Back to Journals » International Journal of General Medicine » Volume 15
Experience and Perspectives in the US on the Evolving Treatment Landscape in Spinal Muscular Atrophy
Authors Ramos-Platt L, Elman L, Shieh PB
Received 1 April 2022
Accepted for publication 10 August 2022
Published 17 September 2022 Volume 2022:15 Pages 7341—7353
DOI https://doi.org/10.2147/IJGM.S369021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Leigh Ramos-Platt,1 Lauren Elman,2 Perry B Shieh3
1Department of Pediatrics, Keck School of Medicine, University of Southern California and Children’s Hospital of Los Angeles, Los Angeles, CA, USA; 2Department of Neurology, University of Pennsylvania, Philadelphia, PA, USA; 3Department of Neurology and Pediatrics, University of California Los Angeles, Los Angeles, CA, USA
Correspondence: Perry B Shieh, Department of Neurology and Pediatrics, University of California Los Angeles, Los Angeles, CA, USA, Email [email protected]
Abstract: Spinal muscular atrophy (SMA) is a rare, progressive neuromuscular disorder that, until recently, was the most common inherited cause of infant mortality. Since 2016, three disease-modifying therapies have emerged, nusinersen, onasemnogene abeparvovec-xioi, and risdiplam, leading to a transformation in the SMA treatment landscape, changes in disease trajectories, and a profound impact on clinical care. This environment poses a challenge to making informed treatment decisions, including initial treatment choice, treatment changes, and potential use of combination therapies as new data emerge. To better understand factors that influence physician-patient decision-making, a roundtable discussion was convened by Biogen (sponsor) with a panel of four US SMA experts. This report shares the panel’s opinions and clinical experiences, with the goals of helping clinicians and people with SMA and their families to better understand the factors influencing real-world treatment decisions and stimulating a broader discussion in the SMA community. The panelists highlighted that patients are often heavily involved in treatment decisions, and physicians must be aware of current data to guide patients in making the best decisions. Thus, in the absence of data from head-to-head treatment comparisons, physicians’ roles include reviewing treatment options and describing what is known of the benefits, challenges, and potential side effects of each therapy with patients and families. For infants and young children, the panelists expressed a sense of urgency for early intervention to minimize motor function loss, whereas the goal for adults is long-term disease stabilization. In the panelists’ experience, factors that influence patients’ decisions to change to an alternative therapy include convenience, administration route, novelty of therapy, and hope for improved function, while reasons for returning to a previous therapy include a perception of decreased efficacy and side effects. Ongoing clinical trials and analyses of real-world experiences should further inform treatment decisions and optimize patient outcomes.
Keywords: nusinersen, onasemnogene abeparvovec-xioi, risdiplam, spinal muscular atrophy
Graphical Abstract:

Plain Language Summary
People with spinal muscular atrophy (SMA), a rare disease caused by a change in the survival motor neuron 1 gene, do not make enough SMN protein to maintain motor neurons, nerve cells that control muscle function. This results in muscle weakness that worsens over time.
Nusinersen, onasemnogene abeparvovec-xioi, and risdiplam are drugs that increase SMN protein. Nusinersen is injected into a space in the lower back (lumbar region) below the end of the spinal cord every 4 months. Onasemnogene abeparvovec-xioi, a gene replacement therapy, is injected once into a vein. Risdiplam is taken by mouth daily. Biogen, the manufacturer of nusinersen, organized a meeting with four SMA doctors to discuss their experiences with SMA treatments and how patients choose a drug.
In the experts’ opinions:
- People with SMA and their families want to be involved in treatment decisions (shared decision-making). To help them, doctors should provide up-to-date information on how well the drugs may work and any possible side effects.
- Infants and children with SMA should begin treatment quickly after diagnosis to minimize loss of motor function and the ability to move.
- For adults with SMA, the primary treatment goal is preventing motor function decline.
- Reasons people with SMA may change to a different drug or return to a previous drug:
- hope for improved function
- convenience
- current drug is not working as expected (decreased efficacy)
- desire to try something new
- side effects.
- Most people changing therapy have been able to keep insurance coverage for their new treatment.
Introduction
Since December 2016, the treatment landscape in spinal muscular atrophy (SMA) has gone through a transformative evolution, with the advancement and approval of multiple disease-modifying treatment options.1–4 Clinicians, patients, and caregivers now face more complex considerations when evaluating opportunities to individualize treatment decisions that address the unique interests and needs of each patient.5–7
SMA is a rare, progressive neuromuscular disorder characterized by muscle weakness and atrophy resulting from irreversible degeneration of the anterior horn motor neurons in the spinal cord and the brain stem nuclei.8,9 SMA occurs in approximately one per 10,000 live births.9 Until this decade, SMA was the most common inherited cause of infant mortality.10 In about 95–98% of patients with SMA, the disease is caused by a deletion in both alleles of the survival motor neuron 1 gene (SMN1) on chromosome 5q; in the remainder, small mutations in SMN1 have been identified.9 In either case, the deletions/mutations lead to reduced SMN protein levels, which results in the degeneration of motor neurons.8,9 Individuals with SMA have one or more copies of SMN2, which is paralogous and nearly identical to SMN1.9 The SMN2 gene differs from SMN1 by 20 nucleotides,11 most significantly, a cytosine to thymine transition in exon 7 of SMN2, which results in aberrant pre-mRNA splicing.12,13 Consequently, full-length SMN2 mRNA is produced in much lower levels, which results in the production of much smaller amounts of fully functional SMN protein than is seen for SMN1 transcripts.12,13
Phenotypes of SMA vary widely, but traditionally have been classified into four primary categories (types I–IV) based on the maximum motor milestone attained.9 The severity of disease has been found to correlate inversely with the number of copies of SMN2.9,14 About 60% of patients have the most severe type, SMA type I, and cannot achieve the motor milestone of sitting without support.9 In addition, they exhibit poor head control, hypotonia, and difficulties with sucking and swallowing, and they never roll over independently.9,15 Before treatments became available, most patients with SMA type I developed severe respiratory failure and did not survive beyond 2 years of age without exceptional measures.9 Patients with SMA type II have a less severe form of the disorder and are able to sit in early disease but cannot stand alone or walk independently.16 Their symptoms generally appear at 6–18 months of life, and up to 70% are alive at age 25 years.9 Patients with SMA type III typically exhibit symptoms at 18 months of age or later and are able to stand and walk independently at some point.16 Type IV is a rare, milder adult-onset form of SMA.9 Patients with type III and IV have normal lifespans.9
In 2016, nusinersen was approved by the US Food and Drug Administration (FDA) as the first disease-modifying therapy for SMA.1,3 Prior to the approval of nusinersen, treatment for SMA was limited to respiratory, nutritional, and orthopedic supportive care to manage comorbidities and improve quality of life, as well as palliative, end-of-life care.1,6,17,18 Nusinersen is a synthetic antisense oligonucleotide designed to bind an intronic sequence downstream of exon 7 to modulate the splicing of SMN2 pre-mRNA.19 This results in more full-length SMN2 transcripts, thereby increasing the production of functional SMN protein. It has been FDA approved for both pediatric and adult patients with SMA and is administered intrathecally at 12 mg every 4 months as maintenance dosing after four loading doses.3 The approved 12-mg dose for nusinersen is the same for all age and weight groups, and the dose was based on results of the ENDEAR and CHERISH studies, which were conducted in children with infantile-onset and later-onset SMA, respectively.3,20,21 Over the last 5 years, two additional therapies have been approved by the FDA for the treatment of patients with SMA. Onasemnogene abeparvovec-xioi was approved in 2019 and is a gene replacement therapy consisting of a replication-deficient adeno-associated virus type 9 (AAV9) capsid containing a functional copy of SMN1.1,22 Onasemnogene abeparvovec-xioi is FDA approved in the United States for pediatric patients who are less than 2 years of age and is administered as a one-time intravenous infusion (dose: 1.1×1014 vector genomes/kg of body weight).4 Risdiplam was approved in 2020 as an orally administered, small-molecule, SMN2 splicing modifier that results in exon 7 inclusion in SMN2 pre-mRNA transcription, thereby increasing SMN protein production.2,23,24 Risdiplam is FDA approved for pediatric and adult patients at daily doses of 0.15 mg/kg for those aged less than 2 months, 0.2 mg/kg for those who are aged 2 months to less than 2 years, 0.25 mg/kg for patients aged 2 years and older and weighing less than 20 kg, and a maximum daily dose of 5 mg for those aged 2 years and older and weighing 20 kg or more.2 An overview of the three therapies is shown in Table 1.
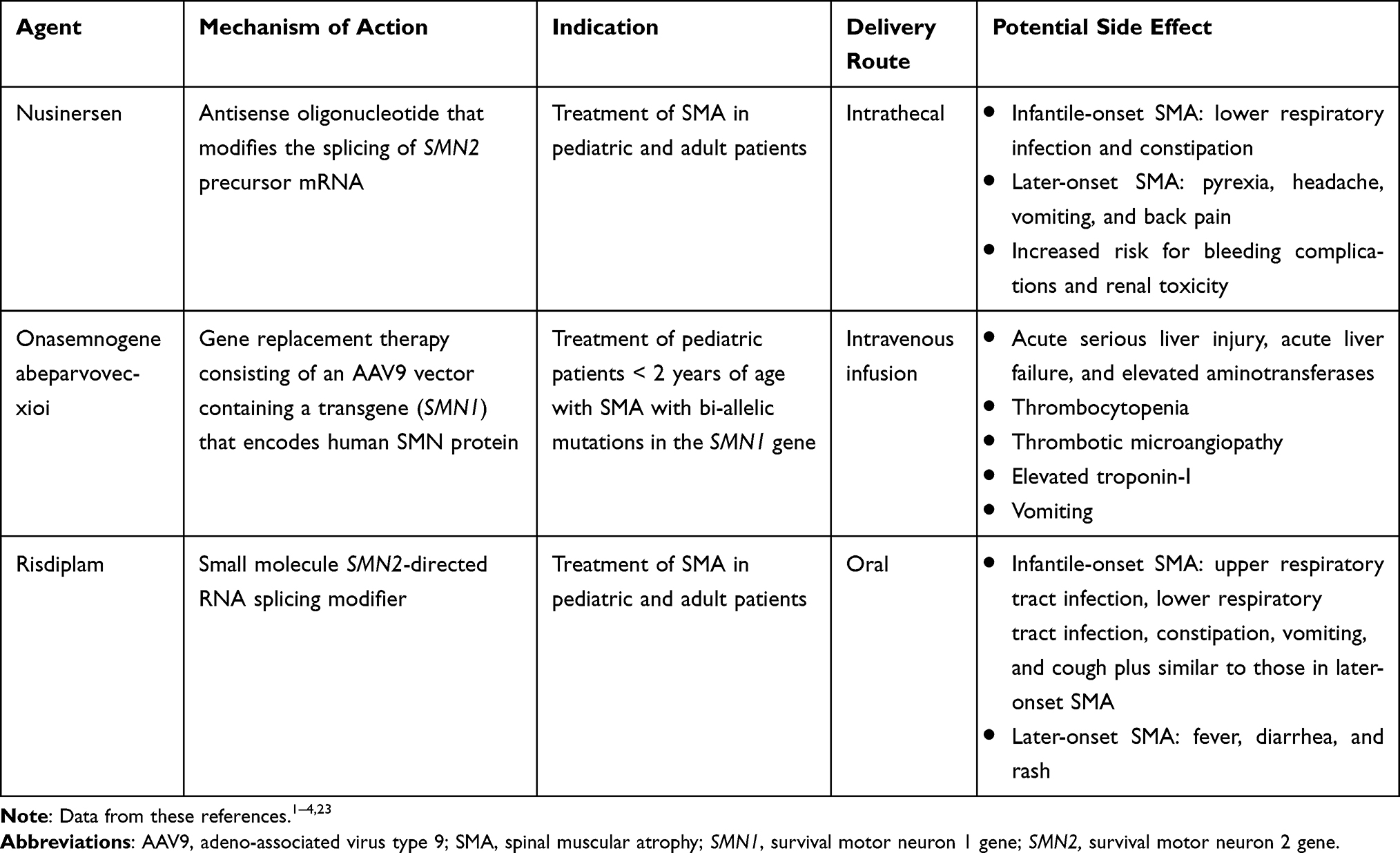 |
Table 1 Currently Available Disease-Modifying Therapies for SMA |
With the introduction of these therapies and the increasing implementation of newborn screening (NBS) programs in the United States and worldwide,25–28 the disease trajectory and clinical care model for patients with SMA has begun to change considerably, as have the phenotypes classically associated with the various types of SMA.5,6 Treated patients with SMA type I are now likely to experience improved motor function and longer survival without ventilatory support (Figure 1).6,20,29,30 SMA type II patients who are treated also may attain greater motor function, and pre-symptomatic treated patients may potentially achieve motor milestone development comparable to healthy children.5,21,31 Treatment decisions include the timing and selection of the initial disease-modifying therapy following a diagnosis of 5q SMA and subsequent questions of whether therapies should be changed or combined. Currently, limited data on combination treatment or therapy changes are available to help inform such decisions;32,33 there are no consensus guidelines on treatment choices, changes of therapy, or combination therapy, and treatment decisions are made on a case-by-case basis.
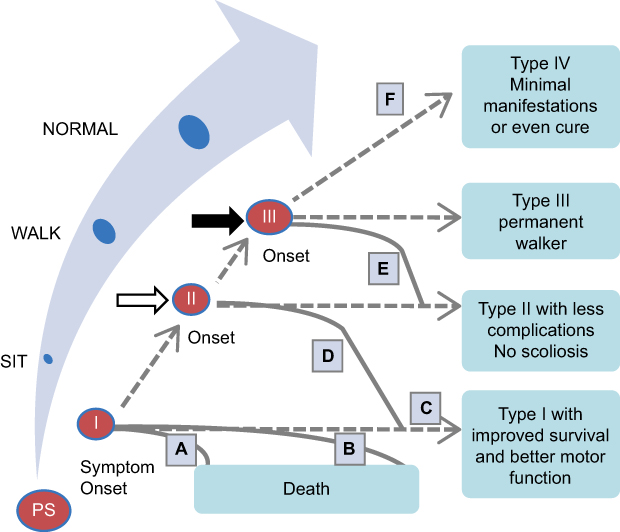 |
Figure 1 Hypothetical changes in SMA phenotypes (Reprinted from Neuromuscul Disord. 27[10] Tizzano EF, Finkel RS. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. 883–889 Copyright 20176 with permission from Elsevier). Ovals represent SMA types, ranging from pre-symptomatic (PS) to type III. A typical type I patient receiving treatment may not be responsive to therapy and, consistent with natural history, not survive past age 2 years. Alternatively, the patient could achieve longer survival or better motor function without a change of SMA type or with a change of SMA type in onset and evolution time. A similar trajectory can be hypothesized for a patient with SMA type II (starting at the white arrow after “sit”) or a patient with SMA type III (starting at the black arrow after “walk”). (A) Parabolic curve, with brief period of maximal motor function followed by rapid decline and death, representing an infant with type I SMA who received palliative care. (B) Plateau phase at maximal motor function, followed by a more gradual decline and eventual death, representing an infant with type I SMA who received proactive nutritional and ventilation support. (C–E) represent increasing levels of response to a therapeutic drug. (C) Sustained plateau phase, without loss of motor function or related feeding and respiratory status. (D) A patient with type I attains sitting (ie, becomes type II), then loses that skill and reverts to type I but is stronger overall and with a better survival. (E) A patient with type I progresses to type II, then attains walking (ie, becomes type III), or type II attains walking, with a plateau phase then a more gradual loss of function back to type II. (F) Functional cure—no motor, respiratory, feeding, or orthopedic impairment, with sustained benefit over time. Abbreviation: SMA, spinal muscular atrophy. |
In the absence of randomized, controlled clinical trial data that address changing or combining therapies, real-world experiences in the clinical setting can serve to inform clinicians with regard to the evolving treatment dynamics in care decisions. Over the last year, there has been a growing interest in understanding the experiences in changing or combining therapies. It is known that some nusinersen patients have changed therapies or returned to nusinersen after subsequent treatment with both onasemnogene abeparvovec-xioi and risdiplam. However, there are limited data on these situations in the peer-reviewed literature.32–37
To begin to better understand the perceptions of the available therapies among healthcare providers, patients, and caregivers in the SMA community, and the decision drivers that influence patient counseling and shared treatment decision-making, including therapy changes and combination therapy, the sponsor convened a virtual roundtable discussion with a panel of SMA-treating clinicians who have treated patients returning to nusinersen after changing to risdiplam. The sponsor organized and guided the roundtable discussion with directed, topical questions, which was followed by additional discussion during development of this paper after the roundtable. This report shares the collective opinions of the participating experts, based on their clinical interactions with patients with SMA, with the hope that these insights may stimulate discussion in the SMA community and enable others, including clinicians, patients, and families, to better understand the factors influencing real-world treatment decisions in the emerging SMA treatment landscape.
Panel Participants
The panel comprised four neurologists who are specialists in neuromuscular diseases at academic institutions and have treated patients with SMA for 13–20 years; these experts, therefore, have extensive experience in working with patients and their families to navigate the treatment options in SMA. The panel included a neurologist (Lauren Ellman [LE]) who treats adult patients with SMA (~40 patients), a pediatric neurologist (Leigh Ramos-Platt [LR-P]; ~70 children with SMA), and two neurologists (Perry B Shieh [PBS], Craig M Zaidman [CMZ]) who treat both pediatric and adult patients with SMA (~70 each). All panelists practice in states where SMA screening is available as part of the general NBS program. The panelists met on June 21, 2021, to discuss their clinical experiences, the challenges they face, and their treatment decisions in the management of patients with SMA; all panelists provided additional input after the roundtable discussion. The topics covered by the panel included treatment initiation, shared treatment decision-making, factors influencing treatment choice, changing therapy, returning to previous therapy, access experience, and future considerations for SMA.
Treatment Initiation
In the panelists’ experience, following a diagnosis of SMA, patients and their caregivers present to the clinic with differing levels of awareness of the treatment options available to them. The physician’s role in the shared decision-making process is to provide education, review the options, and describe what is known about each treatment, including the challenges, the potential side effects, and the experience with each treatment at their center.
The availability of NBS for SMA has helped to increase the visibility and early identification of patients with SMA. In the United States, 47 states have implemented or are in the process of implementing NBS programs for SMA and 97% of newborns are screened for SMA (as of July 1, 2022).38 As of December 29, 2020, 180 infants with SMA have been identified among approximately 2.4 million screened.25 Parents and caregivers of infants with SMA identified through NBS and rapidly referred for neuromuscular consultation may feel unprepared to understand the diagnosis and long-term implications of the disease or render an opinion regarding treatment preferences. These families may need additional counseling to better understand the disease and treatment options before they can arrive at an informed decision. On the other hand, parents and caregivers of infants and children with SMA who have had sufficient time to process the diagnosis and seek out information about the available treatment options often come to the clinic with a strong opinion as to which treatment they would like their child to receive. To obtain information to help them understand their child’s diagnosis of SMA, these parents may have reached out to a patient advocacy group or may have already become acquainted with other families of children with SMA through social media–based community groups. However, while the information garnered through some social media channels may have limitations and potential inaccuracies, the physician is the primary educator in shared decision-making and prescribing medication. It is therefore important to educate physicians who have not previously been involved in treating people with SMA as well as specialists.
Shared Treatment Decision-Making
Infants and Children
In the panelists’ opinions, there is an urgency to treat infants and younger children as soon as possible following a diagnosis of SMA to minimize the loss of motor neurons and, therefore, motor function. It is well established in the literature that early initiation of treatment leads to optimal motor function development and attainment of motor milestones, particularly in those children with SMA types I and II.20,29,39–41
Given the urgency to treat newly diagnosed infants, in patients for whom onasemnogene abeparvovec-xioi is being considered, the panelists have found that there is an emerging interest in bridging the gap between diagnosis and the administration of gene therapy. If access to onasemnogene abeparvovec-xioi is delayed, possibly due to insurance and reimbursement considerations, the panelists believe that an early bridging intervention with nusinersen or risdiplam can be considered to prevent the potential loss of motor neurons. Early intervention (before age 8 months) versus later intervention with onasemnogene abeparvovec-xioi may reduce the adverse effects of the treatment, such as elevated aminotransferases, which appear to be more common in older children42 and could indicate possible liver injury; the onasemnogene abeparvovec-xioi label mandates the administration of systemic corticosteroids before and after the infusion as risk mitigation.4 The panelists also think that recent preclinical data should be considered when contemplating onasemnogene abeparvovec-xioi; long-term toxicity and neuronal degeneration have been reported with gene transfer–mediated overexpression of SMN in animal models.43,44 The clinical relevance of these findings in humans is not known,45 although limited human autopsy data indicate widespread distribution of onasemnogene abeparvovec in peripheral organs and in the central nervous system without the aggregated SMN deposits observed in the murine study.43,46 In the panelists’ experience, the families of young children have generally opted either for gene therapy (if the child is less than 2 years of age) because of its one-time administration or for nusinersen because of its length of use in clinical practice. The panelists noted that older children who are treatment naive are not often seen in the clinic because most older children are already receiving a disease-modifying therapy.
Adults
Although there is a sense of urgency to treat the infant patient population, in adults with SMA who have a limited motor neuron pool, the panelists believe the realistic goal is disease stabilization. The panelists defined this as the prevention of decline and loss of function over time and no progression of respiratory symptoms, which can be assessed both qualitatively and quantitatively, eg, the use of measures such as the North Star Ambulatory Assessment for SMA and the Hammersmith Functional Motor Scale Expanded, with questions such as how far a patient can walk and what is the patient’s chewing ability. This goal is consistent with the hopes expressed by more than half of adult patients with SMA in a recent study on treatment expectations.47
The panelists have found that patients with SMA and their families will have differing levels of understanding of their treatment options and whether a particular treatment may be appropriate, depending on the external sources of information they access (eg, social media/online communities of families and patients with SMA; oversimplified anecdotes vs more robustly reviewed sources of credible scientific information). As an example, some panelists have encountered parents of older children, as well as adults with SMA, who were not aware they did not qualify for treatment with onasemnogene abeparvovec-xioi because they were older than 2 years. This reinforces the need for clinicians to ensure that patients are appropriately informed about the treatment options and their approved uses. Treating physicians must also take an active role in balancing a patient’s personal preferences and considerations (eg, patient age, lifestyle, family situation and status, plans) with the unknown risks of each therapy (eg, irreversible treatment effects, repeated lumbar puncture procedures), while setting realistic treatment goals and expectations for each patient.
Factors Influencing Treatment Choice
In the absence of head-to-head clinical trial data, the panel advised caution when presenting anecdotal data to patients because they may be susceptible to suggestion about the perceived outcomes of one treatment versus another, especially because SMA is a highly heterogeneous disorder in terms of severity and expected outcomes of treatment. The panelists highlighted the importance of the physician in presenting all treatment options in a comprehensive, fair, and balanced manner, explaining the known attributes and potential differentiating features of each therapy. In many instances in US clinics, the patient (or their parent/caregiver proxy) is the one to make the final treatment decision, based on their personal preferences. An overview of some of the topics and considerations that the panelists, other clinicians, patients, and caregivers may encounter when making treatment decisions is shown in Table 2.
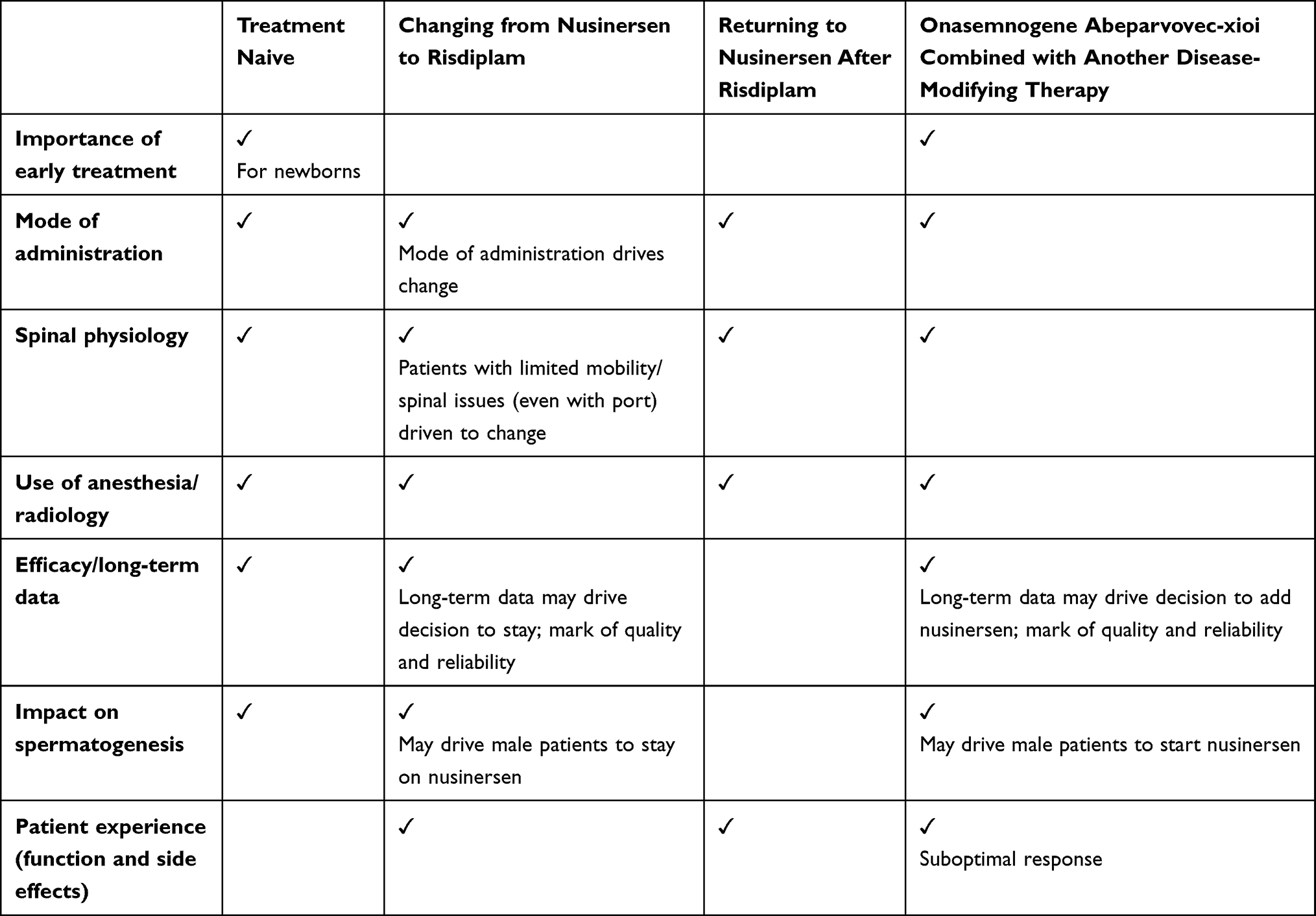 |
Table 2 Topics and Considerations When Making Treatment Decisions |
In the panelist’s experience, convenience appears to be an important factor in personal treatment choice, but the interpretation of “convenience” may differ for each patient. For some patients, the one-time administration of onasemnogene abeparvovec-xioi is the convenient option. For some, intrathecally administered nusinersen every 4 months is preferred because they find adherence to daily dosing to be challenging; others prefer the convenience of a daily oral dose of risdiplam.
Some of the panelists have found that another important aspect of the treatment landscape is the breadth of data and real-world experience that exists for a given therapy; for example, longer-term experience may be the deciding factor in initiating or continuing to receive nusinersen. At the time of the roundtable, real-world data including a 2021 multicenter study in Italy and a 2020 multicenter study in Germany showed treatment with nusinersen for up to 24 and 14 months, respectively, provides improvement or prevention of decline in motor function in children and adults with SMA.48,49
As related to the two approved SMN2 splicing modifiers, the panel’s experience has shown that nusinersen may be more effective in some patients, risdiplam in others. The panelists have heard from colleagues who believe nusinersen and risdiplam may be beneficial for different aspects of the disease because risdiplam is distributed in both the central nervous system and periphery, whereas nusinersen is primarily distributed in the central nervous system. This may account for different patient experiences with the two therapies. This, however, is only a hypothesis, and further research will be needed before any conclusions can be drawn.
Changing Therapy
The panelists noted that the option of an oral therapy has long been of interest to the SMA patient community, particularly those patients with complicated spines or who require sedation or general anesthesia to receive intrathecal nusinersen or who may have experienced lumbar puncture-associated adverse events. The panel noted that the approval of risdiplam in the United States occurred at the height of the COVID-19 pandemic, which resulted in an increase in telemedicine visits replacing some in-person evaluations. Thus, some patients may have changed from nusinersen to risdiplam therapy for the convenience of oral, at-home administration or to avoid the hospital during the COVID-19 pandemic, whether or not they were satisfied with their outcomes with nusinersen. New therapies appeal to some patients regardless of their experience, especially if the new treatment is perceived as more convenient. From the panelists’ experience, these individuals were generally older, female, and non-ambulatory. Some patients have also changed from nusinersen to risdiplam on the basis of recent data describing maintenance of the ability to swallow in infants with SMA type I.29,50 However, the panelists have found that other patients have returned to nusinersen after experiencing worsening bulbar function while on risdiplam. Interestingly, the panelists offered anecdotal information that patients being treated with nusinersen, especially those with intrathecal ports, often opt to remain on nusinersen because the intrathecal administration schedule facilitates treatment adherence and regular checkups with their treating physician.
Safety is an important factor in treatment decision-making. In the panel’s experience, adolescent boys and the families of young boys considering changing to risdiplam often express concerns related to male fertility, prompted by data showing degeneration or necrosis of the testis seminiferous epithelium with associated oligo/aspermia in the epididymis and abnormal sperm parameters in juvenile rats after risdiplam administration.2
Improvements in motor function and other outcome measures are likely to be modest in older patients,51,52 and the panel have found that some patients who are clinically stable on nusinersen have been reluctant to change therapies because of the potential risk of disease progression or of deterioration of previous gains with changing to the new therapy, or because they were waiting for longer-term data on risdiplam.
Returning to Previous Therapy
In the panel’s combined experience, patients rather than the treating physician more often initiated the discussion on returning to nusinersen, their original treatment, after changing to risdiplam. The most common patient-reported reasons for returning to nusinersen were a general sense of feeling less well, a perceived loss in motor function, and increased fatigue while on risdiplam. Some patients initially reported feeling well after changing to risdiplam from nusinersen, with the panelists noting that this was possibly during the period when there was a pharmacokinetic overlap between the two therapies as nusinersen levels decreased. For other patients, a panel member described a decreased ability to sprint (ie, decreased time off the ventilator) after initiating risdiplam that resulted in a need to return to nusinersen. In some patients with SMA, the diminished effectiveness of risdiplam could be related to a potential reduced exposure to risdiplam in older, heavier patients as they receive the maximum daily oral dose of 5 mg.
The panelists discussed anecdotally that some of their patients who switched to risdiplam said they missed the increased strength and decreased fatigue experienced shortly after each nusinersen treatment. The panelists also noted that some patients described a drop-off in energy approximately 3 months after a nusinersen dose, which was not an issue with daily dosing of risdiplam. Additionally, they heard from some patients who reported experiencing a decline in pulmonary function, or gastrointestinal side effects following the change to risdiplam. Unfortunately, due to the COVID-19 pandemic, which caused interruptions in regular in-person clinical assessments, the panelists had few objective endpoints upon which to base treatment decisions.
Medication Access Experience
Consistent with what the panel has heard directly from patients, surveys and interviews with patients with SMA and their families have shown that insurance coverage has been a substantial barrier to treatment in the past.53,54 The panelists noted that the initial efforts to obtain insurance coverage, which was a difficult process for some patients, created a situation in which they were hesitant to go through the process again to change therapy; some adult patients were deterred because they feared loss of coverage. However, this fear generally has not been borne out in the panelists’ practice experiences; most patients who have wanted to change therapy have been able to do so. The panelists felt that the ability to return to the original treatment after changing therapies may be related to the similarity in price between risdiplam and nusinersen, coupled with their different reimbursement mechanisms (nusinersen is often classified as a “medical benefit”, ie, a drug that is administered by a healthcare professional in a clinical setting, and risdiplam is usually classified as a “pharmacy benefit”, ie, is self-administered at home). The panelists have also observed generally good coverage of treatment changes for patients in government insurance programs.
Although therapy changes and returns to previous therapies are being allowed, in the panel’s experience, insurance companies have generally not approved the combination of risdiplam and nusinersen. However, insurance carriers have approved the use of nusinersen or risdiplam after onasemnogene abeparvovec-xioi. Based on the potential for continuous transgene expression by onasemnogene abeparvovec-xioi,55 this is effectively coverage of combination therapy.
Limitations
This paper should not be considered as any form of treatment guidelines, recommendations, or standards of care because no Delphi or other consensus methodology was used to generate its content. Instead, the panelists discussed their experiences and opinions, which may not be transferrable to or representative of encounters in SMA for other healthcare providers, in part because the panel consisted of only four members who have treated only a portion of the individuals who returned to treatment with nusinersen. Therefore, the panelists’ experiences may not reflect those of other healthcare providers whose patients have changed therapies or returned to an earlier therapy. Furthermore, because the panelists practice at academic sites that include multidisciplinary care clinics, they have been able to provide patients with access to a variety of specialists and resources. For example, the panelists have access to interventional radiology, so that intrathecal administration of medication can be performed if needed in patients with complicated spines. In contrast, community-based practices may not have similar resources as readily available for their patients. In addition, the outcomes related to decisions to use combination therapy or return to previous therapy were often self-reported or caregiver reported, have not been quantified clinically, and are subject to recall bias. Finally, the access experiences described here are applicable only in the United States and may vary for patients with other insurance providers and for patients in other countries with different reimbursement processes or other government-funded programs, or where there is no insurance coverage available.
Future Considerations
In the panel’s opinion, a number of questions remain to be addressed that may help guide future treatment and patient management decisions (Table 3). These focus on maximizing outcomes for patients identified in NBS programs and the need for data on treatment changes and combination therapy. Some of these questions may be addressed by ongoing or planned studies of combination treatment, change in therapy, or increases in dose. These studies include DEVOTE (NCT04089566; safety and efficacy of higher dose [28 mg or 50 mg loading doses; 28 mg maintenance dose] nusinersen),56 ASCEND (NCT05067790; risdiplam followed by higher dose [50 mg loading doses; 28 mg maintenance dose] nusinersen),57 RESPOND (NCT04488133; onasemnogene abeparvovec-xioi followed by 12 mg dose nusinersen), JEWELFISH (NCT03032172; onasemnogene abeparvovec-xioi or nusinersen followed by risdiplam), and studies of concurrent therapies that support both SMN production and muscle function (NCT03921528; apitegromab and nusinersen). In addition, observational studies or case series in which children received onasemnogene abeparvovec-xioi preceded or followed by nusinersen have shown combination therapy was well tolerated and resulted in improved outcomes.32,33 Concurrent treatment with risdiplam and nusinersen is also a potential approach as preclinical studies combining an antisense oligonucleotide similar to nusinersen and a small molecule compound RG7800 similar to risdiplam have demonstrated increased survival and improved motor function in a murine model of SMA without overt signs of toxicity.58 However, the combination should be explored with caution and the panelists were unaware of any clinical trials evaluating the efficacy and safety of this approach. Real-world data from SMA disease registries will also fill many data gaps.59
 |
Table 3 Questions for the Future |
Conclusions
As a result of the evolving treatment landscape in SMA and the availability of multiple therapies, the panelists believe that treatment plans can now be individualized to address each patient’s needs and goals. Such individualized treatment plans should be based on shared decision-making between the clinician and the patient and/or caregiver and, over time, may include a change in therapy or a combination of therapies. The panelists have found in their experience that although many patients have preferred to remain on their current therapy, others have expressed interest in changing treatments. Such interest is driven by a variety of factors, including the route of administration, convenience, hope for improved function, and a desire to try something new. If the patient’s goals and expectations on the new therapy are not met, the patient may discuss treatment options with their physician and return to their initial therapy because of, for example, a perception of decreased efficacy or emerging side effects. Ongoing and planned trials may provide additional data to help guide treatment decisions.
Ethics Approval and Informed Consent
This article is based on the opinions provided during a roundtable panel discussion and does not contain any new studies with human participants or animals performed by any of the authors.
Acknowledgments
We thank Dr Craig M Zaidman for his participation in the roundtable discussion and reviewing the first draft of the manuscript (he has served as an advisor to Biogen). Biogen provided funding for medical writing support in the development of this report and reviewed manuscript drafts. Meryl Mandle from Excel Scientific Solutions (Fairfield, CT, USA) provided writing assistance in the development of the first and subsequent drafts based on input from authors, and Jackie Parker and Miranda Dixon from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. The authors had full editorial control of the paper and provided their final approval of all content.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This publication is supported by Biogen (Cambridge, MA, USA). The sponsor was involved in the organization and facilitation of the roundtable discussions. The authors did not receive compensation from Biogen for their participation in the roundtable meeting or for the manuscript development. Financial disclosures, including consulting engagements with Biogen for other SMA-focused activities unrelated to this publication, are provided below.
Disclosure
LR-P: reports honoraria converted to donation to a 3rd non-profit party from Biogen, during the conduct of a study; honoraria for speaking converted to a donation to a third non-profit party from Biogen, outside the submitted work. LE: Advisory boards for Biogen, Roche, and PTC Therapeutics; primary investigator for multiple pharmaceutical trials. PBS: Advisory boards for Biogen; outside of current submitted work; advisory boards for AveXis and Sarepta; clinical trial research contracts with Acceleron, Biogen, Bristol Myers Squibb, Catalyst, Cytokinetics, Ionis Pharmaceuticals, Marathon, Pfizer, PTC, Sanofi-Aventis, Santhera, Sarepta, and Ultragenyx; personal fees from Genentech, Catalyst Grifols, CSL Behring, Alexion, Argenx, Sarepta Therapeutics, grants from Sanofi, Zogenix, Solid Biosciences, Astellas Gene Therapies; grants, personal fees from Pfizer and Novartis Gene Therapies, grants from PTC Therapeutics, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. 2020;21(3):307–315. doi:10.1080/14656566.2019.1704732
2. Genentech, Inc. Evrysdi® (Risdiplam) for Oral Solution. Genentech, Inc; 2022.
3. Biogen. Spinraza (Nusinersen) Injection, for Intrathecal Use. Biogen; 2020.
4. Novartis Gene Therapies. Zolgensma® (Onasemnogene Abeparvovec-Xioi) Suspension, for Intravenous Infusion. AveXis, Inc; 2021.
5. Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy - new phenotypes, new challenges, new implications for care. J Neuromuscul Dis. 2020;7(1):1–13. doi:10.3233/jnd-190424
6. Tizzano EF, Finkel RS. Spinal muscular atrophy: a changing phenotype beyond the clinical trials. Neuromuscul Disord. 2017;27(10):883–889. doi:10.1016/j.nmd.2017.05.011
7. Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355–368. doi:10.1002/ana.24864
8. Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington; 2000.
9. Darras BT, Monani UR, De Vivo DC, et al. Genetic disorders affecting the motor neuron: spinal muscular atrophy. In: Swaiman KF, Ashwal S, Ferriero DM, editors. Swaiman’s Pediatric Neurology.
10. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–2133. doi:10.1016/s0140-6736(08)60921-6
11. Costa-Roger M, Blasco-Pérez L, Cuscó I, Tizzano EF. The importance of digging into the genetics of SMN genes in the therapeutic scenario of spinal muscular atrophy. Int J Mol Sci. 2021;22(16):9029. doi:10.3390/ijms22169029
12. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–6311. doi:10.1073/pnas.96.11.6307
13. Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. doi:10.1093/hmg/8.7.1177
14. Calucho M, Bernal S, Alías L, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208–215. doi:10.1016/j.nmd.2018.01.003
15. Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46(1):1–12. doi:10.1016/j.pediatrneurol.2011.09.001
16. Finkel R, Bertini E, Muntoni F, Mercuri E; ENMC SMA Workshop Study Group. 209th ENMC International Workshop: outcome measures and clinical trial readiness in spinal muscular atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593–602. doi:10.1016/j.nmd.2015.04.009
17. Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet. 2013;6(11):113–120. doi:10.2147/tacg.S53615
18. Wang CH, Finkel RS, Bertini ES, et al.; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027–1049. doi:10.1177/0883073807305788
19. Hoy SM. Nusinersen: first global approval. Drugs. 2017;77(4):473–479. doi:10.1007/s40265-017-0711-7
20. Finkel RS, Mercuri E, Darras BT, et al.; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732. doi:10.1056/NEJMoa1702752
21. Mercuri E, Darras BT, Chiriboga CA, et al.; CHERISH Study Group. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635. doi:10.1056/NEJMoa1710504
22. Hoy SM. Onasemnogene abeparvovec: first global approval. Drugs. 2019;79(11):1255–1262. doi:10.1007/s40265-019-01162-5
23. Dhillon S. Risdiplam: first approval. Drugs. 2020;80(17):1853–1858. doi:10.1007/s40265-020-01410-z
24. Ratni H, Ebeling M, Baird J, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15):6501–6517. doi:10.1021/acs.jmedchem.8b00741
25. Dangouloff T, Vrščaj E, Servais L, Osredkar D; SMA NBS World Study Group. Newborn screening programs for spinal muscular atrophy worldwide: where we stand and where to go. Neuromuscul Disord. 2021;31(6):574–582. doi:10.1016/j.nmd.2021.03.007
26. Jędrzejowska M. Advances in newborn screening and presymptomatic diagnosis of spinal muscular atrophy. Degener Neurol Neuromuscul Dis. 2020;10:39–47. doi:10.2147/dnnd.S246907
27. Chien YH, Chiang SC, Weng WC, et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr. 2017;190:124–129.e121. doi:10.1016/j.jpeds.2017.06.042
28. Waldrop MA, Elsheikh BH. Spinal muscular atrophy in the treatment era. Neurol Clin. 2020;38(3):505–518. doi:10.1016/j.ncl.2020.03.002
29. Baranello G, Darras BT, Day JW, et al.; FIREFISH Working Group. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 2021;384(10):915–923. doi:10.1056/NEJMoa2009965
30. Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, Phase 3 trial. Lancet Neurol. 2021;20(4):284–293. doi:10.1016/s1474-4422(21)00001-6
31. Serra-Juhe C, Tizzano EF. Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors. Eur J Hum Genet. 2019;27(12):1774–1782. doi:10.1038/s41431-019-0415-4
32. Harada Y, Rao VK, Arya K, et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. 2020;62(4):550–554. doi:10.1002/mus.27034
33. Lee BH, Collins E, Lewis L, et al. Combination therapy with nusinersen and AVXS-101 in SMA type 1. Neurology. 2019;93(14):640–641. doi:10.1212/wnl.0000000000008207
34. Mendell JR, Al-Zaidy SA, Lehman KJ, et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021;78(7):834–841. doi:10.1001/jamaneurol.2021.1272
35. D’Silva AM, Holland S, Kariyawasam D, et al. Onasemnogene abeparvovec in spinal muscular atrophy: an Australian experience of safety and efficacy. Ann Clin Transl Neurol. 2022;9(3):339–350. doi:10.1002/acn3.51519
36. Mirea A, Shelby ES, Axente M, et al. Combination therapy with nusinersen and onasemnogene abeparvovec-xioi in spinal muscular atrophy type I. J Clin Med. 2021;10(23):5540. doi:10.3390/jcm10235540
37. Weiß C, Ziegler A, Becker LL, et al. Gene replacement therapy with onasemnogene abeparvovec in children with spinal muscular atrophy aged 24 months or younger and bodyweight up to 15 kg: an observational cohort study. Lancet Child Adolesc Health. 2022;6(1):17–27. doi:10.1016/s2352-4642(21)00287-x
38. Cure SMA. Newborn screening for SMA. Available from: https://www.curesma.org/newborn-screening-for-sma/#implementation-status.
39. De Vivo DC, Bertini E, Swoboda KJ, et al.; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–856. doi:10.1016/j.nmd.2019.09.007
40. Strauss K, Muntoni F, Farrar M, et al. Onasemnogene abeparvovec gene therapy in presymptomatic spinal muscular atrophy (SMA): SPR1NT study update in children with 2 copies of SMN2. Neurology. 2021;96(15 Suppl):4190.
41. Strauss KA, Farrar MA, Swoboda KJ, et al. Onasemnogene abeparvovec-xioi gene-replacement therapy in presymptomatic spinal muscular atrophy: SPR1NT study update. Neurology. 2020;94(15 Suppl):2384.
42. Waldrop MA, Karingada C, Storey MA, et al. Gene therapy for spinal muscular atrophy: safety and early outcomes. Pediatrics. 2020;146(3):e20200729. doi:10.1542/peds.2020-0729
43. Van Alstyne M, Tattoli I, Delestrée N, et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. 2021;24(7):930–940. doi:10.1038/s41593-021-00827-3
44. Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–298. doi:10.1089/hum.2018.015
45. Crawford TO, Sumner CJ. Assuring long-term safety of highly effective gene-modulating therapeutics for rare diseases. J Clin Invest. 2021;131(15):e152817. doi:10.1172/jci152817
46. Thomsen G, Burghes AHM, Hsieh C, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med. 2021;27(10):1701–1711. doi:10.1038/s41591-021-01483-7
47. Osmanovic A, Ranxha G, Kumpe M, et al. Treatment expectations and patient-reported outcomes of nusinersen therapy in adult spinal muscular atrophy. J Neurol. 2020;267(8):2398–2407. doi:10.1007/s00415-020-09847-8
48. Hagenacker T, Wurster CD, Günther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317–325. doi:10.1016/s1474-4422(20)30037-5
49. Pane M, Coratti G, Sansone VA, et al. Type I SMA “new natural history”: long-term data in nusinersen-treated patients. Ann Clin Transl Neurol. 2021;8(3):548–557. doi:10.1002/acn3.51276
50. Darras BT, Masson R, Mazurkiewicz-Bełdzińska M, et al.; FIREFISH Working Group. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021;385(5):427–435. doi:10.1056/NEJMoa2102047
51. Duong T, Wolford C, McDermott MP, et al. Nusinersen treatment in adults with spinal muscular atrophy. Neurol Clin Pract. 2021;11(3):e317–e327. doi:10.1212/cpj.0000000000001033
52. Walter MC, Wenninger S, Thiele S, et al. Safety and treatment effects of nusinersen in longstanding adult 5q-SMA type 3 - a prospective observational study. J Neuromuscul Dis. 2019;6(4):453–465. doi:10.3233/jnd-190416
53. Chen E, Dixon S, Naik R, et al. Early experiences of nusinersen for the treatment of spinal muscular atrophy: results from a large survey of patients and caregivers. Muscle Nerve. 2021;63(3):311–319. doi:10.1002/mus.27116
54. Pacione M, Siskind CE, Day JW, Tabor HK; Pacione M, Siskind CE, Day JW, Tabor HK. Perspectives on Spinraza (nusinersen) treatment study: views of individuals and parents of children diagnosed with spinal muscular atrophy. J Neuromuscul Dis. 2019;6(1):119–131. doi:10.3233/jnd-180330
55. Al-Zaidy SA, Mendell JR. From clinical trials to clinical practice: practical considerations for gene replacement therapy in SMA type 1. Pediatr Neurol. 2019;100:3–11. doi:10.1016/j.pediatrneurol.2019.06.007
56. Day JW, Pascual SIP, Finkel RS, et al. Escalating dose and randomized, controlled study of nusinersen in participants with spinal muscular atrophy (SMA); study design and part A data for the phase 2/3 DEVOTE (232SM203) study to explore high dose nusinersen (2343). Neurology. 2021;96(15 Supplement):2343.
57. Kuntz NL, Hagenacker T, Darras BT, et al. Rationale and design of ASCEND: a phase 3b study evaluating higher dose nusinersen in risdiplam-treated participants with spinal muscular atrophy.
58. Kray KM, McGovern VL, Chugh D, Arnold WD, Burghes AHM. Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic ∆7SMA mice. Neurobiol Dis. 2021;159:105488. doi:10.1016/j.nbd.2021.105488
59. Raynaud S, Viscidi E, Hall S, et al. Utilization of real-world observational data to study safety and effectiveness of spinal muscular atrophy treatments. J Neuromuscul Dis. 2021;8(s1):S71. doi:10.3233/JND-219006
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Disease Modifying Therapies for the Management of Children with Spinal Muscular Atrophy (5q SMA): An Update on the Emerging Evidence
Hjartarson HT, Nathorst-Böös K, Sejersen T
Drug Design, Development and Therapy 2022, 16:1865-1883
Published Date: 16 June 2022