")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Expanded DMPK repeats in dizygotic twins referred for diagnosis of autism versus absence of expanded DMPK repeats at screening of 330 children with autism
Authors Musova Z, Hancarova M, Havlovicova M, Pourova R, Hrdlicka M , Kraus J, Trkova M, Stejkal D, Sedlacek Z
Received 30 May 2016
Accepted for publication 30 June 2016
Published 19 September 2016 Volume 2016:12 Pages 2367—2372
DOI https://doi.org/10.2147/NDT.S113917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Zuzana Musova,1 Miroslava Hancarova,1 Marketa Havlovicova,1 Radka Pourova,1 Michal Hrdlicka,2 Josef Kraus,3 Marie Trkova,4 David Stejskal,4 Zdenek Sedlacek1
1Department of Biology and Medical Genetics, 2Department of Child Psychiatry, 3Department of Child Neurology, Charles University 2nd Faculty of Medicine and University Hospital Motol, 4Gennet, Centre for Fetal Medicine, Prague, Czech Republic
Abstract: Myotonic dystrophy type 1 (DM1) belongs to the broad spectrum of genetic disorders associated with autism spectrum disorders (ASD). ASD were reported predominantly in congenital and early childhood forms of DM1. We describe dizygotic twin boys with ASD who were referred for routine laboratory genetic testing and in whom karyotyping, FMR1 gene testing, and single nucleotide polymorphism array analysis yielded negative results. The father of the boys was later diagnosed with suspected DM1, and testing revealed characteristic DMPK gene expansions in his genome as well as in the genomes of both twins and their elder brother, who also suffered from ASD. In accord with previous reports on childhood forms of DM1, our patients showed prominent neuropsychiatric phenotypes characterized especially by hypotonia, developmental and language delay, emotional and affective lability, lowered adaptability, and social withdrawal. The experience with this family and multiple literature reports of ASD in DM1 on the one side but the lack of literature data on the frequency of DMPK gene expansions in ASD patients on the other side prompted us to screen the DMPK gene in a sample of 330 patients with ASD who were first seen by a geneticist before they were 10 years of age, before the muscular weakness, which may signal DM1, usually becomes obvious. The absence of any DMPK gene expansions in this cohort indicates that targeted DMPK gene testing can be recommended only in ASD patients with specific symptoms or family history suggestive of DM1.
Keywords: autism, myotonic dystrophy type 1, DMPK gene, genetic testing, comorbidity
Introduction
Autism spectrum disorders (ASD) have a significant genetic component, and they are a large heterogeneous group of different disorders which are difficult to distinguish clinically due to their similar phenotype. Autistic features have been associated with many syndromes of known genetic etiology and with de novo and inherited copy number and single nucleotide variants in a magnitude of genes.1 Examples of known genetic disorders that have autism as one of their frequent symptoms include the fragile X syndrome, tuberous sclerosis, Prader–Willi syndrome, and MECP2-associated disorders.2 The deciphering of genetic etiology in a patient with ASD has important consequences for family planning and also for prognosis and management of the patient, including future targeted therapies.
Myotonic dystrophy type 1 (DM1) is also often listed among genetic disorders associated with ASD.2,3 DM1 is an autosomal-dominant disorder with an estimated prevalence of 1/8,000 and a highly variable spectrum of manifestations progressively affecting muscles and many other systems, including the central and peripheral nervous system. It is caused by expansions of an untranslated CTG repeat in the DMPK gene. The length of the expansion influences the severity and age of onset of the disease.4 Four clinical forms of DM1 can be distinguished:4 1) the minimal form with cataract and mild or no muscular manifestations at old age; 2) adult forms mainly presenting with typical progressive muscular dystrophy, muscle weakness, and myotonia; 3) childhood forms with onset between 1 and 20 years of age often manifesting learning disabilities while the neuromuscular symptoms are initially absent; 4) congenital forms with prenatal and neonatal manifestation of severe generalized muscle weakness and hypotonia, hyperreflexia, facial weakness, arthrogryposis, and respiratory insufficiency. Especially the childhood forms of DM1 represent a diagnostic challenge as they initially show only an unspecific psychopathological phenotype with a variable extent of cognitive impairment. Most children with congenital and childhood DM1 show speech and language delay, attention deficit, hyperactivity, or mood and anxiety disorder (reviewed in Echenne and Bassez5 and Ho et al6). Sleep disturbances, muscular weakness, typical facial appearance, cataracts, and heart and gastrointestinal tract problems are usually among the later developing symptoms. Several reports of ASD in children later diagnosed with DM1 exist,7–10 and a large study has shown that up to 50% of children with congenital and childhood forms of DM1 have ASD.11
We report dizygotic twin boys who were referred for laboratory genetic analysis due to autistic features. Karyotyping, fragile X syndrome testing, and single nucleotide polymorphism (SNP) array analysis yielded negative results. At the time of referral and testing of the twins, their father lived separately from the family and his clinical data were not available. In his early forties, he was diagnosed with DM1 and laboratory analysis showed DMPK gene expansions in his genome as well as in the genomes of both twins and their elder brother who also suffered from ASD. This experience and literature reports of autistic features and developmental and speech delay as the first symptoms of DM1 prompted us to screen DMPK expansions in a sample of 330 patients with ASD who were first seen by a geneticist before they reached the age of 10 when the muscular weakness, which may signal DM1, usually becomes obvious.
Materials and methods
Case report of the twins and their family
The currently studied 9-year-old twin boys were born from a second pregnancy of nonconsanguineous Czech parents (at the birth of the twins, the mother and father were 32 and 35 years old, respectively). The delivery was in the 35th week of gestation by cesarean section due to breech position of twin A.
Twin A had a birth weight of 2,150 g (25th percentile) and a height of 44 cm (25th percentile). The adaptation after delivery was uncomplicated (Apgar score 10-10-10), but the newborn had a weak sucking reflex and he failed to thrive. His psychomotor development was delayed, and because of cerebellar signs and central hypotonic syndrome, he has undergone physiotherapy since infancy. He could sit at 12 months and started to walk at 18 months of age. His first words were noted at 3.5 years of age, but his speech was incomprehensible and improved only after intensive speech therapy at 5 years of age. A neurological examination at the age of 3 years suggested ASD, which was then confirmed at 6 years of age. A psychological examination at 5 years of age revealed a mild form of attention deficit hyperactivity disorder (ADHD), a lower degree of emotional maturity, affective lability, moderately increased anxiety, uncertainty, introversion, and specific behavioral features like excessive demand for order and rituals. At the age of 7 years, he was diagnosed with atypical autism with mild or borderline symptomatology using Autism Diagnostic Interview-Revised and the assessment was also supplemented by the Childhood Autism Rating Scale. His behavior was characterized by affective lability, increased anxiety, clinginess, nervousness, lowered adaptability, solitary activities in a child group, impaired reciprocal communication, and reduced eye contact. A combined type of ADHD was observed with agitation, neurotic reactions, impulsivity, easy fatigability, and a short attention span. He had slightly above-average intelligence and comorbid developmental dysphasia. A neurological examination showed mild syndrome of sleep apnea with no signs of myotonia.
Twin B had a birth weight of 2,850 g (75th percentile) and a height of 47 cm (50th percentile). The Apgar score was 10-10-10. He had neonatal jaundice, and was noticeably calm. His food intake was normal, but the switch over to solid food was accompanied by persistent chewing difficulties. His psychomotor development was delayed. Minimal spontaneous movement was observed till the age of 1 year. He started to walk at 17 months of age. His first words were noted at 14 months and stagnation in language development was observed between 1.5 and 2.5 years of age. After intensive speech therapy, he could speak in simple sentences at 4 years of age. Neurological examination revealed expressive dysphasia, poor fine motor skills, psychomotor retardation, autism, and hypotonic syndrome. A psychiatric examination at 3 years of age showed expressive language disorder, delayed speech development, subnormal cognitive development, borderline intellectual disability (ID) (intelligence quotient 70 using the Stanford–Binet Intelligence Scale, 4th Edition), attention deficit, clinginess, and autistic features like rituals, hypersensitivity, social immaturity, and emotional lability. At the age of 4 years, he was diagnosed with moderate-functioning childhood autism with mild-to-moderate symptomatology using Autism Diagnostic Interview-Revised and Childhood Autism Rating Scale. He had a disproportional profile of cognitive skills at the border of mild ID. Due to central sleep apnea and intolerance to physical stress, cardiological examinations were performed at 7 years of age, which confirmed the observations and showed palpitation and a first degree atrioventricular block. A repeated psychological examination at the age of 7 years showed high-functioning autism with borderline normal intelligence, lowered adaptability, negativism, emotional instability, developmental expressive dysphasia, and verbal stereotypes. A neurological examination showed prominent hypotonia, dyspraxia, problems with self-care, enuresis, and encopresis, but no signs of myotonia.
The brother of the twins, who is currently 21 years old, had accelerated early psychomotor and speech development (he has been able to speak fluently since 2 years of age and write in capital letters and read since 3 years of age). In the preschool age, he was diagnosed with ADHD with aggressiveness. The hyperactivity lasted till 12 years of age when it changed to attenuation, fatigue, and hypersomnia. ASD was first considered between 13 and 15 years of age. At the age of 17 years, he started developing cramps in his hands and facial muscles, and at the same time, supraventricular tachycardia and chronic atrial flutter were diagnosed and treated by radiofrequency ablation. Based on the diagnosis in his brothers, a psychological examination was performed at 19 years of age, which showed low-functioning Asperger syndrome with above-average mental performance (intelligence quotient 111) using the Childhood Asperger Syndrome Test, Autism Screening Questionnaire, High-Functioning Autism Spectrum Screening Questionnaire, and Australian Scale for Asperger’s Syndrome. Later examination revealed bipolar affective disorder with aggression. Currently, his phenotype is characterized with prominent asthenic habitus, poor posture, narrow chest, slight ptosis, and thinning hair with frontal baldness.
The twins also have 10- and 13-year-old paternal half-sisters. Their clinical information was not available but it was known that the younger girl was diagnosed with ADHD and her behavior was similar to that of twin B.
The mother of the twins was healthy, but three maternal relatives suffered from schizophrenia, and a nephew of the maternal grandfather of the twins was diagnosed with childhood autism. The clinical data of the twins’ father were missing at the time of ASD testing of the children. He suffered from alcoholism and bipolar affective disorder. At 42 years of age, he was referred by a neurologist for genetic testing of DM1 due to mixed polyneuropathy affecting his upper limbs and chronic neurogenic impairment with myotonic discharges detected on electromyography. A retrospective analysis showed that the first signs, like occasional tongue spasms, prolonged muscle contraction, and inability to relax fingers after exertion, had manifested since the age of 40 years. Alcoholism and bipolar affective disorder (but no muscular symptoms) were reported also in his father, grandfather, and sister, but the clinical data available on these individuals were very sparse. The study of the family was approved by the Ethics Committee of the Charles University 2nd Faculty of Medicine and University Hospital Motol. Written informed consent for participation in the study was obtained from all family members or their legal guardians.
ASD patient cohort
The ASD patient cohort included 330 consecutive patients (272 boys and 58 girls) who were referred for laboratory genetic analysis of ASD at <10 years of age (average age at the first visit was 5.8 years) between December 2005 and March 2010. Only patients with a normal karyotype, no FMR1 gene expansion and no clinically recognizable syndrome were included in the sample. The study of the ASD patient cohort was approved by the Ethics Committee of the Charles University 2nd Faculty of Medicine and University Hospital Motol. Written informed consent was obtained from all participants or their legal guardians.
Laboratory genetic testing for ASD in the twins
Both twins were examined using conventional cytogenetic analysis with standard G-banding and fragile X syndrome testing using the FMR1 TP-PCR kit (Abbott, Abbott Park, IL, USA) (TP-PCR, triplet-repeat primed polymerase chain reaction). Subsequently SNP array testing was performed using HumanCytoSNP-12 BeadChip (~300K resolution; Illumina, San Diego, CA, USA). Due to numerous weakly supported and likely false positive findings in twin B, this analysis was repeated in this patient using a higher-resolution SNP array (HumanOmniExpress-24 BeadChip; ~700K resolution; Illumina).
DMPK gene expansion analysis
Analysis of the CTG repeat in the DMPK gene in the family was performed using triplet-primed PCR on both strands of the repeat according to published protocols.12,13 The triplet-primed PCR products were separated on an ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Long-range PCR employed to assess the size of the expansions used the expand long template PCR system (Roche Diagnostics, Mannheim, Germany) as previously described,14 in buffer 2 with 2M betaine. Screening of DMPK expansions in the ASD patient cohort was performed on one strand using the specific primer located at the 5′ side of the CTG repeat.13
Results
Routine laboratory testing of ASD in the twins, including SNP array analysis, did not show any significant findings. After the clinical diagnosis of suspected DM1 was established in the father, DMPK expansions were revealed first in the father and subsequently also in both twins and their brother. The DMPK genotypes identified in the father, elder brother, twin A, and twin B were 13/130, 5/500, 5/300 and 5/500 CTG repeats, respectively (Figure 1).
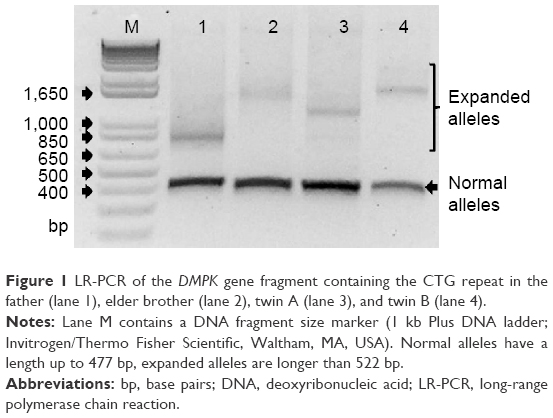 | Figure 1 LR-PCR of the DMPK gene fragment containing the CTG repeat in the father (lane 1), elder brother (lane 2), twin A (lane 3), and twin B (lane 4). |
DMPK expansion testing in the sample of 330 patients with ASD yielded no positive result (all alleles were in the normal range, <35 CTG repeats).
Discussion
We report a case of dizygotic twin boys who were referred for genetic testing of ASD and in whom a routine panel of laboratory analyses yielded no findings. The availability of the clinical data of the father and his diagnosis of suspected DM1 caused a key shift in the deciphering of the genetic basis of the disorder in the family. A DMPK gene expansion and the diagnosis of DM1 were first confirmed in the father and subsequently the same genetic defect was revealed in both twins and their elder brother who also suffered from ASD. The length of the expansion in the father was 130 CTG repeats, which was in accord with the adult form of DM1. The children had longer expansions of 300–550 CTG repeats. This is in agreement with the tendency of the repeat to elongate and cause earlier onset of the disorder and increase its severity in subsequent generations.4 The span of 300–550 CTG repeats corresponds to a wide spectrum of clinical forms of DM1, ranging from congenital to adult forms.15 The clinical signs in the twins and their elder brother are consistent with childhood DM1.
Characteristic muscular signs of DM1, such as muscle weakness, atrophy, and myotonia (which also lead to the typical facial appearance of DM1), are just mildly expressed or absent in childhood and juvenile forms of DM1, and can appear at any age, usually in the second decade of life.4,5 The twins showed prominent neuropsychiatric phenotypes characterized by hypotonia, developmental and language delays, sleep disturbances, fatigue, ID, ASD, and attention deficits, which are often the childhood symptoms of DM1.5,6 However, these symptoms are also associated with many other genetic disorders.
Concerning ASD specifically, several reports described autism in children with DM17–10,16 and a targeted study of 57 children with mainly congenital and childhood forms of DM1 showed that almost half of them had ASD.11 Conversely, the incidence of DM1 among ASD patients was estimated to be <0.2%,3 but focused studies are still missing in the literature. Our analysis of 330 children with ASD, which revealed no DMPK gene expansions in this cohort, strongly supports the rather low rate of DM1 in ASD. As the incidence is so low, the determination of its exact figure would require a very large sample of young ASD patients.
Nevertheless, several studies reported a low occurrence or absence of ASD in DM1 patients: autism was diagnosed in 1/16 patients with juvenile forms;10 in 0/24 patients with childhood and juvenile forms;17 in 0/79 patients with adult forms;18 in 1/17 and 0/15 patients with congenital and infantile/juvenile forms, respectively;16 and in 0/28 patients with childhood and early adult forms.19 When ASD were identified in these studies, they were exclusively present only in congenital and infantile forms of DM1. The main difference between these reports and Ekström et al’s 2008 study,11 which showed a very high incidence of ASD in DM1 patients, was the high proportion of congenital and early childhood forms of DM1 in the latter. However, other factors, like the use of different tests and different historical ASD classification schemes and criteria or random coincidence, can also contribute. Douniol et al’s study in 201219 proposed that DM1 patients did not suffer from ASD but just presented a combination of symptoms that may mimic autism. From the point of view of genetic testing, this has a limited practical impact as these patients are apparently referred for genetic testing due to suspected ASD, notwithstanding the fact as to whether this diagnosis is formally correct or not.
The behavioral phenotypes observed in childhood DM1 patients included social phobia, anxiety, and language delay, while the stereotyped behavior might not be so pronounced.19 Also the patients included in Ekström et al’s 2008 study11 had less severe impairment in the area of restricted repetitive and stereotyped patterns of behavior, interests, and activities. Language delay, anxiety, and introversion with social withdrawal were clearly present in the twins reported here, who, however, also showed remarkable ritual behaviors, stereotypes, and insistence on sameness. In addition to one case of autism identified in Echenne et al’s 2008 study,16 almost a half of their patients with childhood and congenital DM1 showed psychiatric phenotypes, including attention deficit disorders, hyperactivity, conduct disorders, aggression, opposition, anxiety, and depression. Similar to the patients included in Ekström et al’s 2008 study,11 our twin B could also be described as calm, easily fatigued, passive, and with a low pace. Twin A was diagnosed with a mixed type of ADHD, which contrasts with previous reports of prevailing inattention in these patients.19 Our observations thus support the notion that young individuals with DMPK gene expansions show a characteristic wide spectrum of psychiatric phenotypes.
In light of the current understanding of ASD as an extremely heterogeneous large group of different conditions with different genetic etiology,1 it is not surprising that subtypes of ASD with specific genetic determination can have individual ASD symptoms differently pronounced, and that their spectra of symptoms can overlap the spectra of other psychiatric disorders and the continuum of normal population variability. Interestingly, the preponderance of ASD among the congenital and early childhood forms of DM1 may point to a developmental window in which the deleterious role of DMPK expansions, if materialized, can specifically affect the behavioral and cognitive profile of the patient. This may be reminiscent of other genes known to be associated with neurodevelopmental disorders, especially ASD and ID, which affect basic cellular processes like regulation of gene expression; however, the negative phenotypic effect of their defects is limited to early brain development, which is a highly orchestrated and sensitive process.20 Studies in model organisms may suggest that the timing of toxic ribonucleic acid expression during development can affect DM1 severity, with different effects on individual symptoms.21 When considering the causal role of the DMPK expansions for ASD, it has to be recognized that they could also represent only a disease modifier potentiating the expression of another factor, and that the genetic background, which is not fully understood, is likely to play an important role.
In conclusion, the family reported here illustrates the association between DM1 and ASD, and the description of the clinical picture of the patients contributes to the definition of the spectrum of ASD symptoms associated specifically with the DMPK gene expansions. Concurrently, our analysis of the DMPK gene status in a sample of 330 patients with ASD referred for genetic analysis at <10 years of age indicates that DMPK expansions are rather rare among children with autism. Targeted DMPK testing can thus be recommended only in ASD patients with specific symptoms suggestive of DM1 or in patients with a family history of symptoms reminiscent of DM1. The awareness of ASD in children with DMPK expansions is important for the planning of early behavioral training and other supports and interventions, including pharmacotherapy of specific symptoms.
Acknowledgments
We thank the family of the patients for their cooperation. This work was supported by grant NT/14200 from the Czech Ministry of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
De Rubeis S, Buxbaum JD. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet. 2015;24(R1):R24–R31. | ||
Zafeiriou DI, Ververi A, Dafoulis V, Kalyva E, Vargiami E. Autism spectrum disorders: the quest for genetic syndromes. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(4):327–366. | ||
Toriello HV. Approach to the genetic evaluation of the child with autism. Pediatr Clin North Am. 2012;59(1):113–128. | ||
Harley HG, Rundle SA, MacMillan JC, et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet. 1993;52(6):1164–1174. | ||
Echenne B, Bassez G. Congenital and infantile myotonic dystrophy. Handb Clin Neurol. 2013;113:1387–1393. | ||
Ho G, Cardamone M, Farrar M. Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J Clin Pediatr. 2015;4(4):66–80. | ||
Yoshimura I, Sasaki A, Akimoto H, Yoshimura N. A case of congenital myotonic dystrophy with infantile autism. No To Hattatsu. 1989;21(4):379–384. | ||
Blondis TA, Cook E Jr, Koza-Taylor P, Finn T. Asperger syndrome associated with Steinert’s myotonic dystrophy. Dev Med Child Neurol. 1996;38(9):840–847. | ||
Paul M, Allington-Smith P. Asperger syndrome associated with Steinert’s myotonic dystrophy. Dev Med Child Neurol. 1997;39(4):280–281. | ||
Steyaert J, Umans S, Willekens D, et al. A study of the cognitive and psychological profile in 16 children with congenital or juvenile myotonic dystrophy. Clin Genet. 1997;52(3):135–141. | ||
Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):918–926. | ||
Warner JP, Barron LH, Goudie D, et al. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33(12):1022–1026. | ||
Musova Z, Mazanec R, Krepelova A, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A. 2009;149A(7):1365–1374. | ||
Saluto A, Brussino A, Tassone F, et al. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn. 2005;7(5):605–612. | ||
Kamsteeg EJ, Kress W, Catalli C, et al. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet. 2012;20(12):1203–1208. | ||
Echenne B, Rideau A, Roubertie A, Sébire G, Rivier F, Lemieux B. Myotonic dystrophy type I in childhood long-term evolution in patients surviving the neonatal period. Eur J Paediatr Neurol. 2008;12(3):210–223. | ||
Steyaert J, de Die-Smulders C, Fryns JP, Goossens E, Willekens D. Behavioral phenotype in childhood type of dystrophia myotonica. Am J Med Genet. 2000;96(6):888–889. | ||
Kalkman JS, Schillings ML, Zwarts MJ, van Engelen BG, Bleijenberg G. Psychiatric disorders appear equally in patients with myotonic dystrophy, facioscapulohumeral dystrophy, and hereditary motor and sensory neuropathy type I. Acta Neurol Scand. 2007;115(4):265–270. | ||
Douniol M, Jacquette A, Cohen D, et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev Med Child Neurol. 2012;54(10):905–911. | ||
Suliman R, Ben-David E, Shifman S. Chromatin regulators, phenotypic robustness, and autism risk. Front Genet. 2014;5:81. | ||
Gladman JT, Mandal M, Srinivasan V, Mahadevan MS. Age of onset of RNA toxicity influences phenotypic severity: evidence from an inducible mouse model of myotonic dystrophy (DM1). PLoS One. 2013;8(9):e72907. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.