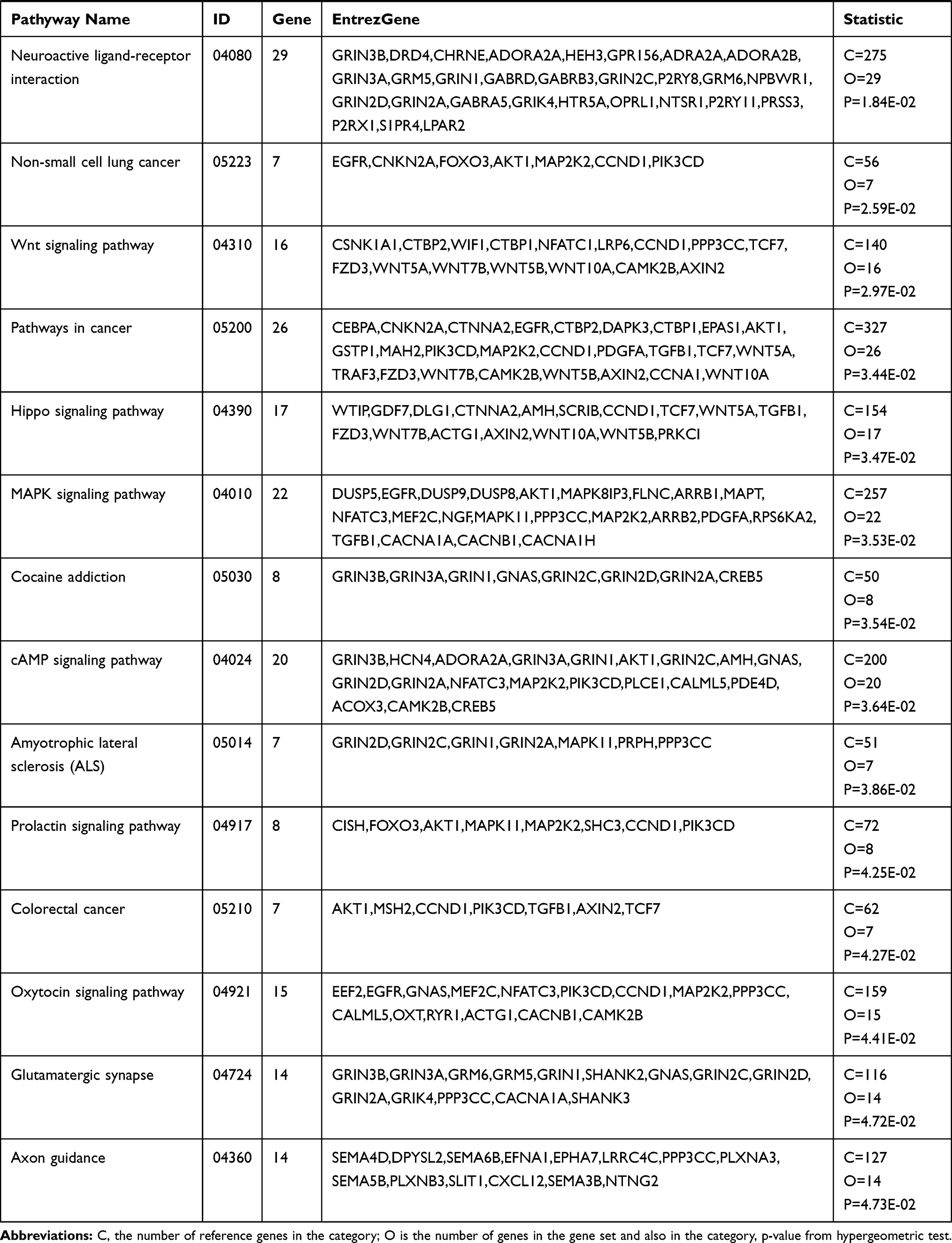 |
Table 4 KEGG Pathway Analysis of Hypermethylated and Hypomethylated Genes |
Discussion
DNA methylation that can regulate gene expression is one of the most important epigenetic modifications in humans. Previous studies have discovered aberrant DNA methylation in HCC tissues rather than APTs.19 The main aims of this research were to study the associations between methylation and CR-HCC and to find new potential biomarkers to diagnose CR-HCC and differs from previous studies in the design and enrolment criteria in the selection of patients for the study.
The two most common forms of aberrant CpG methylation in cancer, namely promoter hypo- or hyper-methylation, may lead to the inappropriate activation of oncogenes or the silencing of tumor suppressor genes (TSGs) while global hypo-methylation causing chromosomal instability, have been widely studied in HCC.20,21 Several recent studies have suggested that aberrant CpG methylation of several tumor suppressor genes such as Ras-association domain family 1 isoform A (RASSF1A), Runt-related transcription factor 3 (RUNX3), E-cadherin, and P16, among others, and cause tumor suppressor gene inactivation in HBV- and HCV-related HCC, implying that epigenetic modification plays an important role in hepatocarcinogenesis.22–25 In the present study, we detected 38,554 differentially methylated CpG sites across the whole genome of CR-HCC and APTs, including 16,701 hypermethylated CpG sites (42.9%) and 21,853 hypomethylated CpG sites (56%). In addition, we selected significant DMRs using stringent criteria. We identified 1728 DMRs, revealing 868 significantly hypermethylated DMRs and 860 significantly hypomethylated DMRs in the CR-HCC group compared with the APT group using MethDiff methods. Our analyses also identified prominent DMR distributions within CpG islands (958), CpG island shores (1051), CpG island shelves (509), and, more rarely, within the 5′ UTR (127) and 3′ UTR (145). Our data also showed that the DMRs were predominantly located within promoter regions (871). Since the promoters in CpG islands generally regulate gene transcription through DNA methylation, DMRs may regulate CR-HCC pathogenesis mainly via the transcriptional inactivation of tumor suppressor genes. It is known that aberrant DNA methylation occurs in CR-HCC tissues, and our results showed that hypermethylation of CpG sites occurs more frequently than hypomethylation. Our results are consistent with those of previous HCC genome-wide methylation studies.26–29
A total of 1129 genes were found to be related to DMRs with high methylation levels in the CR-HCC group, among which methylation occurred in 242 genes in the gene promoter region. A further 962 genes were found to be related to DMRs with low methylation levels in the CR-HCC group, among which methylation occurred in 202 genes in the gene promoter region. Moreover, this study identified the top 30 significantly hyper-and hypo-methylated DMRs and DMR-related genes in CR-HCC tissues compared with APTs, including TIRAP, TFAP2A, CTTN, USP18, ICAM5, and MAP3K6. The real-time qPCR results showed that the expression of HOXB-AS3, HOXB3, and MAP3K6 was downregulated in the CR-HCC tissues compared to the APTs. However, the mRNA expression of TIRAP, SHC3, and CTTN was upregulated in the CR-HCC tissues. The results confirm that the methylation status within the gene structure affects the gene expression in CR-HCC tissues. An increasing number of studies have demonstrated that TLR signaling pathways are modulated by TIR domain-containing adaptors, such as MyD88, TIRAP, and TRIF. TIRAP is specifically involved in the MyD88-dependent pathway via TLR2 and TLR4.30,31 In NSCLC cells, knockdown of TIRAP expression by siRNA suppressed proliferation.32 Cortactin (CTTN) has been identified as a biomarker of cancer metastasis due to its overexpression in various cancers, such as colorectal cancer, head and neck squamous cell carcinoma, among others. Results have suggested that CTTN inhibits the degradation of EGF and promotes the proliferation of CRC cells.33 A previous study has demonstrated that CTTN is a novel HBx-interacting protein and that the HBx-CTTN interaction inhibits cell cycle arrest to enhance cellular proliferation and maintain cellular migration.34 Ubiquitin-specific peptidase 18, also known as UBP43, is an enzyme belonging to the specific ubiquitin protease family; altering USP18 levels affects the stability of the cyclin D1 protein in lung cancer35 with higher levels of USP18 expression influencing KRAS expression in K-Ras-driven murine lung cancers in vivo.36 Of all the identified genes, homeobox A9 (HOXA9) was the most frequently methylated in plasma samples from HCC patients and in HCC tissues, suggesting its potential as a useful HCC biomarker.37 The discovery of specifically hypermethylated genes leading to gene silencing or hypomethylated genes leading to increased transcription is helpful in identifying new factors that are vital for CR-HCC carcinogenesis and progression. Our study of DMRs provides an important association between the aberrant DNA methylation within these sites, which is a vital epigenetic mechanism, and CR-HCC carcinogenesis. These results suggest that the development of CR-HCC is regulated by different genes together with different signaling pathways and their crosstalk and that aberrant DNA methylation is an important event in the pathogenesis of CR-HCC.
The pathways associated with DNA methylation and CR-HCC carcinogenesis were screened by bioinformatic analysis. GO analysis showed that the hypermethylated DMGs were mostly associated with “binding”- and “adhesion” -related molecular functions, including cell adhesion, binding, cell-cell adhesion, plasma-membrane adhesion molecules. “Binding” is another enrichment category of the hypermethylated genes, and it contains “Ion binding” (GO:0043167 P=1.27E-09), “cell adhesion” (GO:0007155, P=1.75E-09), and “DNA binding” (GO:0003677, P= 1.43E-09). Other research has indicated that a suppressive oncogene, namely CDH1, encodes the epithelial cell adhesion molecule E-cadherin which is involved in cell-cell adhesion and cell polarity. The CHD1 gene is known to be an invasion and tumor suppressor gene and its decreased expression due to hypermethylation could promote tumor cell invasion and metastasis.38–40 Jiang L demonstrated that calcium-binding protein 39 (CAB39) promoted the carcinogenesis and development of HCC when CAB39 activated the ERK signaling pathway, suggesting that CAB39 may be useful as a biomarker for the prediction of prognosis as well as the potential of being a novel therapeutic target in clinical practice.41 The hypomethylated DMGs were enriched in the GO categories including “protein binding”, “extracellular ligand-gated ion channel activity”, “regulation of signal transduction”, “cytoskeletal protein binding”. These data show that genes enriched in a number of GO categories are related to the carcinogenesis of CR-HCC and may be useful as specific biomarkers for the diagnosis of CR-HCC.
Furthermore, the results of the KEGG analysis suggested that the hypermethylation-related genes were mainly enriched in the “Rap1 signaling pathway”, “signaling pathways regulating pluripotency of stem cells”, and “Estrogen signaling pathway” in CR-HCC tissues. The hypomethylation-related genes were mainly involved in “colorectal cancer”, “calcium signaling pathway”, “nicotine addiction”, among others. Rap1, a small GTPase, controls various processes, including cell polarity, cell-cell junction formation, and cell adhesion.42 As undifferentiated cells, pluripotent stem cells (PSCs), including embryonic stem cells (ES) and induced pluripotent stem cells (iPS), have the capacity for indefinite self-renewal and are able to generate all cell types of the three germinal layers.43 Human hepatic stem/progenitor cells (hHPCs) from adult liver that resides in the canals of Hering may have a great capacity to proliferate and the ability to differentiate into mature hepatocytes.44 Activation and dysregulated proliferation of hHPCs commonly contribute to the carcinogenesis of HCC in adverse environments. Furthermore, an increasing number of studies have demonstrated that HPCs show strong associations with HCC,45,46 which may provide new directions to study the relationship between methylation and CR-HCC. Other identified pathways such as “Cell cycle” and “Chemokine signaling pathway” are also considered to be important for the development and progression of malignant carcinoma, and the genes involved, together with their methylation patterns, may provide potential novel biomarkers for CR-HCC.
Conclusion
This preliminary work may pave the way for future studies to identify the vital functions of epigenetic mechanisms on CR-HCC carcinogenesis. The present study has several potential limitations, including the relatively small sample size. The sample size was restricted mainly due to the high cost of Methyl-seq, which precludes its application to a large-scale study. Nevertheless, our findings indicate the important role of epigenetic modification on the carcinogenesis of CR-HCC and pave the way for future analyses of a larger sample using a series of biochemical approaches.
Acknowledgments
This research was supported by the 2016 Natural Science Foundation of Anhui Province (Project Number: 1608085QH182)
Disclosure
The authors report no conflicts of interest in this work.
References
1. Toh TB, Lim JJ, Chow EK. Epigenetics of hepatocellular carcinoma. Clin Transl Med. 2019;8(1):13.
2. Zhang W, Chen J, Liu L, Wang L, Liu J, Su D. Prognostic value of preoperative computed tomography in HBV-related hepatocellular carcinoma patients after curative resection. Onco Targets Ther. 2019;12:3791–3804. doi:10.2147/OTT.S199136
3. Kanwal F, Kramer JR, Asch SM, Cao Y, Li L, El-Serag HB. Long-term risk of hepatocellular carcinoma in HCV patients treated with direct acting antiviral agents. Hepatology. 2020;71(1):44–55. doi:10.1002/hep.30823
4. Tian Y, Mok MT, Yang P, Cheng AS. Epigenetic activation of wnt/beta-catenin signaling in NAFLD-Associated hepatocarcinogenesis. Cancers (Basel). 2016;8(8). doi:10.3390/cancers8080076
5. Omichi K, Shindoh J, Yamamoto S, et al. Postoperative outcomes for patients with non-B non-C hepatocellular carcinoma: a subgroup analysis of patients with a history of hepatitis B infection. Ann Surg Oncol. 2015;22(Suppl 3):S1034–S1040. doi:10.1245/s10434-015-4845-0
6. Utsunomiya T, Shimada M, Kudo M, et al. A comparison of the surgical outcomes among patients with HBV-positive, HCV-positive, and non-B non-C hepatocellular carcinoma: a nationwide study of 11,950 patients. Ann Surg. 2015;261(3):513–520. doi:10.1097/SLA.0000000000000821
7. Jun TW, Yeh ML, Yang JD, et al. More advanced disease and worse survival in cryptogenic compared to viral hepatocellular carcinoma. Liver Int. 2018;38(5):895–902. doi:10.1111/liv.13613
8. Lu ZP, Xiao ZL, Yang Z, et al. Hepatitis B virus X protein promotes human hepatoma cell growth via upregulation of transcription factor AP2alpha and sphingosine kinase 1. Acta Pharmacol Sin. 2015;36(10):1228–1236. doi:10.1038/aps.2015.38
9. Kelly AD, Issa JJ. The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr Opin Genet Dev. 2017;42:68–77. doi:10.1016/j.gde.2017.03.015
10. Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017;16(1):29.
11. Weisenberger DJ, Liang G, Lenz HJ. DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies. Oncogene. 2018;37(5):566–577. doi:10.1038/onc.2017.374
12. Puccini A, Loupakis F, Stintzing S, et al. Impact of polymorphisms within genes involved in regulating DNA methylation in patients with metastatic colorectal cancer enrolled in three independent, randomised, open-label clinical trials: a meta-analysis from TRIBE, MAVERICC and FIRE-3. Eur J Cancer. 2019;111:138–147. doi:10.1016/j.ejca.2019.01.105
13. Ma Z, Liu Y, Hao Z, Hua X, Li W. DNA hypermethylation of aurora kinase A in hepatitis C virus positive hepatocellular carcinoma. Mol Med Rep. 2019;20(3):2519–2532.
14. Sang L, Wang XM, Xu DY, Zhao WJ. Bioinformatics analysis of aberrantly methylated-differentially expressed genes and pathways in hepatocellular carcinoma. World J Gastroenterol. 2018;24(24):2605–2616. doi:10.3748/wjg.v24.i24.2605
15. Dou CY, Fan YC, Cao CJ, Yang Y, Wang K. Sera DNA methylation of CDH1, DNMT3b and ESR1 promoters as biomarker for the early diagnosis of hepatitis B virus-related hepatocellular carcinoma. Dig Dis Sci. 2016;61(4):1130–1138. doi:10.1007/s10620-015-3975-3
16. Song MA, Kwee SA, Tiirikainen M, et al. Comparison of genome-scale DNA methylation profiles in hepatocellular carcinoma by viral status. Epigenetics-Us. 2016;11(6):464–474. doi:10.1080/15592294.2016.1151586
17. Li CW, Chiu YK, Chen BS. Investigating pathogenic and hepatocarcinogenic mechanisms from normal liver to HCC by constructing genetic and epigenetic networks via big genetic and epigenetic data mining and genome-wide NGS data identification. Dis Markers. 2018;2018:8635329. doi:10.1155/2018/8635329
18. Borowa-Mazgaj B, de Conti A, Tryndyak V, et al. Gene expression and DNA methylation alterations in the glycine N-methyltransferase gene in diet-induced nonalcoholic fatty liver disease-associated carcinogenesis. Toxicol Sci. 2019;170(2):273–282. doi:10.1093/toxsci/kfz110
19. Udali S, Guarini P, Ruzzenente A, et al. DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin Epigenetics. 2015;7:43.
20. Plass C. Cancer epigenomics. Hum Mol Genet. 2002;11(20):2479–2488. doi:10.1093/hmg/11.20.2479
21. Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1741–1748. doi:10.3748/wjg.14.1741
22. El-Bendary M, Nour D, Arafa M, Neamatallah M. Methylation of tumour suppressor genes RUNX3, RASSF1A and E-cadherin in HCV-related liver cirrhosis and hepatocellular carcinoma. Br J Biomed Sci. 2020;77(1):35–40. doi:10.1080/09674845.2019.1694123
23. Zhang C, Li J, Huang T, et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget. 2016;7(49):81255–81267. doi:10.18632/oncotarget.13221
24. Dong X, He H, Zhang W, Yu D, Wang X, Chen Y. Combination of serum RASSF1A methylation and AFP is a promising non-invasive biomarker for HCC patient with chronic HBV infection. Diagn Pathol. 2015;10:133. doi:10.1186/s13000-015-0317-x
25. Tang H, Yao X, Li C, Liu P, Ma B. Methylation of DAPK, P16 and E-cadherin gene in liver cancer tissues and protein expression level. Int J Clin Exp Pathol. 2016;9(3):3787–3792.
26. Shen J, Wang S, Zhang YJ, et al. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using infinium HumanMethylation 450 BeadChips. Epigenetics-Us. 2013;8(1):34–43. doi:10.4161/epi.23062
27. Ammerpohl O, Pratschke J, Schafmayer C, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2012;130(6):1319–1328. doi:10.1002/ijc.26136
28. Kohles N, Nagel D, Jungst D, Durner J, Stieber P, Holdenrieder S. Prognostic relevance of oncological serum biomarkers in liver cancer patients undergoing transarterial chemoembolization therapy. Tumour Biol. 2012;33(1):33–40. doi:10.1007/s13277-011-0237-7
29. Zheng Y, Huang Q, Ding Z, et al. Genome-wide DNA methylation analysis identifies candidate epigenetic markers and drivers of hepatocellular carcinoma. Brief Bioinform. 2018;19(1):101–108.
30. Jakka P, Bhargavi B, Namani S, Murugan S, Splitter G, Radhakrishnan G. Cytoplasmic linker protein CLIP170 negatively regulates TLR4 signaling by targeting the TLR adaptor protein TIRAP. J Immunol. 2018;200(2):704–714. doi:10.4049/jimmunol.1601559
31. Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16(1):3–9. doi:10.1016/j.smim.2003.10.003
32. Hao S, Li S, Wang J, et al. Phycocyanin exerts anti-proliferative effects through down-regulating TIRAP/NF-kappaB activity in human non-small cell lung cancer cells. Cells-Basel. 2019;8:6.
33. Zhang X, Liu K, Zhang T, et al. Cortactin promotes colorectal cancer cell proliferation by activating the EGFR-MAPK pathway. Oncotarget. 2017;8(1):1541–1554. doi:10.18632/oncotarget.13652
34. Li Y, Fu Y, Hu X, et al. The HBx-CTTN interaction promotes cell proliferation and migration of hepatocellular carcinoma via CREB1. Cell Death Dis. 2019;10(6):405.
35. Guo Y, Chinyengetere F, Dolinko AV, et al. Evidence for the ubiquitin protease UBP43 as an antineoplastic target. Mol Cancer Ther. 2012;11(9):1968–1977. doi:10.1158/1535-7163.MCT-12-0248
36. Mustachio LM, Lu Y, Tafe LJ, et al. Deubiquitinase USP18 loss mislocalizes and destabilizes KRAS in lung cancer. Mol Cancer Res. 2017;15(7):905–914. doi:10.1158/1541-7786.MCR-16-0369
37. Shih YL, Kuo CC, Yan MD, Lin YW, Hsieh CB, Hsieh TY. Quantitative methylation analysis reveals distinct association between PAX6 methylation and clinical characteristics with different viral infections in hepatocellular carcinoma. Clin Epigenetics. 2016;8:41. doi:10.1186/s13148-016-0208-3
38. Gao LM, Xu SF, Zheng Y, et al. Long non-coding RNA H19 is responsible for the progression of lung adenocarcinoma by mediating methylation-dependent repression of CDH1 promoter. J Cell Mol Med. 2019;23(9):6411–6428. doi:10.1111/jcmm.14533
39. Shawky SA, El-Borai MH, Khaled HM, et al. The prognostic impact of hypermethylation for a panel of tumor suppressor genes and cell of origin subtype on diffuse large B-cell lymphoma. Mol Biol Rep. 2019;46(4):4063–4076. doi:10.1007/s11033-019-04856-x
40. Li Z, Guo Z. Comparison of CDH1 gene hypermethylation status in blood and serum among gastric cancer patients. Pathol Oncol Res. 2020;26(2):1057–1062. doi:10.1007/s12253-019-00658-5
41. Jiang L, Yan Q, Fang S, et al. Calcium-binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating extracellular signal-regulated kinase signaling pathway. Hepatology. 2017;66(5):1529–1545. doi:10.1002/hep.29312
42. Moon MY, Kim HJ, Kim MJ, et al. Rap1 regulates hepatic stellate cell migration through the modulation of RhoA activity in response to TGFbeta1. Int J Mol Med. 2019;44(2):491–502.
43. Boheler KR, Joodi RN, Qiao H, et al. Embryonic stem cell-derived cardiomyocyte heterogeneity and the isolation of immature and committed cells for cardiac remodeling and regeneration. Stem Cells Int. 2011;2011:214203. doi:10.4061/2011/214203
44. Tsuchiya A, Kamimura H, Takamura M, et al. Clinicopathological analysis of CD133 and NCAM human hepatic stem/progenitor cells in damaged livers and hepatocellular carcinomas. Hepatol Res. 2009;39(11):1080–1090. doi:10.1111/j.1872-034X.2009.00559.x
45. Hou XJ, Ye F, Li XY, et al. Immune response involved in liver damage and the activation of hepatic progenitor cells during liver tumorigenesis. Cell Immunol. 2018;326:52–59. doi:10.1016/j.cellimm.2017.08.004
46. Li CH, Wang YJ, Dong W, et al. Hepatic oval cell lines generate hepatocellular carcinoma following transfection with HBx gene and treatment with aflatoxin B1 in vivo. Cancer Lett. 2011;311(1):1–10. doi:10.1016/j.canlet.2011.05.035
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.