Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 14
Exocrine Pancreatic Insufficiency in Children – Challenges in Management
Authors Sankararaman S, Schindler T
Received 26 May 2023
Accepted for publication 13 October 2023
Published 26 October 2023 Volume 2023:14 Pages 361—378
DOI https://doi.org/10.2147/PHMT.S402589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Senthilkumar Sankararaman,1 Teresa Schindler2
1Division of Pediatric Gastroenterology, Department of Pediatrics, UH Rainbow Babies & Children’s Hospital / Case Western Reserve University School of Medicine, Cleveland, OH, USA; 2Division of Pediatric Pulmonology, Department of Pediatrics, UH Rainbow Babies & Children’s Hospital, Cleveland, OH, USA
Correspondence: Senthilkumar Sankararaman, Division of Pediatric Gastroenterology, Hepatology & Nutrition, UH Rainbow Babies & Children’s Hospital, Case Western Reserve University SOM, Cleveland, OH, USA, Tel +001 216-844-1765, Fax +001 216-844-8750, Email [email protected]
Abstract: Cystic fibrosis (CF) is the leading etiology for exocrine pancreatic insufficiency (EPI) in children, followed by chronic pancreatitis, Shwachman-Diamond syndrome, and other genetic disorders. Management of EPI in children poses several unique challenges such as difficulties in early recognition, lack of widespread availability of diagnostic tests and limited number of pediatric-specific pancreatic centers. Pancreatic enzyme replacement therapy is the cornerstone of EPI management and in young children difficulties in administering pancreatic enzymes are frequently encountered. Patients with EPI also should be screened for fat-soluble vitamin deficiencies and receive appropriate supplementation. Among disorders with EPI in children, CF is the relatively well-studied condition, and most management recommendations for EPI in children come from expert consensus and conventional practice guidelines. The impact of EPI can be greater in children given their high metabolic demands and rapid growth. Early diagnosis and aggressive management of EPI prevent consequences of complications such as malnutrition, fat-soluble vitamin deficiencies, and poor bone health and improve outcomes. Management by multi-disciplinary team is the key to success.
Keywords: exocrine pancreatic insufficiency, EPI, children, PERT, pancreatic enzymes
Introduction
Exocrine pancreatic insufficiency (EPI) is defined as inadequate secretion of pancreatic enzymes (function of acini) and/or sodium bicarbonate (ductal function) resulting in suboptimal gastrointestinal (GI) absorption of nutrients.1 Pancreas has a huge functional reserve and frank steatorrhea does not occur until approximately 90% of pancreatic tissue is destroyed with pancreatic enzymes reduced to 5–10% of normal physiological quantities.2,3 Even though the pancreas participates in the digestion of all macronutrients, lipase is predominantly affected in EPI resulting in impaired absorption of fat and fat-soluble vitamins.2 In the early stages, EPI can be difficult to diagnose due to its rarity and non-specific GI symptoms.
The common causes of EPI in adults include chronic pancreatitis, cystic fibrosis (CF), diabetes mellitus, inflammatory bowel disease (IBD), and pancreatic cancer.1,4–7 EPI in adults is relatively well described and various societal guidelines exist for EPI management in adults.8–11 Even though there are many similarities in EPI between children and adults, there are key differences in the etiology and also specific challenges exist in managing EPI in young children. Cystic fibrosis (CF) is the most common and well-studied condition causing EPI in children and most treatment recommendations for EPI are inferred from expert consensus-based CF management guidelines. Outside CF, manifestations of EPI may be difficult to recognize due to its rarity, and limited availability of pediatric-specific pancreatic centers. There is also paucity of high-quality literature regarding management of EPI in this population.
In this review, we detail all the disorders which present with EPI during infancy and childhood and their management. We also highlight the challenges involved in the diagnosis of EPI in the pediatric age group with specific focus on the difficulties encountered in the administration of pancreatic enzyme replacement therapy (PERT) in children.
Causes of EPI
Primary Causes of EPI
EPI could be primary with decreased pancreatic parenchyma or secondary to non-pancreatic causes. Primary pancreatic causes include several genetic syndromes or chronic pancreatitis resulting in pancreatic parenchymal loss of volume and function. After CF, chronic pancreatitis and Shwachman-Diamond syndrome (SDS) are the known primary causes of EPI in children.12–14 Other genetic syndromes such as Johanson-Blizzard syndrome (JBS), Pearson-marrow syndrome, Jeune syndrome, pancreatic aplasia/hypoplasia, and deficiency of isolated pancreatic enzymes are rarely encountered13,15,16 (Table 1 and Table 2). Unlike adults, pancreatectomy for treatment of pancreatic neoplasia resulting in EPI is relatively uncommon in children.12
 |
Table 1 Causes of Exocrine Pancreatic Insufficiency in Children |
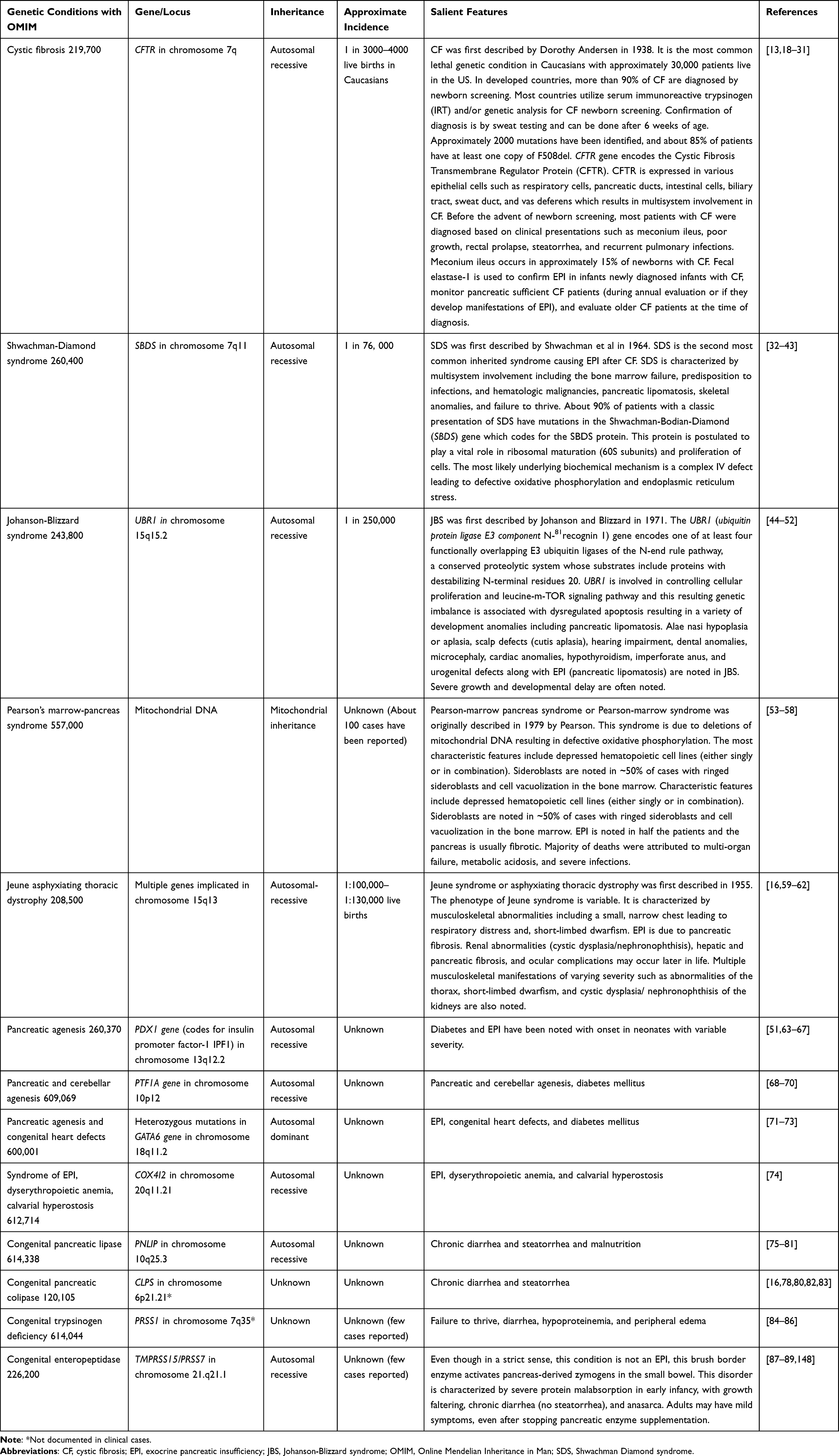 |
Table 2 Inherited Syndromes Which Causes Exocrine Pancreatic Insufficiency During Infancy:15,21 |
Secondary Causes of EPI
Secondary pancreatic insufficiency could result from lower stimulation of pancreatic secretions in duodenal mucosal inflammatory disorders such as refractory celiac disease or IBD with duodenitis, which may result in reduced release of cholecystokinin (CCK) from the damaged intestinal cells2,14 (Table 1). Other causes of secondary pancreatic insufficiency include inappropriate mixing of pancreatic secretions with intestinal contents in conditions such as surgeries involving the upper gastrointestinal tract or inactivation of pancreatic lipase and colipase due to excessive acidity of the intestinal contents (Table 1).
Transient pancreatic insufficiency, a relatively poorly understood entity, is noted in severe malnutrition, post-viral infection state, or after pancreatic trauma.91,92 A decrease in pancreatic amylase and lipase activities is often encountered in infants likely due to maturational delay of the digestive system and may not cause any symptoms of malabsorption as the lipase present in breast milk and salivary amylase have compensatory activities.93,94
Clinical Presentation of EPI
Steatorrhea is noted in severe EPI and is usually reported by caregivers as bulky, malodorous, oily stools in the infant’s diaper.13 Similar to adults, older children and adolescents may manifest steatorrhea as large bulky, oily stools, which may be difficult to flush. Other symptoms of EPI include chronic loose stools, weight loss or poor weight gain, voracious appetite, bloating, and excessive flatulence.17 Malnutrition and poor bone health are also frequently encountered.95 In patients with undiagnosed EPI or patients without fat-soluble vitamin supplementation, deficiencies of fat-soluble vitamins (A, D, E, and K) can also be noted.15 In the early stages, EPI is difficult to diagnose due to rarity and non-specific GI symptoms. As children are actively growing, EPI has more severe consequences with significant impairment on growth. Also, the symptoms of EPI overlap with other GI conditions resulting in malabsorption such as celiac disease, inflammatory bowel diseases, small intestinal bacterial overgrowth and giardiasis. The constellation of EPI symptoms and extra-pancreatic manifestations in other genetic conditions may help prompt the clinician for an EPI workup (Table 2).
Diagnosis of EPI
Indirect Pancreatic Function Tests
Pancreatic function tests are widely classified as indirect or direct tests (Table 3). Indirect tests are mostly stool-based, noninvasive, and less reliable in the early stages of EPI. Fecal fat screening by Sudan red staining (>2.5 droplets/high-power field) involves microscopic analysis of fecal fat. This test is not recommended as it is nonspecific for EPI and can be positive in other causes of malabsorption or as a result of rapid gut transit in young children.17
 |
Table 3 Pancreatic Function Tests |
Co-efficient of fat absorption (CFA) is the gold standard test for fat malabsorption which is defined as the ratio between ingested fat minus excreted fat in the stool)/ingested fat and expressed as a percentage.12 Normal value of CFA for patients older than 6 months of age is ≥93%, whereas in infants less than 6 months of age, ≥85% is normal92,96,97 This difference in CFA in young infants is due to mild physiologic reduced intestinal fat absorption due to delay in maturation. This test should be performed on a defined fat diet and a 72-hour stool fat collection with strict documentation of intake and stools excreted.12 A high-fat diet consisting of 100 g of fat per day is recommended for adolescents and adults and 2 g/kg in younger children.17 This test is not specific for EPI but will be positive in any condition involving fat malabsorption.13 CFA testing is cumbersome to perform, stool sample collection is unpleasant for patients and families and, a strict log of fat intake is time-consuming.13
Fecal elastase-1, an ELISA-based test for the human enzyme, is often useful to screen for EPI and has utility under selective circumstances. Compared to CFA, fecal elastase-1 has lower sensitivity (~25%) but higher specificity (96.4%) for EPI.98,99 Stool sampling for elastase-1 testing is commonly used because it is non-invasive. The assay tests for human elastase, whereas pancreatic enzyme replacement therapy (PERT) products are porcine-based, so patients can continue taking PERT even when repeating elastase testing. Fecal elastase-1 <200 μg/gm of stool is indicative of EPI. A value of 100–200 μg/gm is classified as mild to moderate EPI, and <100 μg/gm is indicative of severe EPI and associated with steatorrhea.100,101 Reducing the cutoff from 200 to 100 μg/gm increases the specificity of EPI but lowers the sensitivity.102 A solid or semi-solid stool should be used to quantify fecal elastase-1 to avoid a false positive test for EPI resulting from stool dilution. Fecal elastase-1 is not helpful if isolated with enzyme deficiency is suspected.98,99
13C-mixed triglyceride breath test is noninvasive but is not commonly available in all countries and is also challenging to administer in infants and young children.17,103
Direct Pancreatic Function Tests
Direct (stimulatory) pancreatic function tests such as classical double-balloon Dreiling tube (oroduodenal tube) test are rarely performed in children due to their invasiveness, discomfort, and radiation. More frequently, endoscopic testing with CCK or secretin stimulation is performed.2,92,98,104 Direct tests have higher sensitivity and specificity but are expensive and not available at all centers. Here, measurement of pancreatic enzymes and bicarbonate via endoscopic collection and testing can be done but mostly at tertiary centers. The testing should be carefully interpreted (isolated enzyme deficiency) in children younger than two years of age as enzyme activities mature with age.105
Radiological Tests
Ultrasound is often the first radiological test used to evaluate the pancreatic anatomy but computer tomography (CT), or magnetic resonance cholangiopancreatography (MRCP), specifically secretin-enhanced MRCP, are more helpful to better delineate the pancreatic anatomy such as pancreatic atrophy, pancreatic lipomatosis and evidence of chronic pancreatitis such as pancreatic ductal disruptions, parenchymal calcifications106,107 (Figure 1A-D). In young children, the CT or MRCP testing may require deep sedation or anesthesia. Also, the availability and interpretation of s-MRCP is limited to academic centers. In addition to anatomic details, endoscopic ultrasound (EUS) is also helpful in procuring biopsy in cases when a tissue diagnosis is needed. The availability of EUS with advanced endoscopist is also limited to select pediatric academic centers.
 |
Figure 1 (A) Computed tomography (axial view) of a 8-year-old patient with cystic fibrosis showing pancreatic atrophy and pancreatic calcifications (white arrow). (B) Magnetic resonance cholangiopancreatography (axial view) of an infant with exocrine pancreatic insufficiency from Pearson syndrome showing pancreatic atrophy with fatty replacement (white arrow). (C) Computed tomography (axial view) of a 5-year-old patient with exocrine pancreatic insufficiency from tropical calcific pancreatitis showing extensive pancreatic calcifications (white arrow). (D) Magnetic resonance enterography (coronal view) of a 4-year-old patient with exocrine pancreatic insufficiency showing annular pancreas (white arrow) and pancreatic hypoplasia with small body and tail of the pancreas (black arrow). |
Management of EPI
PERT is the mainstay of EPI treatment along with fat-soluble vitamin supplementation. Dosage recommendations of PERT in children are predominantly inherited from expert recommendations of CF management. Both European and North American CF guidelines outlined PERT dosage for different age groups.96,108–111
Currently, the PERT dose is based on age and quantity/quality of food intake. Another way to dose is based on the fat content of the food.96,108 PERT doses are calculated either based on body weight or on the amount of fat ingested. Calculation of PERT based on fat intake is considered more physiological, accurate, and superior but cumbersome and difficult for some patients and families.112 In infants, 2000–4000 lipase units/120 mL of infant formula (about 1600 lipase units/gram of fat ingested per day) or per breastfeeding is recommended with a maximum daily dose of 10,000 lipase units/day.96,108–111 For children 1–4 years of age, a starting dose of 1000 lipase units/kg for meals and 500 lipase units/kg for snacks is recommended.96,108 Similarly, for children >4 years of age, recommended starting dose is 500 lipase units/kg/meal and 250 lipase units/kg per snack with a dose of 1000–2500 lipase units/kg/meal or 2000–4000 lipase units/gram of dietary fat with a daily maximum 10,000 lipase units/kg/day.96,108
A lower effective dose can be started and slowly increased for response with a daily dose not exceeding the daily maximum 10,000 lipase units/kg/day or 2500 lipase units/kg/meal.96,108 This maximum dosage of PERT in CF evolved in the 1990s in both the US and Europe as a result of the development of a complication known as fibrosing colonopathy in CF.110,113,114 The median dose consumed by children who developed fibrosing colonopathy was 13,393 USP lipase units/kg/day.110,113,114 In the 1990s, PERT were often over-filled and dosage higher than 6000 lipase/kg/meal was thought to be the contributing factor for fibrosing colonopathy.109,115 With the current daily recommendation of maximum dosage of PERT, the cases of fibrosing colonopathy were no longer being reported.116
There is little evidence-based literature on how to provide PERT via the enteral feeding tube.117,118 If patients can take PERT orally, experts recommend administration of PERT at the beginning of bolus feeds (or any feeds lasting less than 3 hours duration).118 For feeds lasting longer than 3 hours, the estimated PERT dose, based on the fat content of the enteral feeding, can be divided and given every 3 hours.118 For continuous night time feeds, a conventional practice is to provide PERT capsules 50% orally at the beginning and the remaining 50% at the end of nocturnal enteral feedings.118,119
RelizorbTM (immobilized lipase cartridge) was approved by the FDA for adults with EPI in 2015, for children over five years of life in 2017, and recently got approved for use in children 2 and older. This cartridge connects in-line with enteral tube feedings and provides continuous fat absorption through the course of night-time tube feedings. Studies have shown increased omega-3 fatty acid levels and decreased subjective symptoms of malabsorption.120,121 One cartridge is recommended for 500 mL of feeds with a maximum of two in-line cartridges.120,121
The Cystic Fibrosis Foundation (CFF) does not recommend using generic, non-proprietary PERT.96 Similarly, CFF recommends against using over the counter PERT as they are not tested for efficacy or safety.96 Registered dietitian is an indispensable member of the EPI care team and provides general guidance regarding enzyme adjustments based on the fat content of foods and beverages. In patients with non-CF EPI, a lower range of the above mentioned doses are generally enough to successfully manage symptoms of EPI and achieve optimal nutritional status.
CF (EPI)-specific fat-soluble vitamins are recommended for patients with EPI.122 Serum levels of fat-soluble vitamins should be evaluated at diagnosis, monitored approximately 3–6 months after initiating fat-soluble vitamin supplementation (or after dose modification), and thereafter every year.32,96 The doses of PERT and fat-soluble vitamins should be regularly monitored by a dietitian with EPI expertise and doses should be adjusted to prevent deficiencies. Adjustment of dose of additional fat-soluble vitamin supplementation is warranted based on the levels.32,96
Specific Challenges of PERT Administration in Children
Treatment with PERT poses unique challenges in infants and young children. For infants and young children who are unable to swallow capsules, encapsulated microbead or microsphere PERT formulations can be used. The enzymes are administered by opening the capsules and sprinkling the contents onto soft food mixtures with a pH of 4.5 or less (eg, applesauce, banana, sweet potatoes, or pureed apricot) and fed via a small spoon before each feeding.13 PERT microbeads should not be mixed with food with a pH >7 (eg, milk) for a prolonged period of time as their enteric coating will dissolve, exposing the enzymes which will be inactivated by the exposure to gastric acid.13 The beads should not be chewed or crushed. Acid suppression medications (H2 receptor antagonists or proton pump inhibitors) can be used to enhance the efficacy.13 To prevent oral mucosal ulceration from the digestive enzymes, parents are advised to ensure that no beads remain in the baby’s mouth after feeding by sweeping the mouth after PERT administration. In breastfed babies, enzymes left on the breast may also cause ulcerations. Also, infants can develop perianal irritation from faster transit of pancreatic enzymes and use a barrier cream to prevent this complication.
However, in infants, the maximum daily dose may be transiently exceeded. The data from the CFF registry revealed that the mean dosage per feeding in infants was approximately 1500 lipase units/kg/feed (range 641–3653 lipase units/kg/feed), and as most infants are fed every 2–3 hours, the total daily maximum was easily surpassed at least for a short period with no further documented increase in fibrosing colonopathy cases.110
Poor weight gain and persistence of other subjective EPI symptoms can be encountered despite adequate PERT dosage.112 As children are growing, frequent assessments of dosing adjustments should be done based on their weight gain. Also, assessment of adherence and administration technique is recommended before dosing changes. PERT administration in children may pose unique challenges in the school environment.112 PERT should be taken just before or during the meal. Many schools may recommend PERT be administered at the nurse’s office before going to the cafeteria and if the time interval is long (>30 minutes), PERT may not be effective.112 Due to decreased secretion of bicarbonate from the pancreas, gastric chyme may not be sufficiently neutralized, leading to inadequate dissolution of enteric coating and precipitation of bile acid. A trial of acid suppression or a PERT product with bicarbonate can be utilized in patients with persistent symptoms.123,124 If the response to PERT is inadequate with continued steatorrhea with or without poor weight gain, further evaluation is required125 (Table 4).
 |
Table 4 Factors to Consider for Poor Response to PERT and Persistence of EPI Symptoms |
Individual Syndromes Related to EPI in Children
Cystic Fibrosis
In CF, the pancreatic involvement starts in utero and continues into childhood.13,18 CFTR is highly expressed in pancreatic duct epithelium and controls anion secretion into the lumen.13,19 CFTR dysfunction impedes fluid and anion (Cl− and HCO3−) secretion, which results in acidic luminal contents with thick secretions leading to obstruction of ducts and eventual destruction of the pancreas resulting in EPI.13,19 These viscid secretions plugging pancreatic ducts have been documented even in preterm infants. This destructive process results in an increase in the serum trypsinogen levels, which is the basis for IRT in CF newborn screening test.19 The pathophysiology of pancreatic insufficiency in CF also occurs due to mechanisms such as inhibition of endocytosis in acinar cells, prevailing inflammatory milieu, and imbalance in membrane lipids in CF-regulated cells.20 The defect in HCO3− secretion in CF also results in acidic duodenal fluid. This leads to the irreversible inactivation of lipase and other pancreatic enzymes.17 About 85% of CF patients are pancreatic insufficient.21 Nearly two-thirds of these infants have EPI at birth and another 15–20% develop EPI by school age.22,126,127
Mutations (either homozygous or compound heterozygous) classified as I–III and VI are associated with EPI status, and usually, IV and V have pancreatic sufficiency status.19,23 Pancreatic sufficient CF patients are more prone to acute pancreatitis (approximately 10–15%), and repeated episodes of pancreatitis may eventually lead to insufficiency.128 In CF, multiple factors are implicated as causes of malnutrition, namely decreased intake (reduced appetite, nausea, abdominal pain interfering with intake), maldigestion or malabsorption, excessive energy expenditure (inflammatory catabolism), increased losses, and the CFTR gene defect itself.24 The presence of CF-related liver disease and CF-related diabetes also further exacerbates malnutrition. In children, CF has more severe consequences with impairment of growth.24
There is a direct relationship between nutritional status and improved pulmonary function and subsequently survival.129 In the 1960s and 1970s, low fat diets were commonly utilized to manage the symptoms of fat malabsorption which resulted in severe growth failure.129 An elegant study by Corey et al compared children followed by Toronto and Boston CF centers revealed that Toronto children had improved growth and survival compared to children followed in Boston.129 The prime difference between these centers is, patients in Toronto had high energy, high fat diet whereas in Boston, children were fed high energy, fat restricted diet.129 The introduction of enteric coated enzymes in the form of microspheres in 1979 revolutionized the management of EPI.25 The introduction of newborn screening of CF has tremendously improved the nutrition and overall status of CF children than those diagnosed later with symptoms.130 Owing to these major interventions (early diagnosis, initiation of aggressive nutritional management and improvements in respiratory management), the median predicted survival published in 2021 was 52 years which has increased from ~28 years in the early 1990s.21(Figure 2)
 |
Figure 2 Median predicted survival age of patients with cystic fibrosis from the year 1990 to 2021 in five-year increments (reproduced with permission from the Cystic Fibrosis Foundation). Notes: Cystic fibrosis patients under care at CF Foundation-accredited care centers in the United States, who consented to have their data entered. Cystic Fibrosis Foundation Patient Registry, 2021 Annual Data Report, Bethesda, Maryland, ©2022 Cystic Fibrosis Foundation. |
PERT, along with high fat diet and CF (EPI)-specific vitamins is the mainstay of nutritional management of CF. The Cystic Fibrosis Foundation (CFF) recommends PERT for all infants with two CFTR mutations associated with PI and in infants with stool elastase <200 μg/gm of stool or coefficient of fat absorption (CFA) <85% (in infants <6 months of age), or infants with unequivocal manifestations of malabsorption.96 In pancreatic sufficient CF patients with recurrent episodes of pancreatitis, stool elastase testing is recommended to screen for EPI.108
For infants and children who are not gaining weight, feeding difficulties due to oral aversion should be evaluated. Oral aversion could be due to immature oral skills or behavioral feeding refusal. In a study involving children with CF and EPI, the behavioral and aggressive nutrition intervention improved energy intake and height for age Z score, but not weight for age Z score in preschoolers 2–6 years of age.131 Failure to thrive in CF infants needs additional evaluation for salt deficiency, essential fatty acid deficiency, and zinc deficiency. Short term zinc supplementation (1 mg elemental zinc/kg/day in divided doses) can be provided empirically in infants who have difficulty gaining weight.96 Hyponatremic dehydration can occur in infants and children with CF due to increased losses of salt in sweat, and low salt content of commercial baby foods.96,132 Salt supplementation is recommended to prevent deficiency; 1/8th teaspoon of table salt (~2.5 mEq) per day for less than 6 months of age, increasing to 1/4th teaspoon of table salt per day for greater than 6 months of age.96 In adults with CF, the BMI targets for men is 23 kg/M2 for men and greater than 22 kg/M2 for women. In children, due to their active growth, age and gender-specific anthropometric percentiles are used. For infants and children less than two years, the target anthropometric measures include weight for length greater/equal to 50th percentile and for children 2–18 years, BMI > or equal to 50th percentile for healthy controls of same age and gender.108,125 Specific pediatric CF guidelines include glucose tolerance testing around 10 years of age to screen for CFRD and also, bone mineral density screening recommended at 8–10 years of age.108
The European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) and the European Cystic Fibrosis Society (ECFS) guidelines and the CFF guidelines recommend providing 110–200% of energy requirements in patients with CF to maintain nutritional status with the targeted anthropometric measurements.108 A variety of high-calorie oral supplements are available on the market which are often recommended for CF patients who are having trouble achieving weight gain targets. Supplemental gastrostomy tube feeds (as a nocturnal infusion) are often recommended in children when oral intake is not adequate to provide the needed calories.120,121 About 10–15% of CF patients in the US require tube feeding.21,120
Ivacaftor, a small-molecule potentiator of CFTR utilized in gating mutations, has been shown to improve pancreatic function in some patients.133–137 Patients 2 years and older with at least one mutation of F508del are now eligible for the triple combination elexacaftor-tezacaftor-ivacaftor, and their efficacy on pancreatic function in early years of life remains elusive.138,139
Shwachman-Diamond Syndrome
SDS is characterized by EPI, bone marrow failure, and a predisposition towards myelodysplasia or acute leukemia (mainly acute myeloid leukemia).33–36 Most patients with SDS present in infancy with EPI symptoms or with recurrent infections.37 The presentation is variable with nearly half of the patients present with steatorrhea and neutropenia.33,36 Neutropenia is the most common hematological abnormality.33 A positive family history, hypocellular marrow, and other congenital anomalies are often noted.33–35
Pancreatic insufficiency is due to a decrease in pancreatic acinar cell mass and symptoms are present in 90% of patients during infancy.35–37,140 Here, the pancreatic ductal architecture and islets are unaffected and pancreatic acini are replaced by fat.38 Similarly, the islet cells are unaffected. Radiological tests such as ultrasound, CT, or MRCP could reveal pancreatic lipomatosis.44
Given the rarity, most management guidelines are a result of expert consensus opinion.36 Prior to the advent of genetic analysis, the diagnosis of SDS was essentially based on bone marrow and pancreatic abnormalities. Dror and colleagues outlined the current diagnostic criteria which incorporates biallelic SBDS gene mutations along with hematological findings (neutropenia which may be cyclic or chronic, anemia, thrombocytopenia, hypocellular bone marrow), pancreatic insufficiency (reduced trypsinogen in children <3 years of age and reduced isoamylase >3 years of age), and pancreatic lipomatosis on imaging, reduced stool elastase or elevated fecal fat excretion based on CFA).32,37 Skeletal abnormalities include short stature with or without metaphyseal dysplasia, and rib cage abnormalities.36,37,141 Hepatomegaly and elevated liver enzymes are noted in many patients.32,39 Other findings such as skeletal anomalies such as neurocognitive problems, height <3rd percentile, and first-degree family member with SDS.32 Recurrent infections such as pneumonia, otitis media, bone infections, and sepsis are frequently noted. Nearly half the patients may present with failure to thrive.39
Management by a multidisciplinary team is recommended, and the essential members of the team include hemato-oncologist, gastroenterologist, dietician, geneticist, endocrinologist, orthopedic specialist, physical and occupational therapist, social worker, and pharmacist. Many SDS patients have an age-related improvement in pancreatic impairment over time and beyond four years, nearly 50% will stop requiring PERT.35–37 Neutropenia rarely assumes clinical significance and responds well to low doses of granulocyte colony stimulating factor (G-CSF). Many SDS patients have responded excellently to stem cell transplantation.
Johanson-Blizzard Syndrome
Most patients exhibit characteristic phenotypical features such as alae nasi hypoplasia or aplasia, scalp defects (cutis aplasia), hearing impairment, dental anomalies, microcephaly, cardiac anomalies, hypothyroidism, imperforate anus, and urogenital defects along with EPI.45–49 Most patients have severe growth and developmental delay.45–47 Similar to SDS, patients with Johanson-Blizzard syndrome have a primary failure of pancreatic acinar development and are replaced by fat and connective tissue.16,50,51 Unlike CF, the pancreatic ductular secretion of anions and fluids is unaffected.51 If pancreatic insufficiency is not aggressively treated, infants may succumb to malnutrition and overwhelming infections.46
Pearson-Marrow Pancreas Syndrome
EPI is noted in half the patients and unlike, SDS the pancreas is fibrotic.53 The exact pathogenesis for EPI is unclear.54 The pancreas is normal in-utero and pancreatic damage occurs postnatally. Due to defects in oxidative phosphorylation, mitochondrial energy production is impaired which may render the pancreas prone to damage by reactive oxygen species.16 Patients with this disorder have poor prognosis and the majority of deaths are secondary to multi-organ failure, metabolic acidosis, and severe infections.55 Kearns-Sayre syndrome is a similar but milder phenotype predominantly involving muscles with progressive dysfunction.16,56
Jeune Syndrome
Jeune syndrome or asphyxiating thoracic dystrophy was first described in 1955.59 It is a rare autosomal recessive disorder with a global incidence estimated at 1:100,000 to 1:130,000 live births. The disease phenotype is variable. It is characterized by musculoskeletal abnormalities including a small, narrow chest leading to respiratory distress and, short-limbed dwarfism.60 EPI is due to pancreatic fibrosis.61,62 Renal abnormalities (cystic dysplasia/nephronophthisis), hepatic and pancreatic fibrosis, and ocular complications may occur later in life.16,60
Isolated Enzymes Deficiencies
Isolated deficiencies of lipase, colipase, trypsinogen, and enterokinase have been described.75 Enterokinase is found on the intestinal brush border and is responsible for the activation of trypsinogen and chymotrypsinogen and hence its deficiency could lead to protein malabsorption. The deficiency of trypsinogen or enterokinase is characterized by failure to thrive, hypoalbuminemia, and edema. Isolated amylase deficiency is usually due to a maturational defect in young children and resolves by 2–3 years of age and does not produce clinical symptoms as the functional capacity of salivary amylase is enough to compensate well to prevent symptoms.142
Pancreatic Agenesis
Genes causing pancreatic agenesis are rare and described in Table 2. Patients with partial agenesis or hypoplasia remain asymptomatic given the high pancreatic functional reserve.51
Chronic Pancreatitis Leading to EPI
In adults, most patients have CP secondary to alcoholism and biliary tract diseases. In children and adolescents, majority of CP are associated with genetic abnormalities (mutations such as PRSS1, CFTR, SPINK1, and CTRC), biliary obstruction (eg, pancreas divisum, sphincter of Oddi dysfunction, gallstones), and drug toxicity or side-effects12,143 (Table 1). In a multi-center evaluation by the International Study Group of Pediatric Pancreatitis: In Search for a Cure (INSPPIRE) consortium, 26–30% of children with acute recurrent pancreatitis or CP were found to have EPI which is similar to adults (33%).143–145 Alcohol and tobacco were noted in about 50% of adults as risk factors, whereas in children, alcohol and tobacco consumption were associated with 1% and 7% respectively.143 In the study by Schwarzenberg and colleagues, 18% of patients developed EPI within six years of their first episode of acute pancreatitis.143 Tropical chronic pancreatitis is a rare condition noted in south India, Sri Lanka, and other tropical countries in which children often have recurrent episodes of pancreatitis, which leads to pancreatic insufficiency.146 The exact etiology is unclear, but malnutrition, nutrient deficiencies, dietary cyanogen toxicity, and an underlying genetic predisposition have been proposed.146
In another INSPPIRE study, within six years of the initial acute pancreatitis episode, the cumulative proportion with EPI was noted as 18% (95% CI: 12.4–25.6%).147 EPI patients may present clinically with manifestations of malabsorption or can be subclinical.92 Many experts recommend PERT if fecal elastase is <100 μg/g or 72-hour fecal fat (coefficient of fat absorption >15% in infants less than six months and >7% for patients greater than 6 months).145,148 PERT dosages in CP management in children are adopted from CF guidelines.148 For children younger than four years, 1000 lipase units/kg/meal and for children older than four years, 1000–2500 lipase units/kg/meal.148 For snacks, half the PERT dose is recommended.148 Similar to CF management guidelines, the daily maximum dose of <10,000 lipase units/kg is recommended to prevent fibrosing colonopathy.114
Conclusions
Despite many similarities, etiological conditions causing EPI in children are unique from adults and specific challenges exist in the management of EPI in children. Clinicians should have a high index of suspicion for early diagnosis and aggressive management to optimize the clinical outcome. Children with EPI should be evaluated frequently for nutritional assessments. The literature regarding the management of EPI in children is based on experience gained from CF management, and evidence-based management of pther causes of EPI in children is limited. Pancreatic enzyme replacement therapy (PERT) is the cornerstone of EPI treatment along with fat-soluble vitamin supplementation. PERT can either be dosed based on the age and diet or more accurately based on the fat content of the food, (2000–4000 lipase units/gram fat). The PERT dosage can be slowly titrated for response with a daily dose not exceeding 10,000 lipase units/kg/day or 2500 lipase units/kg/meal. Multidisciplinary intervention is the key to successful management and favorable outcomes.
Funding
There is no funding to report.
Disclosure
SS has no disclosures; TS has the following to declare: Chiesi (advisor and speaker’s bureau), AbbVie (speaker’s bureau), and Nestlé (advisor).
References
1. Löhr JM, Dominguez-Munoz E, Rosendahl J, et al. United European Gastroenterology evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis (HaPanEU). United Eur Gastroenterol j. 2017;5(2):153–199. doi:10.1177/2050640616684695
2. Bitton S, Pettei MJ. Exocrine pancreatic insufficiency. Pediatrics in Review. 2016;37(2):85–87. doi:10.1542/pir.2015-0084
3. DiMagno EP, Go VL, Summerskill W. Relations between pancreatic enzyme outputs and malabsorption in severe pancreatic insufficiency. N Eng J Med. 1973;288(16):813–815. doi:10.1056/NEJM197304192881603
4. Capurso G, Traini M, Piciucchi M, Signoretti M, Arcidiacono PG. Exocrine pancreatic insufficiency: prevalence, diagnosis, and management. Clin Exp Gastroenterol. 2019;129–139. doi:10.2147/CEG.S168266
5. Singh VK, Haupt ME, Geller DE, Hall JA, Diez PMQ. Less common etiologies of exocrine pancreatic insufficiency. World J Gastroenterol. 2017;23(39):7059. doi:10.3748/wjg.v23.i39.7059
6. Dominguez-Munoz JE, Drewes AM, Lindkvist B, et al. Recommendations from the United European Gastroenterology evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis. Pancreatology. 2018;18(8):847–854. doi:10.1016/j.pan.2018.09.016
7. Shimizu K, Ito T, Irisawa A, et al. Evidence-based clinical practice guidelines for chronic pancreatitis 2021. J Gastroenterol. 2022;57(10):709–724. doi:10.1007/s00535-022-01911-6
8. Phillips ME, Hopper AD, Leeds JS, et al. Consensus for the management of pancreatic exocrine insufficiency: UK practical guidelines. BMJ open Gastroenterol. 2021;8(1):e000643. doi:10.1136/bmjgast-2021-000643
9. Toouli J, Biankin AV, Oliver MR, Pearce CB, Wilson JS, Wray NH. Management of pancreatic exocrine insufficiency: Australasian Pancreatic Club recommendations. Med j Australia. 2010;193(8):461–467. doi:10.5694/j.1326-5377.2010.tb04000.x
10. Sabater L, Ausania F, Bakker OJ, et al. Evidence-based guidelines for the management of exocrine pancreatic insufficiency after pancreatic surgery. Ann Surg. 2016;264(6):949–958. doi:10.1097/SLA.0000000000001732
11. Gardner TB, Adler DG, Forsmark CE, Sauer BG, Taylor JR, Whitcomb DC. ACG clinical guideline: chronic pancreatitis. Official j Am Coll Gastroenterol ACG. 2020;115(3):322–339. doi:10.14309/ajg.0000000000000535
12. Forsmark CE. Diagnosis and management of exocrine pancreatic insufficiency. Curr Treat Options Gastroenterol. 2018;16(3):306–315. doi:10.1007/s11938-018-0186-y
13. Uc A, Fishman DS. Pancreatic disorders. Pediatric Clin. 2017;64(3):685–706. doi:10.1016/j.pcl.2017.01.010
14. Gheorghe C, Seicean A, Saftoiu A, et al. Romanian guidelines on the diagnosis and treatment of exocrine pancreatic insufficiency. J Gastrointestin Liver Dis. 2015;24(1):117–123. doi:10.15403/jgld.2014.1121.app
15. Sankararaman S, Schindler T, Sferra TJ. Management of Exocrine Pancreatic Insufficiency in Children. Nutrition Clin Practice. 2019;34(S1):S27–S42. doi:10.1002/ncp.10388
16. Stormon MO, Durie PR. Pathophysiologic basis of exocrine pancreatic dysfunction in childhood. J Pediatr Gastroenterol Nutr. 2002;35(1):8–21. doi:10.1097/00005176-200207000-00004
17. Taylor CJ, Chen K, Horvath K, et al. ESPGHAN and NASPGHAN report on the assessment of exocrine pancreatic function and pancreatitis in children. J Pediatr Gastroenterol Nutr. 2015;61(1):144–153. doi:10.1097/MPG.0000000000000830
18. Sturgess JM. Structural and developmental abnormalities of the exocrine pancreas in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1984;3:S55–66. doi:10.1097/00005176-198400031-00011
19. Wilschanski M, Durie PR. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut. 2007;56(8):1153–1163. doi:10.1136/gut.2004.062786
20. Freedman SD, Blanco P, Shea JC, Alvarez JG. Mechanisms to explain pancreatic dysfunction in cystic fibrosis. Med Clin North Am. 2000;84(3):657–664. doi:10.1016/S0025-7125(05)70248-0
21. Cystic Fibrosis Foundation, 2021 Patient Registry Annual Data Report. Available from: https://www.cff.org/Research/Researcher-Resources/Patient-Registry.
22. Guy-Crotte O, Carrère J, Figarella C. Exocrine pancreatic function in cystic fibrosis. Eur J Gastroenterol Hepatol. 1996;8(8):755–759.
23. Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. doi:10.1016/0092-8674(93)90353-R
24. Gaskin K. Nutritional care in children with cystic fibrosis: are our patients becoming better? Eur J Clin Nutr. 2013;67(5):558–564. doi:10.1038/ejcn.2013.20
25. Altman K, McDonald CM, Michel SH, Maguiness K. Nutrition in cystic fibrosis: from the past to the present and into the future. Pediatr Pulmonol. 2019;54(S3):S56–S73. doi:10.1002/ppul.24521
26. Southern KW, Munck A, Pollitt R, et al. A survey of newborn screening for cystic fibrosis in Europe. J Cystic Fibrosis. 2007;6(1):57–65. doi:10.1016/j.jcf.2006.05.008
27. Salvatore D, Buzzetti R, Baldo E, et al. An overview of international literature from cystic fibrosis registries. Part 3. Disease incidence, genotype/phenotype correlation, microbiology, pregnancy, clinical complications, lung transplantation, and miscellanea. J Cystic Fibrosis. 2011;10(2):71–85. doi:10.1016/j.jcf.2010.12.005
28. Hollander FM, de Roos NM, Heijerman HG. The optimal approach to nutrition and cystic fibrosis: latest evidence and recommendations. Curr Opin Pulm Med. 2017;23(6):556–561. doi:10.1097/MCP.0000000000000430
29. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am j Dis Children. 1938;56(2):344–399. doi:10.1001/archpedi.1938.01980140114013
30. Gorter R, Karimi A, Sleeboom C, Kneepkens C, Heij H. Clinical and genetic characteristics of meconium ileus in newborns with and without cystic fibrosis. J Pediatr Gastroenterol Nutr. 2010;50(5):569–572. doi:10.1097/MPG.0b013e3181bb3427
31. Cade A, Walters M, McGinley N, et al. Evaluation of fecal pancreatic elastase‐1 as a measure of pancreatic exocrine function in children with cystic fibrosis. Pediatr Pulmonol. 2000;29(3):172–176. doi:10.1002/(SICI)1099-0496(200003)29:3<172::AID-PPUL3>3.0.CO;2-1
32. Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman‐Diamond syndrome. Ann N Y Acad Sci. 2011;1242(1):40–55. doi:10.1111/j.1749-6632.2011.06349.x
33. Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman–Diamond syndrome: update from the North American Shwachman–Diamond syndrome registry. J Pediatr. 2014;164(4):866–870. doi:10.1016/j.jpeds.2013.11.039
34. Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman–Diamond syndrome: an update. Hematology/Oncol Clin. 2013;27(1):117–128. doi:10.1016/j.hoc.2012.10.003
35. Mack DR, Forstner GG, Wilschanski M, Freedman MH, Durie PR. Shwachman syndrome: exocrine pancreatic dysfunction and variable phenotypic expression. Gastroenterology. 1996;111(6):1593–1602. doi:10.1016/S0016-5085(96)70022-7
36. Nelson AS, Myers KC. Diagnosis, treatment, and molecular pathology of Shwachman-Diamond syndrome. Hematology/Oncol Clin. 2018;32(4):687–700. doi:10.1016/j.hoc.2018.04.006
37. Ginzberg H, Shin J, Ellis L, et al. Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pediatr. 1999;135(1):81–88. doi:10.1016/S0022-3476(99)70332-X
38. Cipolli M. Shwachman-Diamond syndrome: clinical phenotypes. Pancreatology. 2001;1(5):543–548. doi:10.1159/000055858
39. Ikuse T, Kudo T, Arai K, et al. Shwachman–Diamond syndrome: nationwide survey and systematic review in Japan. Pediatrics Int. 2018;60(8):719–726. doi:10.1111/ped.13601
40. Goobie S, Popovic M, Morrison J, et al. Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7. Am J Human Genetics. 2001;68(4):1048–1054. doi:10.1086/319505
41. Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman–Diamond syndrome. Nat Genet. 2003;33(1):97–101. doi:10.1038/ng1062
42. Shwachman H, Diamond LK, Oski FA, Khaw K-T. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65(5):645–663. doi:10.1016/S0022-3476(64)80150-5
43. Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917–929. doi:10.1101/gad.623011
44. Nijs E, Callahan MJ, Taylor GA. Disorders of the pediatric pancreas: imaging features. Pediatr Radiol. 2005;35(4):358–373. doi:10.1007/s00247-004-1326-1
45. Johanson A, Blizzard R. A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J Pediatr. 1971;79(6):982–987. doi:10.1016/S0022-3476(71)80194-4
46. Civelek Z, Urganci N, Usta M, Celik M. A rare cause of pancreatic insufficiency; Johanson Blizzard Syndrome. J Pak Med Assoc. 2018;68(5):801–803.
47. Almashraki N, Abdulnabee MZ, Sukalo M, Alrajoudi A, Sharafadeen I, Zenker M. Johanson-Blizzard syndrome. World J Gastroenterol. 2011;17(37):4247. doi:10.3748/wjg.v17.i37.4247
48. Zenker M, Mayerle J, Lerch MM, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat Genet. 2005;37(12):1345–1350. doi:10.1038/ng1681
49. Scheers I, Berardis S. Congenital etiologies of exocrine pancreatic insufficiency. Front Pediatrics. 2022;10:909925. doi:10.3389/fped.2022.909925
50. Jones NL, Hofley PM, Durie PR. Pathophysiology of the pancreatic defect in Johanson-Blizzard syndrome: a disorder of acinar development. J Pediatr. 1994;125(3):406–408. doi:10.1016/S0022-3476(05)83286-X
51. Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology. 2007;132(2):745–762. doi:10.1053/j.gastro.2006.12.054
52. Liu S, Wang Z, Jiang J, et al. Severe forms of Johanson-Blizzard syndrome caused by two novel compound heterozygous variants in UBR1: clinical manifestations, imaging findings and molecular genetics. Pancreatology. 2020;20(3):562–568. doi:10.1016/j.pan.2020.01.007
53. Reynolds E, Byrne M, Ganetzky R, Parikh S. Pediatric single large-scale mtDNA deletion syndromes: the power of patient reported outcomes. Mol Genet Metab. 2021;134(4):301–308. doi:10.1016/j.ymgme.2021.11.004
54. Yoshimi A, Ishikawa K, Niemeyer C, Grünert SC. Pearson syndrome: a multisystem mitochondrial disease with bone marrow failure. Orphanet J Rare Dis. 2022;17(1):1–11. doi:10.1186/s13023-022-02538-9
55. Pronman L, Rondinelli M, Burkardt DDC, Velayuthan S, Khalili AS, Bedoyan JK. Pearson syndrome: a rare cause of failure to thrive in infants. Clin Pediatr (Phila). 2019;58(7):819–824. doi:10.1177/0009922819834285
56. Rötig A, Bourgeron T, Chretien D, Rustin P, Munnich A. Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum Mol Genet. 1995;4(8):1327–1330. doi:10.1093/hmg/4.8.1327
57. Pearson HA, Lobel JS, Kocoshis SA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95(6):976–984. doi:10.1016/S0022-3476(79)80286-3
58. Rotig A, Colonna M, Bonnefont J, et al. Mitochondrial DNA deletion in Pearson’s marrow/pancreas syndrome. Lancet. 1989;333(8643):902–903. doi:10.1016/S0140-6736(89)92897-3
59. Jeune BC, Carron R. Dystrophie thoracique asphyxiante de caractère familial. Arch Fr Pediatr. 1955;12:866–891.
60. De Vries J, Yntema J, Van Die C, Crama N, Cornelissen E, Hamel B. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. Eur J Pediatr. 2010;169(1):77. doi:10.1007/s00431-009-0991-3
61. Karjoo M, Koop CE, Cornfeld D, Holtzapple PG. Pancreatic exocrine enzyme deficiency associated with asphyxiating thoracic dystrophy. Arch Dis Child. 1973;48(2):143. doi:10.1136/adc.48.2.143
62. Turkel S, Diehl E, Richmond J. Necropsy findings in neonatal asphyxiating thoracic dystrophy. J Med Genet. 1985;22(2):112–118. doi:10.1136/jmg.22.2.112
63. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106–110. doi:10.1038/ng0197-106
64. Thomas IH, Saini NK, Adhikari A, et al. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF‐1 Pro63fsX60 mutation. Pediatr Diabetes. 2009;10(7):492–496. doi:10.1111/j.1399-5448.2009.00526.x
65. Nicolino M, Claiborn KC, Senée V, Boland A, Stoffers DA, Julier C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes. 2010;59(3):733–740. doi:10.2337/db09-1284
66. Wright NM, Metzger DL, Borowitz SM, Clarke WL. Permanent neonatal diabetes mellitus and pancreatic exocrine insufficiency resulting from congenital pancreatic agenesis. Am j Dis Children. 1993;147(6):607–609. doi:10.1001/archpedi.1993.02160300013005
67. Winter WE, Maclaren NK, Riley WJ, Toskes PP, Andres J, Rosenbloom AL. Congenital pancreatic hypoplasia: a syndrome of exocrine and endocrine pancreatic insufficiency. J Pediatr. 1986;109(3):465–468. doi:10.1016/S0022-3476(86)80119-6
68. Sellick GS, Barker KT, Stolte-Dijkstra I, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301–1305. doi:10.1038/ng1475
69. Al‐Shammari M, Al‐Husain M, Al‐Kharfy T, Alkuraya FS. A novel PTF1A mutation in a patient with severe pancreatic and cerebellar involvement. Clin Genet. 2011;80(2):196–198. doi:10.1111/j.1399-0004.2010.01613.x
70. Hoveyda N, Shield JP, Garrett C, et al. Neonatal diabetes mellitus and cerebellar hypoplasia/agenesis: report of a new recessive syndrome. J Med Genet. 1999;36(9):700–704.
71. Allen HL, Flanagan SE, Shaw-Smith C, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44(1):20–22. doi:10.1038/ng.1035
72. Balasubramanian M, Shield J, Acerini C, et al. Pancreatic hypoplasia presenting with neonatal diabetes mellitus in association with congenital heart defect and developmental delay. Am J Med Genet A. 2010;152(2):340–346. doi:10.1002/ajmg.a.33194
73. Yorifuji T, Kawakita R, Hosokawa Y, Fujimaru R, Yamaguchi E, Tamagawa N. Dominantly inherited diabetes mellitus caused by GATA6 haploinsufficiency: variable intrafamilial presentation. J Med Genet. 2012;49(10):642–643. doi:10.1136/jmedgenet-2012-101161
74. Shteyer E, Saada A, Shaag A, Kidess R, Revel-Vilk S, Elpeleg O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Human Genetics. 2009;84(3):412–417. doi:10.1016/j.ajhg.2009.02.006
75. Behar DM, Basel-Vanagaite L, Glaser F, et al. Identification of a novel mutation in the PNLIP gene in two brothers with congenital pancreatic lipase deficiency. J Lipid Res. 2014;55(2):307–312. doi:10.1194/jlr.P041103
76. Sheldon W. Congenital pancreatic lipase deficiency. Arch Dis Child. 1964;39(205):268. doi:10.1136/adc.39.205.268
77. Figarella C, De Caro A, Leupold D, Poley J. Congenital pancreatic lipase deficiency. J Pediatr. 1980;96(3):412–416. doi:10.1016/S0022-3476(80)80683-4
78. Ligumsky M, Granot E, Branski D, Stankiewicz H, Goldstein R. Isolated lipase and colipase deficiency in two brothers. Gut. 1990;31(12):1416–1418. doi:10.1136/gut.31.12.1416
79. Figarella C, Negri GA, Sarles H. Presence of colipase in a congenital pancreatic lipase deficiency. Biochimica et Biophysica Acta. 1972;280(1):205–210. doi:10.1016/0005-2760(72)90227-5
80. Ghishan FK, Moran JR, Durie PR, Greene HL. Isolated congenital lipase—colipase deficiency. Gastroenterology. 1984;86(6):1580–1582. doi:10.1016/S0016-5085(84)80175-4
81. Szabó A, Xiao X, Haughney M, Spector A, Sahin-Tóth M, Lowe ME. A novel mutation in PNLIP causes pancreatic triglyceride lipase deficiency through protein misfolding. Biochim Biophys Acta. 2015;1852(7):1372–1379. doi:10.1016/j.bbadis.2015.04.002
82. Hildebrand H, Borgström B, Bekassy A, Erlanson-Albertsson C, Helin I. Isolated co-lipase deficiency in two brothers. Gut. 1982;23(3):243–246. doi:10.1136/gut.23.3.243
83. Davis RC, Xia Y, Mohandas T, Schotz MC, Lusis AJ. Assignment of the human pancreatic colipase gene to chromosome 6p21. 1 to pter. Genomics. 1991;10(1):262–265. doi:10.1016/0888-7543(91)90509-D
84. Townes PL. Trypsinogen deficiency disease. J Pediatr. 1965;66(2):275–285. doi:10.1016/S0022-3476(65)80184-6
85. Townes PL, Bryson MF, Miller G. Further observations on trypsinogen deficiency disease: report of a second case. J Pediatr. 1967;71(2):220–224. doi:10.1016/S0022-3476(67)80076-3
86. Townes PL. Trypsinogen deficiency and other proteolytic deficiency disease. Bi Defects OAS. 1972;7(2):95–101.
87. Holzinger A, Maier EM, Bück C, et al. Mutations in the proenteropeptidase gene are the molecular cause of congenital enteropeptidase deficiency. Am J Human Genetics. 2002;70(1):20–25. doi:10.1086/338456
88. Haworth J, Gourley B, Hadorn B, Sumida C. Malabsorption and growth failure due to intestinal enterokinase deficiency. J Pediatr. 1971;78(3):481–490. doi:10.1016/S0022-3476(71)80231-7
89. Ghishan FK, Lee P, Lebenthal E, Johnson P, Bradley CA, Greene HL. Isolated congenital enterokinase deficiency: recent findings and review of the literature. Gastroenterology. 1983;85(3):727–731. doi:10.1016/0016-5085(83)90033-1
90. Hadorn B, Tarlow M, Lloyd J, Wolff O. Intestinal enterokinase deficiency. Lancet. 1969;1(7599):812–813. doi:10.1016/S0140-6736(69)92071-6
91. Bartels RH, Meyer SL, Stehmann TA, Bourdon C, Bandsma RH, Voskuijl WP. Both exocrine pancreatic insufficiency and signs of pancreatic inflammation are prevalent in children with complicated severe acute malnutrition: an observational study. J Pediatr. 2016;174:165–170. doi:10.1016/j.jpeds.2016.04.013
92. Freeman AJ, Maqbool A, Bellin MD, et al. Medical Management of Chronic Pancreatitis in Children: a Position Paper by the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Pancreas Committee. J Pediatr Gastroenterol Nutr. 2021;72(2):324–340. doi:10.1097/MPG.0000000000003001
93. Hadorn B, Zoppi G, Shmerling D, Prader A, McIntyre I, Anderson CM. Quantitative assessment of exocrine pancreatic function in infants and children. J Pediatr. 1968;73(1):39–50. doi:10.1016/S0022-3476(68)80037-X
94. Freed LM, York CM, Hamosh P, Mehta NR, Hamosh M. Bile salt-stimulated lipase of human milk: characteristics of the enzyme in the milk of mothers of premature and full-term infants. J Pediatr Gastroenterol Nutr. 1987;6(4):598–604. doi:10.1097/00005176-198707000-00019
95. Haaber AB, Rosenfalck A, Hansen B, Hilsted J, Larsen S. Bone mineral metabolism, bone mineral density, and body composition in patients with chronic pancreatitis and pancreatic exocrine insufficiency. Int J Pancreatol. 2000;27(1):21–27. doi:10.1385/IJGC:27:1:21
96. Borowitz D, Robinson KA, Rosenfeld M, et al. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155(6):S73–S93. doi:10.1016/j.jpeds.2009.09.001
97. Rings EH, Minich DM, Vonk RJ, Stellaard F, Fetter WP, Verkade HJ. Functional development of fat absorption in term and preterm neonates strongly correlates with ability to absorb long-chain fatty acids from intestinal lumen. Pediatr Res. 2002;51(1):57–63. doi:10.1203/00006450-200201000-00011
98. Horvath K, Mehta DI, Hill ID. Assessment of exocrine pancreatic function during endoscopy in children. J Pediatr Gastroenterol Nutr. 2019;68(6):768–776. doi:10.1097/MPG.0000000000002230
99. Daftary A, Acton J, Heubi J, Amin R. Fecal elastase-1: utility in pancreatic function in cystic fibrosis. J Cystic Fibrosis. 2006;5(2):71–76. doi:10.1016/j.jcf.2006.01.005
100. Khan A, Vege SS, Dudeja V, Chari ST. Staging exocrine pancreatic dysfunction. Pancreatology. 2022;22(1):168–172. doi:10.1016/j.pan.2021.11.005
101. Walkowiak J, Sands D, Nowakowska A, et al. Early decline of pancreatic function in cystic fibrosis patients with class 1 or 2 CFTR mutations. J Pediatr Gastroenterol Nutr. 2005;40(2):199–201. doi:10.1097/00005176-200502000-00022
102. Vanga RR, Tansel A, Sidiq S, El-Serag HB, Othman MO. Diagnostic performance of measurement of fecal elastase-1 in detection of exocrine pancreatic insufficiency: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2018;16(8):1220–1228. e1224. doi:10.1016/j.cgh.2018.01.027
103. Domínguez-Muñoz JE, Nieto L, Vilariño M, Lourido MV, Iglesias-García J. Development and diagnostic accuracy of a breath test for pancreatic exocrine insufficiency in chronic pancreatitis. Pancreas. 2016;45(2):241–247. doi:10.1097/MPA.0000000000000434
104. Patel N, Sellers ZM, Grover A, et al. Endoscopic pancreatic function testing (ePFT) in children: a position paper from the NASPGHAN pancreas committee. J Pediatr Gastroenterol Nutr. 2021;72(1):144–150. doi:10.1097/MPG.0000000000002931
105. Cruz LA, Parniczky A, Mayhew A, et al. Utility of direct pancreatic function testing in children. Pancreas. 2017;46(2):177–182. doi:10.1097/MPA.0000000000000724
106. Swensson J, Zaheer A, Conwell D, Sandrasegaran K, Manfredi R, Tirkes T. Secretin-enhanced MRCP: how and why—AJR expert panel narrative review. AJR Am J Roentgenol. 2021;216(5):1139. doi:10.2214/AJR.20.24857
107. Hopson P, Smadi Y, Mehta V, Patel S, Mehta D, Horvath K. Assessment of exocrine pancreatic function in children and adolescents with direct and indirect testing. Front Pediatrics. 2022;10:908542. doi:10.3389/fped.2022.908542
108. Turck D, Braegger CP, Colombo C, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutrition. 2016;35(3):557–577. doi:10.1016/j.clnu.2016.03.004
109. Borowitz DS, Grand RJ, Durie PR. Use of pancreatic enzyme supplements for patients with cystic fibrosis in the context of fibrosing colonopathy. J Pediatr. 1995;127(5):681–684. doi:10.1016/S0022-3476(95)70153-2
110. Borowitz D, Gelfond D, Maguiness K, Heubi JE, Ramsey B. Maximal daily dose of pancreatic enzyme replacement therapy in infants with cystic fibrosis: a reconsideration. J Cystic Fibrosis. 2013;12(6):784–785. doi:10.1016/j.jcf.2013.05.011
111. Gelfond D, Heltshe SL, Skalland M, et al. Pancreatic enzyme replacement therapy use in infants with cystic fibrosis diagnosed by newborn screening. J Pediatr Gastroenterol Nutr. 2018;66(4):657. doi:10.1097/MPG.0000000000001829
112. Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in cystic fibrosis. J Cystic Fibrosis. 2017;16:S70–S78. doi:10.1016/j.jcf.2017.06.011
113. Smyth RL, Smyth A, Lloyd D, van Velzen D, Heaf DP. Strictures of ascending colon in cystic fibrosis and high-strength pancreatic enzymes. Lancet. 1994;343(8889):85–86. doi:10.1016/S0140-6736(94)90817-6
114. FitzSimmons SC, Burkhart GA, Borowitz D, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Eng J Med. 1997;336(18):1283–1289. doi:10.1056/NEJM199705013361803
115. Smyth R, O’Hea U, Burrows E, et al. Fibrosing colonopathy in cystic fibrosis: results of a case-control study. Lancet. 1995;346(8985):1247–1251. doi:10.1016/S0140-6736(95)91860-4
116. Konrad J, Eber E, Stadlbauer V. Changing paradigms in the treatment of gastrointestinal complications of cystic fibrosis in the era of cystic fibrosis transmembrane conductance regulator modulators. Paediatr Respir Rev. 2022;42:9–16. doi:10.1016/j.prrv.2020.12.001
117. Schwarzenberg SJ, Hempstead SE, McDonald CM, et al. Enteral tube feeding for individuals with cystic fibrosis: cystic Fibrosis Foundation evidence-informed guidelines. J Cystic Fibrosis. 2016;15(6):724–735. doi:10.1016/j.jcf.2016.08.004
118. Saxby PC, Kench A, King S, Crowder T, van der Haak N. CFNGAG. Nutrition Guidelines for Cystic Fibrosis in Australia and New Zealand. In: Bell SC, editor.Thoracic Society of Australia and New Zealand.Sydney;2017.
119. Schwarzenberg SJ, Borowitz D. Challenging barriers to an option for improved provision of enteral nutrition. J Cystic Fibrosis. 2019;18(4):447–449. doi:10.1016/j.jcf.2019.06.002
120. Freedman S, Orenstein D, Black P, et al. Increased fat absorption from enteral formula through an in-line digestive cartridge in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2017;65(1):97–101. doi:10.1097/MPG.0000000000001617
121. Stevens J, Wyatt C, Brown P, Patel D, Grujic D, Freedman SD. Absorption and safety with sustained use of RELiZORB evaluation (ASSURE) study in patients with cystic fibrosis receiving enteral feeding. J Pediatr Gastroenterol Nutr. 2018;67(4):527. doi:10.1097/MPG.0000000000002110
122. Available from: https://www.cff.org/Life-With-CF/Daily-Life/Fitness-and-Nutrition/Nutrition/Getting-Your-Nutrients/Vitamin-Comparison-Chart-for-CF-Specific-Multivitamins.pdf.
123. Gelfond D, Ma C, Semler J, Borowitz D. Intestinal pH and gastrointestinal transit profiles in cystic fibrosis patients measured by wireless motility capsule. Dig Dis Sci. 2013;58(8):2275–2281. doi:10.1007/s10620-012-2209-1
124. Konstan MW, Accurso FJ, Nasr SZ, Ahrens RC, Graff GR. Efficacy and safety of a unique enteric-coated bicarbonate-buffered pancreatic enzyme replacement therapy in children and adults with cystic fibrosis. Clin Investig (Lond). 2013;3(8):723. doi:10.4155/cli.13.62
125. van der Haak N, King SJ, Crowder T, Kench A, Painter C, Saxby N. Highlights from the nutrition guidelines for cystic fibrosis in Australia and New Zealand. J Cystic Fibrosis. 2020;19(1):16–25. doi:10.1016/j.jcf.2019.05.007
126. Couper R, Corey M, Durie P, Forstner G, Moore D. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr. 1995;127(3):408–413. doi:10.1016/S0022-3476(95)70072-2
127. Sergeev V, Chou FY, Lam GY, Hamilton CM, Wilcox PG, Quon BS. The extrapulmonary effects of cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Ann Am Thorac Soc. 2020;17(2):147–154. doi:10.1513/AnnalsATS.201909-671CME
128. De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics. 2005;115(4):e463–e469. doi:10.1542/peds.2004-1764
129. Corey M, McLaughlin F, Williams M, Levison H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41(6):583–591. doi:10.1016/0895-4356(88)90063-7
130. Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics. 2001;107(1):1–13. doi:10.1542/peds.107.1.1
131. Kumar S, Ooi CY, Werlin S, et al. Risk factors associated with pediatric acute recurrent and chronic pancreatitis: lessons from INSPPIRE. JAMA Pediatr. 2016;170(6):562–569. doi:10.1001/jamapediatrics.2015.4955
132. Özçelik U, Göçmen A, Kiper N, Coşkun T, Yilmaz E, Özgüç M. Sodium chloride deficiency in cystic fibrosis patients. Eur J Pediatr. 1994;153(11):829–831. doi:10.1007/BF01972892
133. Nichols A, Davies J, Jones D, Carr S. Restoration of exocrine pancreatic function in older children with cystic fibrosis on ivacaftor. Paediatr Respir Rev. 2020;35:99–102. doi:10.1016/j.prrv.2020.04.003
134. Davies JC, Wainwright CE, Sawicki GS, et al. Ivacaftor in infants aged 4 to< 12 months with cystic fibrosis and a gating mutation. Results of a two-part Phase 3 clinical trial. Am J Respir Crit Care Med. 2021;203(5):585–593. doi:10.1164/rccm.202008-3177OC
135. Megalaa R, Gopalareddy V, Champion E, Goralski JL. Time for a gut check: pancreatic sufficiency resulting from CFTR modulator use. Pediatr Pulmonol. 2019;54(8):E16–E18. doi:10.1002/ppul.24353
136. Munce D, Lim M, Akong K. Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr Pulmonol. 2020;55(12):3381–3383. doi:10.1002/ppul.25065
137. Ritivoiu M-E, Drăgoi CM, Matei D, et al. Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics. 2023;15(1):162. doi:10.3390/pharmaceutics15010162
138. Davies JC, Sermet-Gaudelus I, Naehrlich L, et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J Cystic Fibrosis. 2021;20(1):68–77. doi:10.1016/j.jcf.2020.07.023
139. Kapouni N, Moustaki M, Douros K, Loukou I. Efficacy and safety of elexacaftor-tezacaftor-ivacaftor in the treatment of cystic fibrosis: a systematic review. Children. 2023;10(3):554. doi:10.3390/children10030554
140. Thompson AS, Giri N, Gianferante DM, et al. Shwachman Diamond syndrome: narrow genotypic spectrum and variable clinical features. Pediatr Res. 2022;92(6):1671–1680. doi:10.1038/s41390-022-02009-8
141. McLennan TW, Steinbach HL. Shwachman’s syndrome: the broad spectrum of bony abnormalities. Radiology. 1974;112(1):167–173. doi:10.1148/112.1.167
142. Mehta DI, Wang HH, Akins RE, Wang L, Proujansky R. Isolated pancreatic amylase deficiency: probable error in maturation. J Pediatr. 2000;136(6):844–846. doi:10.1016/S0022-3476(00)96936-1
143. Schwarzenberg SJ, Uc A, Zimmerman B, et al. Chronic pancreatitis: pediatric and adult cohorts show similarities in disease progress despite different risk factors. J Pediatr Gastroenterol Nutr. 2019;68(4):566–573. doi:10.1097/MPG.0000000000002279
144. Schwarzenberg SJ, Bellin M, Husain SZ, et al. Pediatric chronic pancreatitis is associated with genetic risk factors and substantial disease burden. J Pediatr. 2015;166(4):890–896. e891. doi:10.1016/j.jpeds.2014.11.019
145. Abu-El-Haija M, Hornung L, Ellery K, et al. Bone health in children with recurrent and chronic pancreatitis: a multi-center cross sectional analysis. Pancreatology. 2023. doi:10.1016/j.pan.2023.08.006
146. Mohan V, Premalatha G, Pitchumoni C. Tropical chronic pancreatitis: an update. J Clin Gastroenterol. 2003;36(4):337–346. doi:10.1097/00004836-200304000-00012
147. Liu QY, Abu-El-Haija M, Husain SZ, et al. Risk factors for rapid progression from acute recurrent to chronic pancreatitis in children: report from INSPPIRE. J Pediatr Gastroenterol Nutr. 2019;69(2):206–211. doi:10.1097/MPG.0000000000002405
148. Párniczky A, Abu-El-Haija M, Husain S, et al. EPC/HPSG evidence-based guidelines for the management of pediatric pancreatitis. Pancreatology. 2018;18(2):146–160. doi:10.1016/j.pan.2018.01.001
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.