Back to Journals » Cancer Management and Research » Volume 12
Evodiamine Exerts Anticancer Effects Against 143B and MG63 Cells Through the Wnt/β-Catenin Signaling Pathway
Authors Yang S ![]() , Chen J, Tan T, Wang N, Huang Y, Wang Y, Yuan X, Zhang P, Luo J, Luo X
, Chen J, Tan T, Wang N, Huang Y, Wang Y, Yuan X, Zhang P, Luo J, Luo X
Received 11 November 2019
Accepted for publication 25 March 2020
Published 28 April 2020 Volume 2020:12 Pages 2875—2888
DOI https://doi.org/10.2147/CMAR.S238093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Rudolph Navari
Shengdong Yang,1 Jin Chen,2 Tao Tan,1 Nan Wang,1 Yanran Huang,1 Yuping Wang,1 Xiaohui Yuan,3 Ping Zhang,3 Jinyong Luo,3 Xiaoji Luo1
1Department of Orthopedics, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 2Department of Dermatology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3Key Laboratory of Clinical Diagnosis of Education Ministry, College of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, People’s Republic of China
Correspondence: Xiaoji Luo
Department of Orthopaedics, The First Affiliated Hospital of Chongqing Medical University, No 1 YouYi Road, Yuzhong, Chongqing 400016, People’s Republic of China
Tel +86 2389012820 Fax +86 2389012820 Email [email protected]
Jinyong Luo
Key Laboratory of Clinical Diagnosis of Education Ministry, College of Laboratory Medicine, Chongqing Medical University, 1 Yixueyuan Road, Chongqing 400016, People’s Republic of China
Tel +86 2368485239 Fax +86 2368485239 Email [email protected]
Background: Osteosarcoma is the most common primary malignant bone neoplasm and is associated with abysmal prognosis. There are limitations of current treatment methods. Therefore, developing new agents to treat osteosarcoma is exceptionally urgent.
Aim: This study aimed to evaluate the anticancer effects of evodiamine (EVO) on osteosarcoma cells and, meanwhile, to investigate the underlying mechanisms involved.
Materials and Methods: The effect of EVO on the proliferation of osteosarcoma was detected by MTT assay, crystal violet assay and colony formation assay. The effects of EVO on the migration and invasion of osteosarcoma were detected by wound-healing assay and transwell assay. The effect of EVO on apoptosis of osteosarcoma was measured by Hoechst 33258 staining and cell cycle assay. The protein expression levels were detected by Western blotting assay. The activity of Wnt/β-Catenin signaling pathway was detected by luciferase reporter assay and Western blotting assay.
Results: According to MTT, crystal violet and colony formation assay results, EVO significantly inhibited the cell proliferation in a dose-dependent manner. Hoechst 33258 staining assay revealed that EVO induced cell apoptosis in a concentration-dependent manner. Moreover, EVO inhibited the migration and invasion of the osteosarcoma cells. Mechanistic studies revealed that EVO suppresses metastatic through suppressing epithelial–mesenchymal transition (EMT) as indicated by elevating the expression of epithelial marker E‐cadherin and reducing the expression of mesenchymal markers N‐cadherin and vimentin, as well as EMT transcription factors Snail and MMPs. Subsequently, EVO induced cell cycle arrest at the G2/M phase that correlated with reduced levels of cyclin D1 protein, while the apoptotic effects of EVO were associated with the upregulation of Bax and Bad and a decrease in Bcl-2 protein levels. Furthermore, EVO exerted the anticancer effects by suppressing Wnt/β-catenin signal pathway in osteosarcoma cells.
Conclusion: In summary, EVO exhibited potent anticancer effects against human osteosarcoma cells and promoted apoptosis through suppressing Wnt/β-catenin signaling pathway. These results indicated that EVO may be regarded as a new approach for osteosarcoma treatment.
Keywords: evodiamine, osteosarcoma, anticancer, Wnt/β-catenin
Introduction
Osteosarcoma is the most common primary malignant bone neoplasm, which predominantly occurs among children and young adults.1 According to the recent data from the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) program, the incidence rate of osteosarcoma in the United States between 0 and 19 years of age from 2012 to 2016 has been 5.6%.2 It is associated with a high tendency of local invasion and early pulmonary metastasis, which leads to the poor prognosis of osteosarcoma.3 Moreover, the five-year overall survival rate of metastatic osteosarcoma patients is less than 20%.4 Due to the application of surgery, adjuvant chemotherapy and radiotherapy for osteosarcoma management, the long-term survival rate for localized osteosarcoma has risen to 60–70%.5,6 However, the development of therapeutic resistance and presentation of various severe toxic side effects restrict the administration of chemotherapy.7 Accordingly, the exploration of novel and efficient anticancer agents for osteosarcoma is urgently required.
In the past decades, many naturally derived compounds have attracted considerable attention for their anticancer effects.8,9 Evodiamine (EVO) is a famous alkaloid with a quinazolinocarboline skeleton, which was isolated from Evodia ruraecarpa.10 The biological activities of EVO have been widely investigated, including anti-obesity, anti-inflammatory, anti-atherosclerotic, neuroprotective, and anticancer effects.10 Among them, the anticancer activity of EVO with the multitargeting molecule is attractive. Previous studies evaluated the anticancer effects of EVO in a variety of cancer cell lines.11 The anticancer effects of EVO in cancer cells were related to the induction of apoptosis, as well as inhibition of proliferation, migration, cell cycle progression, and angiogenesis by affecting multitargets.12 EVO inhibited the proliferation of non-small cell lung cancer A549 cells through decreasing the activity of AKT/nuclear factor-κB (NF-κB) and Sonic hedgehog/GLI family zinc finger 1 (SHH/GLI1) signaling pathways.13 It was reported that EVO downregulated cell viability and inhibited cell cycle progression in human hepatocellular carcinoma (HCC) HepG2 cells by decreasing the p-Akt level and increasing the levels of apoptotic protein Bax, cleaved-caspase-3 and cleaved-PARP (poly ADP-ribose polymerase).14 EVO was reported to downregulate migration and upregulate apoptosis by inactivating phosphorylation of extracellular signal-regulated kinase (p-ERK) and activating p38 mitogen-activated protein kinase (MAPK) in human breast cancer MDA-MB-231 cells.15 EVO induced the apoptosis of human colorectal carcinoma cells COLO-205 via the upregulation of p53 and Bax/Bcl-2 ratio, as well as decreasing mitochondrial transmembrane potential.16 Through inhibition of expressions of β-catenin and VEGFa, EVO was shown to exert anticancer effects on HCCs (HepG2, SMMC-7721, H22) by downregulating angiogenesis.17 Similarly, recent studies reported that EVO inhibited the proliferation of human osteosarcoma 143B cells through inactivation of the PTEN/P13k/Akt pathway.18 Evidences indicated that EVO also induced growth inhibition and inactivated the migration and invasion of osteosarcoma U2OS cells by inactivating Raf/MEK/ERK signaling pathway.19 In the present study, we examined the anticancer activity and the related mechanism of EVO in human osteosarcoma cells 143B and MG63. Our results not only confirmed the previous findings but also revealed that EVO could exert anticancer effects through suppressing Wnt/β-catenin signaling pathway in cancer cells.
Materials and Methods
Cell Culture and Treatment
The osteosarcoma cell lines 143B and MG63 were provided by Dr Tongchuan He (University of Chicago, USA), which originate from the America Type Culture Collection (ATCC, USA). Cells were cultured in high glucose Dulbecco Modified Eagle Medium (DMEM; Hyclone™, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone™, ThermoFisher Scientific, Waltham, MA, USA) and 100 U/mL of penicillin-streptomycin (Gibco, Invitrogen, Carlsbad, CA, USA), which maintained in a humidified atmosphere of 5% CO2 incubator at 37°C. Osteosarcoma cells propagating in the dishes were washed with Phosphate-Buffered Saline (PBS; Gibco, Invitrogen, Carlsbad, CA, USA) and treated with 0.25% trypsin-EDTA (ThermoFisher Scientific, Waltham, MA, USA) to subculture. Cells in the exponential phase were collected for subsequent experiments.
The compound EVO was purchased from the Chengdu Herbpurify CO., LTD (Chengdu, Sichuan, China), with purity over 98%. Stock solutions of EVO were prepared in dimethyl sulfoxide (DMSO; Sigma, St Louis, MO, USA) at the concentration of 80mM, which would be diluted with fresh medium immediately before application. Cells were treated with different concentrations of EVO for 24h, 48h and 72h as desired, and an equal volume of DMSO (final concentration ≤0.1%) was added to the experimental control (DM group) with only culture medium adding as negative control (NC group).
MTT Assay
Cell viability and growth were assessed using an MTT assay. MTT powder was purchased from Sigma Chemical Co. (St Louis, MO, USA). Briefly, the cultured 143B or MG63 osteosarcoma cells were transplanted into 96-well at the appropriate seeding density of 5×103 cells/well and treated with predesigned concentration of EVO (0.5, 1, 2, 4, 6, 8, 16 and 32 μM for 143B cells; 0.25, 0.5, 1, 2, 4, 6, 8 and 16 μM for MG63 cells) or DMSO. The cells only cultured with the medium were set as a negative control group. After cultured for 24, 48 and 72 hours at 37°C, cells were incubated with 20μL MTT (5 mg/mL) for an additional 4h at 37°C, then the medium was replaced with 100μL 100% dimethylsulfoxide (DMSO) in each well, following which the plates were then agitated on a plate shaker for 10 min before measurement. The absorbance was measured at a wavelength of 490 nm on a microplate reader.
Crystal Violet Assay
The osteosarcoma cells were seeded in the 24-well plate with a density of 2.5×104 cells/well and treated with predesigned concentrations of EVO (1, 2, 4 and 8 μM for 143B cells; 0.5, 1, 2 and 4 μM for MG63 cells) or DMSO. The cells only cultured with the medium were set as a negative control group. After incubation for 24, 48 and 72 hours, the cells were washed carefully with PBS, fixed with 4% paraformaldehyde solution for 30 min. Then, the cells were stained with 0.5% crystal violet dye solution (Beyotime, Shanghai, China) under dark conditions for 10 min, after that the cells were carefully washed with distilled water to remove the redundant crystal violet solution and dried at room temperature. The images were obtained using an image scanner (Epson Perfection V200 Photo, China). For quantification analysis, a 200μL 20% acetic acid solution was added to the well to dissolve the crystal violent, the plate was shaken for 20 min. The absorbance was estimated at 590nm on a microplate reader.
Colony Formation Assay
The osteosarcoma cell lines 143B and MG63 (500 cells/well) were seeded into 6-well plates and treated with a predesigned concentration of EVO (0.5, 1, 2 and 4 μM for 143B cells; 0.25, 0.5, 1 and 2 μM for MG63 cells) or DMSO. This is to determine the long-term effect of EVO exposure. The cells that were only cultured with the medium was set as a negative control group. After incubated at 37°C for 7 days, the cells were washed by PBS, fixed with 4% paraformaldehyde solution and stained with crystal violet for counting. Finally, the colonies were scored and photographed. The colony of more than 50 cells was counted by ImageJ software (National Institutes of Health, USA).
Wound-Healing Assay
Osteosarcoma cells were seeded into 6-well plates. After the cells reached 80–90% confluence, wounds were created with a sterile 10 μL pipette tip. After washing with PBS, cells were treated EVO (1, 2, 4 and 8 μM for 143B cells; 0.5, 1, 2 and 4 μM for MG63 cells) or DMSO in serum-free DMEM and incubated at 37 °C for 24 hours. Images of wounds at 0h, 12h and 18h for 143B cells while 0, 12 and 24 hours for MG63 cells after scratching were obtained using an inverted microscope (ECLIPSE Ti, Nikon, Japan) at x200 magnification. The wound areas at different time points were measured with the ImageJ software (National Institutes of Health, USA).
Transwell Assay
For the invasion assay, the top transwell chambers (24-well insert; 8-μm pore size, Millipore, USA) were coated with Matrigel matrix (BD Biosciences, USA) according to the manufacturer’s instructions. For the migration assay, transwell chambers without Matrigel were used. In both assays, cells (6×105/mL) were seeded onto the upper chamber in 500µL of serum-free medium containing EVO (1, 2, 4 and 8 μM for 143B cells; 0.5, 1, 2 and 4 μM for MG63 cells) or DSMO, and 500µL medium supplemented with 10% FBS was used as a chemo-attractant in the lower chamber. After incubation for 48h at 37°C, the cells that did not migrate or invade through the pores were removed by a cotton swab. Filters were fixed with 4% paraformaldehyde solution for 20 min and stained with 0.1% crystal violet for 10min. The migrated or invaded cells were then visualized and counted from 5 randomly selected fields (x200 magnification).
Hoechst 33258 Staining Assay
A Hoechst Staining Kit was purchased from Solarbio Science & Technology Co., Ltd (Beijing, China). The cells were seeded in a 24-well plate with density of 2.5×104 cells/well. After treated with predesigned concentrations of EVO (1, 2, 4 and 8 μM for 143B cells; 0.5, 1, 2 and 4 μM for MG63 cells) or DMSO for 48h, the cells were fixed in 4% paraformaldehyde solution for 10 min, washed with PBS, stained with 200μL Hoechst 33258 per well for 30 min under dark condition and then washed twice with PBS. Finally, the stained cells were visualized at a magnification of 200× and counted from 5 randomly selected fields using a fluorescence microscope (ECLIPSE Ti, Nikon, Japan).
Cell Cycle Assay
Osteosarcoma cells were cultured in 6cm dishes and treated with EVO (2, 4 and 8 μM for 143B cells) or DMSO for 48h. Then, the cells were collected, fixed with 75% ethanol at 4°C overnight, and stained with PI (Beyotime, Shanghai, China) for 15 min at room temperature in the dark. The stained cells were immediately analyzed by flow cytometer (CytoFLEX, Beckman Coulter, Fullerton, CA, USA) and data were analyzed by FlowJo 7.6 software.
Luciferase Assay
The p-BGluc TCF/LEF (TOP-luc) was generously donated by Dr Tongchuan He (University of Chicago, USA). The cells proliferated to 70% confluence after 24 hours of culture. The 143B cells were transfected with the reporter construct using the Lipofectamine 2000 reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s protocol. Then, cells were grown in fresh medium and treated with various concentrations of EVO (0.5, 1, 2, 4, 8 and 16 μM for 143B cells) or DMSO only for 24 hours. The amounts of luciferase were measured with the Luciferase Reporter Assay System Kit (New England Biolabs, USA) according to the manufacturer’s protocol. The relative transcriptional activity was converted into fold induction above the vehicle control value.
Western Blot Assay
Total protein was extracted from cells treated with EVO or DMSO by using the mammalian protein extraction reagent RIPA (Beyotime, China). The protein concentration was measured using BCA (bicinchoninic acid) protein assays (Beyotime, China). Protein samples were boiled with 6× SDS loading buffer for 5 min, separated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, USA). Membranes were blocked in 5% bovine serum albumin (BSA; Solarbio, Beijing, China) for 2h and then incubated with diluted specific primary antibodies overnight at 4°C. β-Actin was purchased from Zoonbio Technology (Beijing, China), while PCNA, β-catenin, c-Myc, cyclin D1, GSK-3β, Bcl-2, Bad, Bax, Caspase-3, cleaved Caspase-3, PARP, cleaved-PARP, MMP-2, MMP-7, MMP-9, vimentin, E-cadherin, N-cadherin and Snail were purchased from Cell Signaling Technology (Danvers, CO, USA). Primary antibodies were detected using horseradish peroxidase-conjugated secondary antibodies. The bands were visualized using an enhanced chemiluminescent kit (Millipore, USA) according to the manufacturer’s protocol. The protein bands were pictured and analyzed by using the ChemiDoc MP Imaging System and Image Lab Software (Bio-Rad, California, USA).
Statistical Analysis
All data represent at least three independent experiments and are presented as the mean ± SD. Differences among groups were analyzed by Single-factor variance analysis with the least significant difference test using SPSS 24.0 software (SPSS Inc., Chicago, IL, USA). A value of P<0.05 was considered to indicate a statistically significant difference.
Results
EVO Inhibits the Growth of 143B and MG63 Cells
To examine the antiproliferation effects of EVO on osteosarcoma, cell viability was measured at 24, 48 and 72 hours, respectively, after 143B and MG63 cells treated with EVO at the indicated concentration of EVO and the experimental control group was treated with DMSO while the nothing adding was set as negative control (NC) group by MTT assay. As shown in Figure 1A and B, the proliferation rate was significantly lower in EVO-treated groups than the NC group and DMSO group. Additionally, the proliferation rate was decreased in a time- and dose-dependent manner. Based on these data, we chose 1, 2, 4 and 8 µM for 143B cells and 0.5, 1, 2 and 4 µM for MG63 cells as the working concentrations for the subsequent experiment. Also, we further confirmed with the antiproliferation effects of EVO by using crystal violet assays. As shown in Figure 1C–F, the results were similar to the MTT assays. Both proliferation assays indicated that EVO could effectively suppress the growth of osteosarcoma cell lines. Furthermore, we conducted the colony formation assays to verify whether EVO could inhibit the carcinogenic property of osteosarcoma cells. Figure 1G–J exhibited that EVO could inhibit tumorigenesis in a dose-dependent manner, which was consistent with the antiproliferation finding. As shown in Figure 1K and L, EVO also significantly suppressed the expression of proliferating cell nuclear antigen (PCNA), a marker for the assessment of osteosarcoma growth.
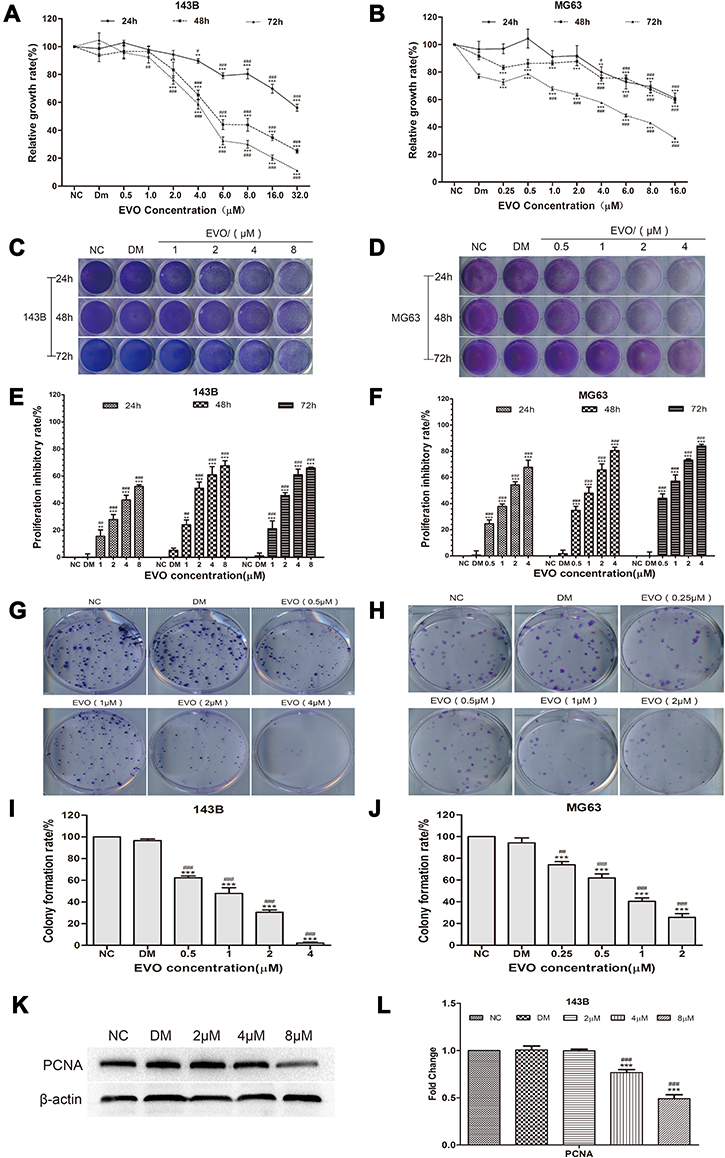 |
Figure 1 EVO inhibits the growth of 143B and MG63 cells. Human osteosarcoma cell lines, (A) 143B and (B) MG63 cells were treated with EVO with various dosages and the cell viabilities were examined by MTT assay. The data are presented as the mean ± SD (n=3, each group). **p<0.01, ***p<0.001 vs NC group; #p<0.05, ##p<0.01, ###p<0.001 vs DMSO group. (C and D) The crystal violet staining result in 143B and MG63 cells were seeded in 24-well plates and treated with different concentrations of EVO or DMSO, followed by crystal violet staining at the indicated time-points. (E and F) Quantitative results of crystal violet staining in 143B and MG63 cells. The data are presented as the mean ± SD (n=3, each group). **p<0.01, ***p<0.001 vs NC group; #p<0.05, ##p<0.01, ###p<0.001 vs DMSO group. (G and H) 500 cells/well were seeded in plates and the medium was changed every 4 days. After 14 days, the plates were stained for the formation of cell colonies with crystal violet dye. (I and J) The number of colonies was counted. The data are presented as the mean ± SD (n=3, each group). ***p<0.001 vs NC group; ##p<0.01, ###p<0.001 vs DMSO group. (K and L) Western blot assay results show the PCNA protein level in 143B cells with β-actin as loading control. The quantitative results are presented as the mean ± SD (n=3, each group). ***p<0.001 vs NC group; ###p<0.001 vs DMSO group. |
EVO Induced Cell Cycle Arrest at the G2/M Phase
Cell cycle arrest and apoptosis induction are two major causes of cell proliferative inhibition. In order to examine the effect of EVO on the cell cycle progression of osteosarcoma cells, cells were treated with or without EVO, and percentages of cells at the different cell cycle phases were measured using a flow cytometric analysis via PI staining. As shown in Figure 2A and B, EVO significantly (p<0.001) increases the percentage of cells at the G2/M phase and decreased the G0/G1 and S phase of the cell cycle in a concentration-dependent manner. This result indicated that EVO treatment effectively mediated G2/M cell cycle arrest in 143B cells.
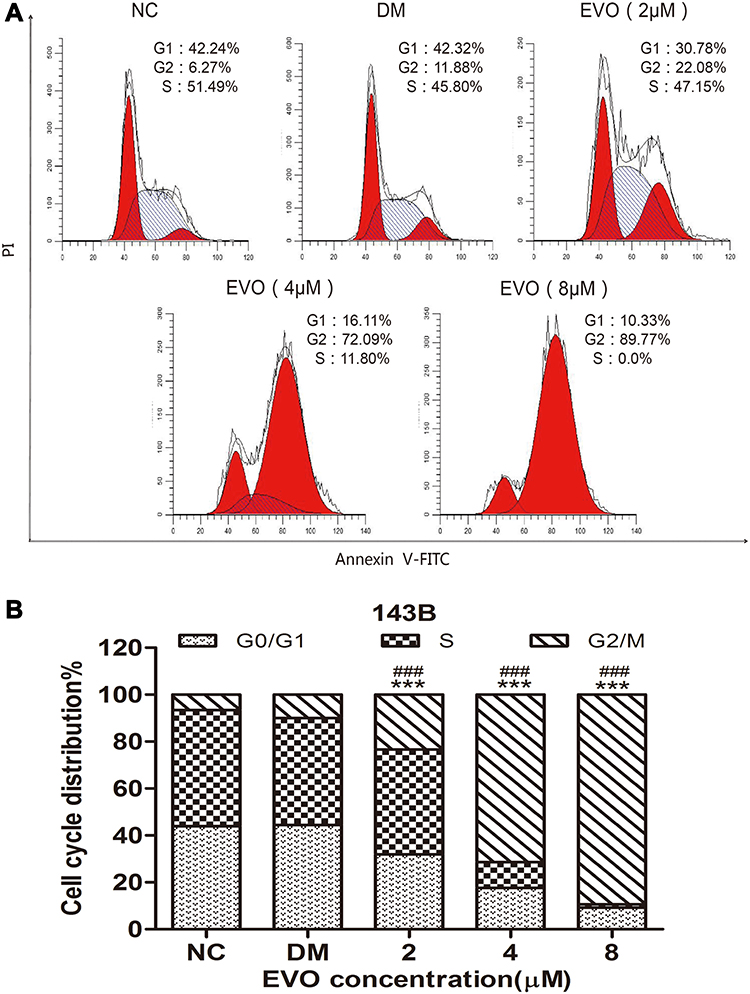 |
Figure 2 EVO induced cell cycle arrest at the G2/M phase. (A) The representative FACS plots displayed differences in cell cycle phases of 143B cells treated with different concentrations of EVO or DMSO. (B) Bar graphs show that EVO led to cell cycle arrest at the G2/M phase in 143B cells. The data are presented as the mean ± SD (n=3, each group). ***p<0.001 vs NC group; ###p<0.001 vs DMSO group. |
EVO Induces the Apoptosis of 143B and MG63 Cells by Activating the Mitochondrial Apoptotic Pathway
The above results indicated that inhibition of the cell cycle progression might be an upstream event leading to apoptosis. We next investigated whether EVO induced apoptosis in 143B and MG63 osteosarcoma cells. We evaluated apoptosis via Hoechst 33258 staining assay and Western blot analysis for detecting apoptotic marker caspase-3 and PARP protein level. The images acquired by fluorescence microscopy revealed that with the increase of EVO dose, a growing number of cells exhibited the apoptotic morphological characteristics, such as nuclear condensation and apoptotic bodies (Figure 3A–D). Also, the protein expression of caspase-3, cleaved caspase-3, PARP, cleaved PARP increased at 24 hours in a concentration-dependent manner. Altogether, these results suggest that EVO can induce apoptosis in osteosarcoma cells. Furthermore, we detected the expression levels of the Bcl-2 family proteins, which play a crucial role in regulating the mitochondria-dependent pathway of apoptosis. As shown in Figure 3E and F, Bcl-2 was downregulated, while Bax and Bad expression were upregulated in a dose-dependent manner following EVO treatment. Therefore, the results indicated that EVO induced apoptosis through the mitochondrial pathway.
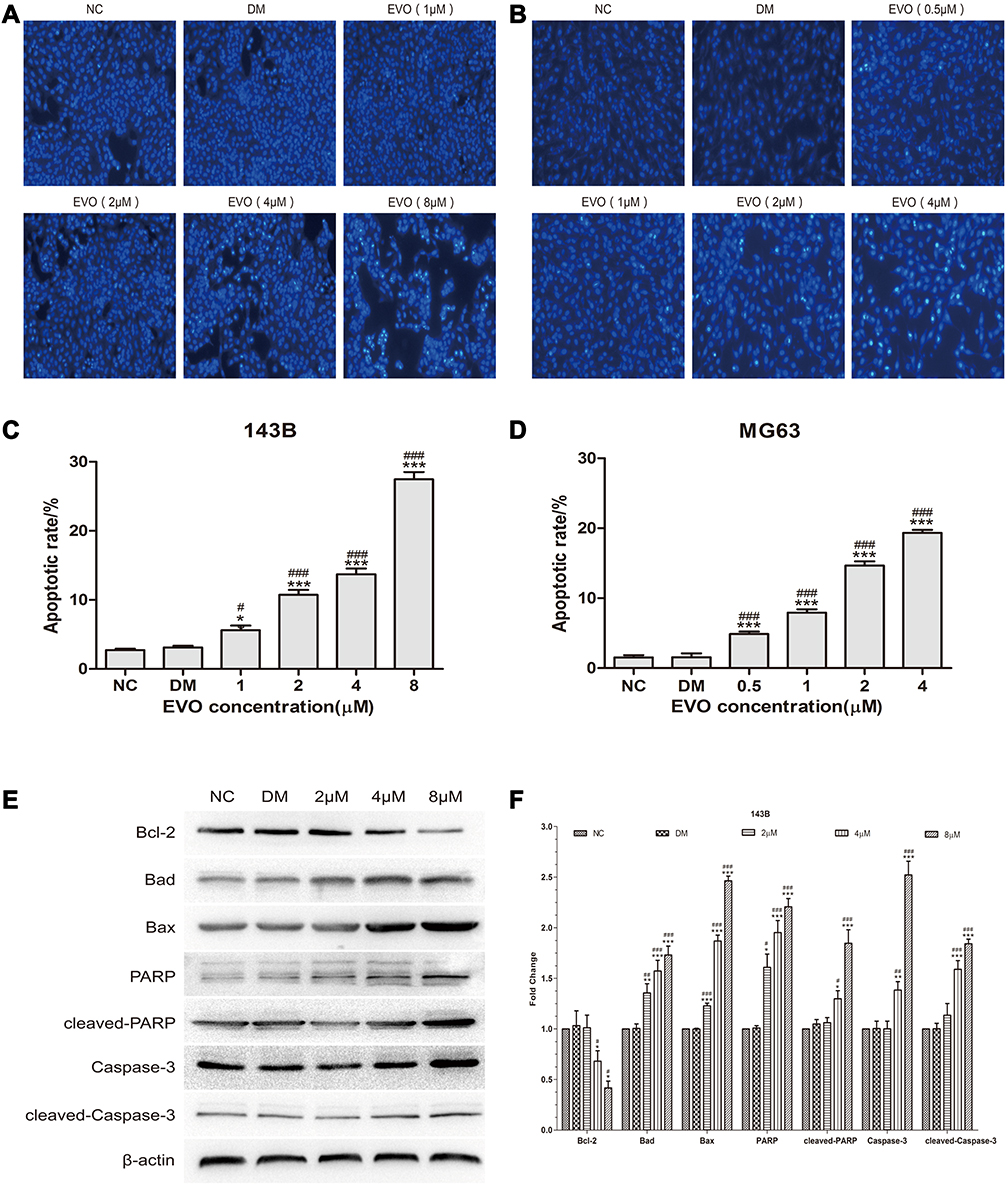 |
Figure 3 EVO induces the apoptosis of 143B and MG63 cells by activating the mitochondrial apoptotic pathway. (A and B) 143B and MG63 cells were treated with different concentrations of EVO or DMSO for 24 hours, and then stained with Hoechst 33,258. (C and D) Statistical analysis for the apoptosis assay. The data are presented as the mean ± SD (n=3, each group). *p<0.05, ***p<0.001 vs NC group; #p<0.05, ###p<0.001 vs DMSO group. (E and F) Cell apoptosis‐related proteins including caspase-3, cleaved caspase-3, PARP, cleaved-PARP, Bcl-2, Bad and Bax were analyzed by Western blotting. β-Actin was served as a loading control. The data are presented as the mean ± SD (n=3, each group). *p<0.05, **p<0.01, ***p<0.001 vs NC group; #p<0.05, ##p<0.01, ###p<0.001 vs DMSO group. |
EVO Suppresses the Migration and Invasion of 143B and MG63 Cells
Subsequently, the effect of EVO on the metastatic ability of osteosarcoma cells was investigated by wound-healing and transwell assays. The results of wound-healing assay showed that the scratches in EVO-treated cells were remarkably larger than that of in control cells both at 12 and 24 hours (Figure 4A–D). Meanwhile, transwell migration assay indicated that EVO significantly suppressed the migration rates of osteosarcoma cells in comparison to their corresponding control cells (Figure 4E–H). In addition, matrigel invasion assay proved that EVO suppressed the invasiveness of osteosarcoma cells compared with their corresponding control cells (Figure 4I–L). These observations strongly confirm that EVO significantly inhibited the migration and invasion of 143B and MG63 cells in a concentration-dependent manner. It is well accepted that epithelial–mesenchymal transition (EMT) is essential to tumor invasiveness and migration. Therefore, we investigated the expression of EMT-related markers through Western blotting. The results revealed that EVO caused a marked increase in E-cadherin, but significantly downregulated N-cadherin and Vimentin, as well as EMT transcription factors Snail (Figure 4M and N), which indicated that EVO suppresses the process of EMT in osteosarcoma cells. Matrix metalloproteinases (MMPs) are important proteolytic enzymes able to degrade the extracellular matrix and basement membrane and serve critical roles in the invasion process. As determined by Western blot assay, EVO treatment significantly decreased the expression of MMP-2, MMP-7 and MMP-9 proteins (Figure 4M and N). Accordingly, the above data reveal that EVO represses the invasion and migration abilities of osteosarcoma cells via the suppression of EMT and the downregulation of MMPs expression.
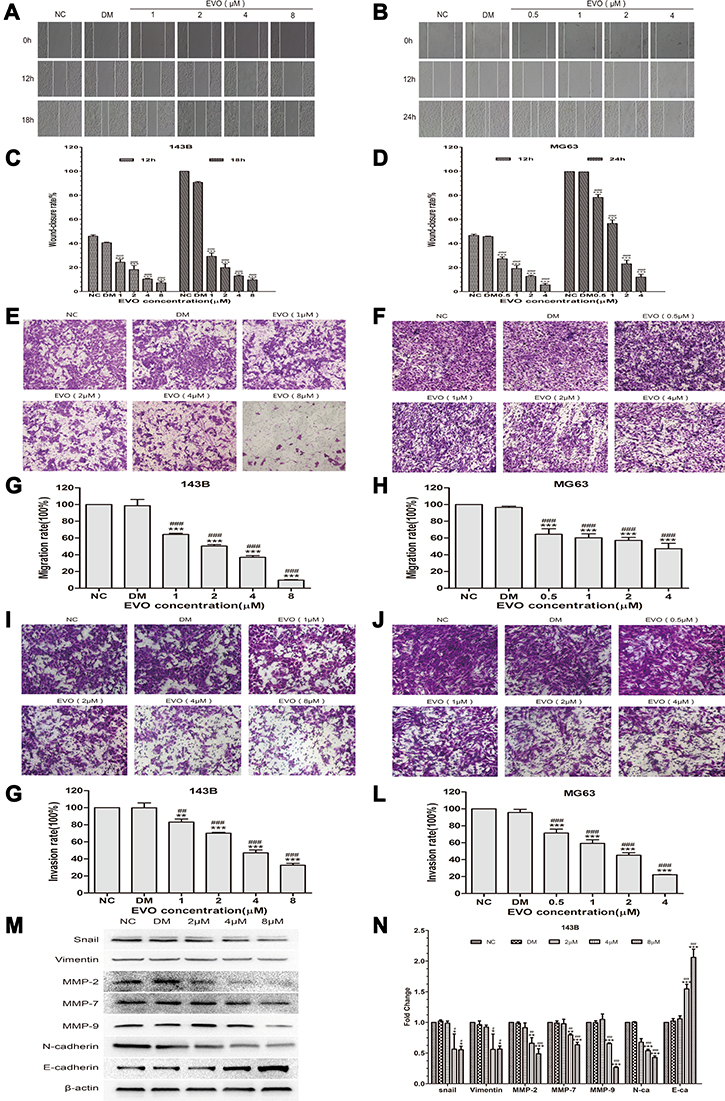 |
Figure 4 EVO suppresses the migration and invasion of 143B and MG63 cells. (A and B) The wound-healing assay showed the migratory abilities of 143B and MG63 cells treated with different concentrations of EVO or DMSO. Photographs were respectively obtained at 0h, 12h and 18h for 143B cells while 0, 12 and 24 hours for MG63 cells. (C and D) Representative statistical plots are presented. The data are presented as the mean ± SD (n=3, each group). ***p<0.001 vs NC group; ###p<0.001 vs DMSO group. (E and F) Representative figure of 143B and MG63 cells treated with different concentrations of EVO or DMSO in the transwell migration assay. (G and H) The amounts of migrated cells were counted under five randomly visual fields. The data are presented as the mean ± SD (n=3, each group). ***p<0.001 vs NC group; ###p<0.001 vs DMSO group. (I and J) Representative figure of 143B and MG63 cells treated with different concentrations of EVO or DMSO in the transwell matrigel invasion assay. (K and L) The amounts of invaded cells were counted under five randomly visual fields. The data are presented as the mean ± SD (n=3, each group). **p<0.01, ***p<0.001 vs NC group; ##p<0.01, ###p<0.001 vs DMSO group. (M and N) EMT-related markers including E-cadherin, vimentin and N-cadherin; EMT transcription factors Snail and MMPs including MMP-2, MMP-7 and MMP-9 in 143B cells treated with different concentrations of EVO or DMSO were analyzed by Western blotting. β-Actin was served as a loading control. The quantitative data are presented as the mean ± SD (n=3, each group). *p<0.05, **p<0.01, ***p<0.001 vs NC group; #p<0.05, ##p<0.01, ###p<0.001 vs DMSO group. |
EVO Inhibits the Wnt/β-Catenin Signaling Pathway
To further investigate the underlying mechanism of anticancer effects of EVO on osteosarcoma cells, we next examined the Wnt/β-catenin signaling pathway, which is important for the action of chemotherapeutic drugs. Firstly, we found the protein level of Wnt pathway effector β-catenin was downregulated in EVO-treated cells compared to the control group cells (Figure 5A and B). Thus, we hypothesized that EVO might involve in cancer cell growth and metastasis via the Wnt/β-catenin signaling pathway. The stability of β-catenin in cells is tightly regulated by the Axin/APC/GSK3β complex. Phosphorylation of β-catenin by GSK-3β results in its degradation, which leads to the inactivation of Wnt/β-catenin signaling. Thus, we explored whether EVO can affect the expression of GSK-3β, the negative regulator of Wnt/β-catenin signaling. We found that its protein expression level was markedly increased in response to EVO (Figure 5A and B). Subsequently, we used the TOP-Luc luciferase reporter gene system, which contains TCF/LEF-responsive elements to analyze the TCF/LEF transcription factor activity of Wnt signaling (Figure 5C). The result indicated that EVO suppressed TCF/LEF transcription factor activity in osteosarcoma cells. Moreover, we found that the Wnt target proteins cyclin D1 and c-Myc were also downregulated in EVO-treated osteosarcoma cells (Figure 5A and B). These data indicated that the suppression of Wnt/β-catenin signaling pathway was mainly involved in EVO-induced anticancer effects in osteosarcoma cells.
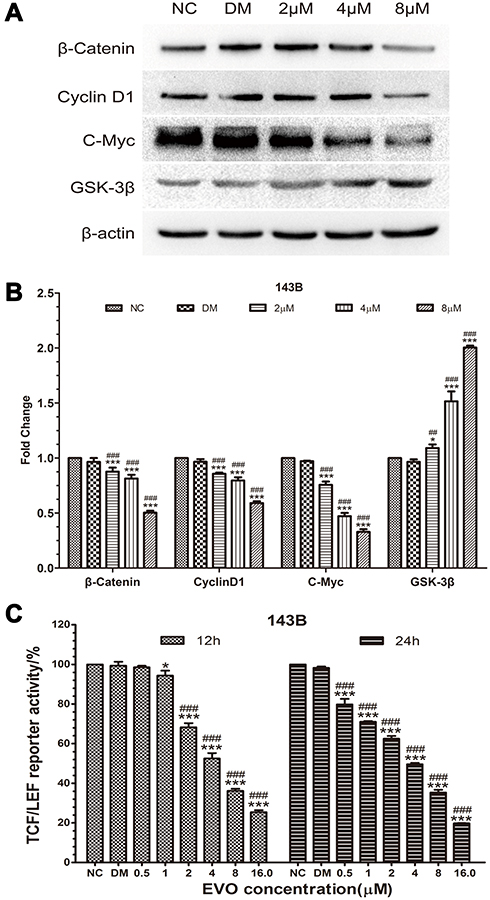 |
Figure 5 EVO inhibits the Wnt/β-catenin signaling pathway. (A and B) The protein levels of β-catenin, GSK-3β, and downstream targets including c-Myc and cyclin D1 were examined in 143B cells treated with different concentrations of EVO or DMSO. β-Actin was served as a loading control. The quantitative data are presented as the mean ± SD (n=3, each group). *p<0.05, ***p<0.001 vs NC group; ##p<0.01, ###p<0.001 vs DMSO group. (C) The effect of EVO on the Wnt/β-catenin signaling pathway was determined using the TOP-Luc luciferase reporter gene system. The relative reporter activities were exhibited as the mean ± SD (n=3, each group). *p<0.05, ***p<0.001 vs NC group; ###p<0.001 vs DMSO group. |
Discussion
In recent years, natural Chinese compounds, especially plant-derived compounds, have received extensive attention as sources of new drugs for anticancer therapy.20 EVO, a quinolone alkaloid, is traditionally used for headache or stomach ache in China.21 However, its effects on bone tumors and its potential mechanisms are unclear. In the present study, we demonstrated that EVO inhibited cell proliferation, migration and invasion, induced cycle arrest and cell apoptosis of osteosarcoma cells in a dose-dependent manner. Mechanistic studies revealed that cell cycle arrest at the G2/M phase may be correlated with reduced levels of cyclin D1 protein, while the apoptotic effects of EVO were associated with the upregulation of Bax and Bad and a decrease in Bcl-2 protein levels. Moreover, EVO suppresses metastatic through suppressing EMT as indicated by elevating the expression of epithelial marker E‐cadherin and reducing the expression of mesenchymal markers N‐cadherin and vimentin, as well as EMT transcription factors Snail and MMPs. Furthermore, EVO exerted the anticancer effects by suppressing Wnt/β-catenin signal pathway in osteosarcoma cells.
Proliferating cell nuclear antigen (PCNA) is a crucial factor in DNA synthesis and repair, initially characterized as the auxiliary protein of DNA polymerases δ andε22 PCNA is involved in a wide range of cellular processes including DNA replication, nucleotide-excision repair, mismatch repair, the cell cycle and apoptosis.23 PCNA expression provides a reliable marker of proliferative activity a wide range of human neoplasia, which also well known is overexpressed in osteosarcoma tissues.24 In this study, we found that PCNA was remarkably downregulated by EVO in a dose-dependent manner, which further confirmed that EVO exerted an antiproliferation role in osteosarcoma cells.
Cell cycle is one of the crucial mechanisms underlying the regulation of tumor growth. Cell growth is defined by several genetically defined checkpoints to ensure its coordinated progression through the different stages of the cell cycle and to monitor DNA integrity. The cell cycle is driven by the alternative expression or degradation of different cyclins, which exert their function by binding to different CDKs.25 Cyclin D1 is activated by binding to CDK4/6 during the cell cycle progression.26,27 Cell-cycle arrest in response to stress is integral to the maintenance of genomic integrity and several anticancer compounds carry out their effects by arresting cell cycle.28 Alterations in the machinery that controls the G2/M transition are frequently observed in many types of human cancers.29 The analysis of the cell cycle with different concentrations of EVO in 143B cells revealed a higher number of cells at the G2/M phase, whereas the number of cells in the G0/G1 and S phase decreased compared with the untreated cells. These results indicate that EVO inhibits cell proliferation via G2/M phase arrest in a dose-dependent manner. The accumulation of G2/M phase cells resulting from EVO treatment led us to determine the expression level of cell cycle regulators. We also found that EVO suppressed the expression of cyclin D1. These data indicated that EVO induced G2/M arrest may be correlated with reducing levels of cyclin D1 protein, which resulted in a growth blockade on osteosarcoma cells.
Natural products typically exhibit their anticancer activities by not only cell-cycle arrest but also triggering apoptosis. Cells undergo apoptosis through the extrinsic pathway (death receptor pathway) or intrinsic pathway (the mitochondrial pathway).30 Mitochondria play a crucial role in apoptotic regulation and the mitochondrial dysfunction that occurs during apoptosis.31 Caspases are a family of proteolytic enzymes that function to mediate apoptosis via programmed cell death. Of these, caspase-3 is an intracellular protease that is critical in apoptosis and works early in the apoptotic process.32 It is reported that caspase-3 activation resulted in the cleavage of PARP, leading to overactivated, the catalytic receptor process will consume a large amount of ATP and NAD+ in the cell, and eventually energy depletion leads to cell death.33,34 The Bcl-2 family of proteins promotes cell death by modulating the mitochondrial release of pro-apoptotic factors and acts as a critical life-death decision point within the common pathway of apoptosis.35 This protein family includes antiapoptotic (eg, Bcl-2) and pro-apoptotic (eg, Bax and Bad) factors. With decreased Bcl-2 and increased Bax and Bad, mitochondrion is stimulated to release cytochrome c, which in turn activates caspase-3, ultimately leading to apoptosis.36 In this study, we revealed that after incubation with EVO, a dose-dependent increase of caspase-3, cleaved-caspase3, PARP and cleaved PARP was detected. Moreover, we observed downregulation of pro-apoptotic marker, Bax and Bad, and upregulation of antiapoptotic Bcl-2 proteins in EVO-treated osteosarcoma cells. Therefore, our results suggest that mitochondrial apoptosis induction and G2/M phase cell cycle arrest both contributed to the anticancer activity of EVO.
Besides, we also explored the antimetastatic effect of EVO on osteosarcoma cells. The wound-healing assay and transwell assay revealed that EVO suppressed the invasion and migration ability of osteosarcoma cells. Osteosarcoma cells possess highly invasive properties by undergoing EMT, a unique phenotypic switch. EMT plays a critical role in carcinoma metastasis, which is characterized by inhibition of epithelial molecule E-cadherin and gaining of mesenchymal markers, such as N-cadherin, vimentin and fibronectin.37,38 MMPs are critical proteolytic enzymes with the capacity to degrade the extracellular matrix and basement membrane and play a critical role in the invasion process.39 MMPs are key modulators of various biological processes, including the EMT process, cancer, angiogenesis, skeletal formation, inflammation and cell migration.40 E-cadherin, a transmembrane glycoprotein, can regulate cell-cell adhesion. It is a significant epithelial marker and its expression is reduced in cells that have undergone EMT.41 N-cadherin is a calcium-dependent adhesion molecule and in the presence of Ca,2+ which resists hydrolysis by protease and promotes tumor cell metastasis.42 Evidence has established the expression and activation of Snail transcriptional factor during EMT in E-cadherin suppression and cancer progression.43,44 Vimentin is a type III intermediate filament protein with a molecular weight of 57 kDa and is expressed in non-epithelial cells, particularly in mesenchymal cells.45 Our study found that EVO could significantly downregulate the Snail and epithelial marker (E-cadherin), meanwhile upregulated of mesenchymal markers (N-cadherin, vimentin, MMP-2, MMP-7 and MMP-9) in osteosarcoma cells. Therefore, for the first time, our study demonstrated that the suppressive effect of EVO on osteosarcoma cell invasion and migration may be related to EMT.
Deregulation of Wnt signaling has been implicated in the development and pathogenesis of a wide range of cancers, including osteosarcoma.46,47 In the canonical Wnt/β-catenin signaling, Wnt ligands bind to the dual receptor complex comprised frizzled and low-density lipoprotein receptor-related protein 5/6 (LRP5/6).48,49 This leads to inactivation of the β-catenin destruction complex, Axin/APC/GSK-3β, thus relieving the critical mediator β-catenin from its constitutive proteasomal degradation.50 β-Catenin subsequently accumulates in the cytoplasm and translocates into the nucleus, where it associates with TCF/LEF family transcription factors to alter the expression of downstream target genes.51 In the present study, we explored the effect of EVO on GSK-3β, a negative regulator of Wnt/β-catenin signaling, and found that the total GSK-3β protein level was significantly elevated in response to the treatment with EVO. Moreover, our data indicate that downregulation of β-catenin protein in OS cells may result from the degradation initialized by GSK-3β, at least. Furthermore, we found that EVO reduced the fluorescent expression of TCF/LEF fluorescent reporter plasmids, which suggests that EVO may regulate osteosarcoma cells by suppressing the Wnt/β-Catenin signaling pathway. c-Myc, MMPs and cyclin D1, the downstream targets of Wnt, were downregulated. The c-Myc is a multifunctional oncogene involved in cell growth, proliferation, tumorigenesis, and so is frequently upregulated in various types of cancer cells. One of the key biological functions of c-Myc is to promote cell-cycle progression in several cancers.52 Also, Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1.53 A new function of Wnt/Snail-mediated tumor progression was recently found.54 In the presence of Wnt signaling, GSK-3β is unable to phosphorylate two of its known targets, β-catenin and Snail, and therefore stabilizes these two molecules in the nucleus.44 Therefore, by blocking the activity of GSK-3β, Wnt can stabilize the level of Snail and β-catenin to induce EMT and cancer metastasis.
In summary, we have demonstrated that EVO exhibited potent antiproliferation and antimetastatic effects against human osteosarcoma cells through suppressing Wnt/β-catenin signaling pathway (Figure 6). Based on these findings, EVO is a therapeutic agent worthy of further development as a clinical trial candidate for treating osteosarcoma.
 |
Figure 6 An illustration of how EVO exerts anticancer activity in osteosarcoma cells through the suppressing Wnt/β-catenin signaling pathway. |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13.
2. Howlader N, Noone AM, Krapcho M, et al. (eds). SEER Cancer Statistics Review, 1975–2016, National Cancer Institute: Bethesda. Available from: https://seer.cancer.gov/csr/1975_2016/. based on November 2018 SEER data submission, posted to the SEER web site. April, 2019.
3. Hayden JB, Hoang BH. Osteosarcoma: basic science and clinical implications. Orthop Clin North Am. 2006;37(1):1–7. doi:10.1016/j.ocl.2005.06.004
4. He JP, Hao Y, Wang XL, et al. Review of the molecular pathogenesis of osteosarcoma. Asian Pac J Cancer Prev. 2014;15(15):5967–5976. doi:10.7314/APJCP.2014.15.15.5967
5. Saraf AJ, Fenger JM, Roberts RD. Osteosarcoma: accelerating progress makes for a hopeful future. Front Oncol. 2018;8:4. doi:10.3389/fonc.2018.00004
6. Simpson E, Brown HL. Understanding osteosarcomas. JAAPA. 2018;31(8):15–19. doi:10.1097/01.JAA.0000541477.24116.8d
7. Biazzo A, De Paolis M. Multidisciplinary approach to osteosarcoma. Acta Orthop Belg. 2016;82(4):690–698.
8. Seca AML, Pinto D. Plant secondary metabolites as anticancer agents: successes in clinical trials and therapeutic application. Int J Mol Sci. 2018;19(1):263. doi:10.3390/ijms19010263
9. Bhattacharya S, Muhammad N, Steele R, Kornbluth J, Ray RB. Bitter melon enhances natural killer-mediated toxicity against head and neck cancer cells. Cancer Prev Res (Phila). 2017;10(6):337–344. doi:10.1158/1940-6207.CAPR-17-0046
10. Yu H, Jin H, Gong W, Wang Z, Liang H. Pharmacological actions of multi-target-directed evodiamine. Molecules. 2013;18(2):1826–1843. doi:10.3390/molecules18021826
11. Jiang J, Hu C. Evodiamine: a novel anti-cancer alkaloid from Evodia rutaecarpa. Molecules. 2009;14(5):1852–1859. doi:10.3390/molecules14051852
12. Hu X, Li D, Chu C, et al. Antiproliferative effects of alkaloid evodiamine and its derivatives. Int J Mol Sci. 2018;19(11):11. doi:10.3390/ijms19113403
13. Lin L, Ren L, Wen L, Wang Y, Qi J. Effect of evodiamine on the proliferation and apoptosis of A549 human lung cancer cells. Mol Med Rep. 2016;14(3):2832–2838. doi:10.3892/mmr.2016.5575
14. Yang F, Shi L, Liang T, et al. Anti-tumor effect of evodiamine by inducing Akt-mediated apoptosis in hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;485(1):54–61. doi:10.1016/j.bbrc.2017.02.017
15. Du J, Wang XF, Zhou QM, et al. Evodiamine induces apoptosis and inhibits metastasis in MDA-MB-231 human breast cancer cells in vitro and in vivo. Oncol Rep. 2013;30(2):685–694. doi:10.3892/or.2013.2498
16. Yang ZG, Chen AQ, Liu B. Antiproliferation and apoptosis induced by evodiamine in human colorectal carcinoma cells (COLO-205). Chem Biodivers. 2009;6(6):924–933. doi:10.1002/cbdv.200800256
17. Shi L, Yang F, Luo F, et al. Evodiamine exerts anti-tumor effects against hepatocellular carcinoma through inhibiting β-catenin-mediated angiogenesis. Tumor Biol. 2016;37(9):12791–12803. doi:10.1007/s13277-016-5251-3
18. Meng ZJ, Wu N, Liu Y, et al. Evodiamine inhibits the proliferation of human osteosarcoma cells by blocking PI3K/Akt signaling. Oncol Rep. 2015;34(3):1388–1396. doi:10.3892/or.2015.4084
19. Zhou Y, Hu J. Evodiamine induces apoptosis, G2/M cell cycle arrest, and inhibition of cell migration and invasion in human osteosarcoma cells via Raf/MEK/ERK signalling pathway. Med Sci Monit. 2018;24:5874–5880. doi:10.12659/MSM.909682
20. Liao JF, Chiou WF, Shen YC, Wang GJ, Chen CF. Anti-inflammatory and anti-infectious effects of Evodia rutaecarpa (Wuzhuyu) and its major bioactive components. Chin Med. 2011;6(1):6. doi:10.1186/1749-8546-6-6
21. Gavaraskar K, Dhulap S, Hirwani RR. Therapeutic and cosmetic applications of Evodiamine and its derivatives–A patent review. Fitoterapia. 2015;106:22–35. doi:10.1016/j.fitote.2015.07.019
22. Park SY, Jeong MS, Han CW, Yu HS, Jang SB. Structural and functional insight into proliferating cell nuclear antigen. J Microbiol Biotechnol. 2016;26(4):637–647. doi:10.4014/jmb.1509.09051
23. Choe KN, Moldovan GL. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol Cell. 2017;65(3):380–392. doi:10.1016/j.molcel.2016.12.020
24. Park HR, Park YK. Expression of p53 protein, PCNA, and Ki-67 in osteosarcomas of bone. J Korean Med Sci. 1995;10(5):360–367. doi:10.3346/jkms.1995.10.5.360
25. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093. doi:10.1242/dev.091744
26. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl). 2016;94(12):1313–1326. doi:10.1007/s00109-016-1475-3
27. Wiman KG, Zhivotovsky B. Understanding cell cycle and cell death regulation provides novel weapons against human diseases. J Intern Med. 2017;281(5):483–495.
28. Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23(36):9408–9421. doi:10.1200/JCO.2005.01.5594
29. Stark GR, Taylor WR. Control of the G2/M transition. Mol Biotechnol. 2006;32(3):227–248. doi:10.1385/MB:32:3:227
30. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
31. Goldar S, Khaniani MS, Derakhshan SM, Baradaran B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J Cancer Prev. 2015;16(6):2129–2144. doi:10.7314/APJCP.2015.16.6.2129
32. Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai). 2005;37(11):719–727. doi:10.1111/j.1745-7270.2005.00108.x
33. Kuzhandaivel A, Nistri A, Mladinic M. Kainate-mediated excitotoxicity induces neuronal death in the rat spinal cord in vitro via a PARP-1 dependent cell death pathway (Parthanatos). Cell Mol Neurobiol. 2010;30(7):1001–1012. doi:10.1007/s10571-010-9531-y
34. Xu P, Cai X, Zhang W, et al. Flavonoids of Rosa roxburghii Tratt exhibit radioprotection and anti-apoptosis properties via the Bcl-2(Ca(2+))/Caspase-3/PARP-1 pathway. Apoptosis. 2016;21(10):1125–1143. doi:10.1007/s10495-016-1270-1
35. Mohan V, Agarwal R, Singh RP. A novel alkaloid, evodiamine causes nuclear localization of cytochrome-c and induces apoptosis independent of p53 in human lung cancer cells. Biochem Biophys Res Commun. 2016;477(4):1065–1071. doi:10.1016/j.bbrc.2016.07.037
36. Stevens M, Oltean S. Modulation of the apoptosis gene Bcl-x function through alternative splicing. Front Genet. 2019;10:804. doi:10.3389/fgene.2019.00804
37. Xiang J, Fu X, Ran W, Wang Z. Grhl2 reduces invasion and migration through inhibition of TGFβ-induced EMT in gastric cancer. Oncogenesis. 2017;6(1):e284. doi:10.1038/oncsis.2016.83
38. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35(4):645–654. doi:10.1007/s10555-016-9648-7
39. Wang A, Jin C. Knockdown of HE4 suppresses aggressive cell growth and malignant progression of ovarian cancer by inhibiting the JAK/STAT3 pathway. Biol open. 2019;8(9):bio043570.
40. Huang T, Chen QF, Chang BY, Shen LJ, Li W. TFAP4 promotes hepatocellular carcinoma invasion and metastasis via activating the PI3K/AKT signaling pathway. Dis markers. 2019;2019:7129214.
41. Zhang LN, Zhao L, Yan XL, Huang YH. Loss of G3BP1 suppresses proliferation, migration, and invasion of esophageal cancer cells via Wnt/β‐catenin and PI3K/AKT signaling pathways. J Cell Physiol. 2019;234(11):20469–84.
42. Wang W, Li Q, Yang T, et al. Anti-cancer effect of Aquaporin 5 silencing in colorectal cancer cells in association with inhibition of Wnt/β-catenin pathway. Cytotechnology. 2018;70(2):615–624. doi:10.1007/s10616-017-0147-7
43. Zheng YJ, Zhao JY, Liang TS, et al. Long noncoding RNA SMAD5-AS1 acts as a microRNA-106a-5p sponge to promote epithelial mesenchymal transition in nasopharyngeal carcinoma. FASEB J. 2019;33(11):12915–28.
44. Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13(9):963–972. doi:10.2174/15680096113136660102
45. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68(18):3033–3046. doi:10.1007/s00018-011-0735-1
46. Danieau G, Morice S, Redini F, Verrecchia F, Royer BB. New insights about the Wnt/beta-catenin signaling pathway in primary bone tumors and their microenvironment: a promising target to develop therapeutic strategies? Int J Mol Sci. 2019;20(15):3751. doi:10.3390/ijms20153751
47. Lin CH, Ji T, Chen CF, Hoang BH. Wnt signaling in osteosarcoma. Adv Exp Med Biol. 2014;804:33–45.
48. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. doi:10.1016/j.ctrv.2017.11.002
49. Zhou Y, Wang T, Hamilton JL, Chen D. Wnt/β-catenin signaling in osteoarthritis and in other forms of arthritis. Curr Rheumatol Rep. 2017;19(9):53. doi:10.1007/s11926-017-0679-z
50. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016
51. Chen C, Tian A, Zhao M, Ma X. Adenoviral delivery of VHL suppresses bone sarcoma cell growth through inhibition of Wnt/beta-catenin signaling. Cancer Gene Ther. 2019;26(3–4):83–93.
52. Dong H, Hu J, Wang L, et al. SOX4 is activated by C-MYC in prostate cancer. Med Oncol. 2019;36(11):92.
53. Eischen CM, Packham G, Nip J, et al. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene. 2001;20(48):6983–6993. doi:10.1038/sj.onc.1204892
54. Lee SY, Jeon HM, Ju MK, et al. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012;72(14):3607–3617. doi:10.1158/0008-5472.CAN-12-0006
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.