Back to Journals » OncoTargets and Therapy » Volume 11
Evodiamine activates cellular apoptosis through suppressing PI3K/AKT and activating MAPK in glioma
Authors Wang R, Deng D, Shao N, Xu Y, Xue L, Peng Y ![]() , Liu Y, Zhi F
, Liu Y, Zhi F ![]()
Received 26 October 2017
Accepted for publication 18 January 2018
Published 2 March 2018 Volume 2018:11 Pages 1183—1192
DOI https://doi.org/10.2147/OTT.S155275
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Rong Wang,1,2,* Danni Deng,2,* Naiyuan Shao,1 Yuan Xu,2 Lian Xue,2 Ya Peng,1 Yatian Liu,3 Feng Zhi1,2
1Department of Neurosurgery, The First People’s Hospital of Changzhou, Changzhou, Jiangsu, China; 2Modern Medical Research Center, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China; 3Department of Radiation Oncology, Jiangsu Cancer Hospital, Jiangsu Institute of Cancer Research, Nanjing Medical University Affiliated Cancer Hospital, Nanjing, Jiangsu, China
*These authors contributed equally to this work
Background: Glioblastoma multiforme (GBM) is the most malignant primary tumor of the central nervous system and is associated with a very poor prognosis. No further improvements in outcomes have been reported since radiotherapy-temozolomide therapy was introduced. Therefore, developing new agents to treat GBM is important.
Aim: This study aimed to evaluate the anti-tumor effect of evodiamine (Evo) on GBM cells, and to determine the underlying mechanisms involved.
Results: According to MTT assay results, Evo significantly inhibited the cell proliferation in a time- and dose-dependent manner. Fluorescence microscopy and flow cytometry analyses revealed that Evo induced cell apoptosis in a concentration-dependent manner. Moreover, Evo induced reactive oxygen species (ROS) production and mitochondrial membrane potential (MMP) disruption. Finally, Evo induced apoptosis in cancer cells by suppressing PI3K/AKT signaling and inducing MAPK phosphorylation (p38 and JNK, but not ERK) to regulate apoptotic proteins (Bax, Bcl-2, Cytochrome c, Caspase-3, and PARP).
Conclusion: In summary, Evo inhibits cell proliferation by inducing cellular apoptosis via suppressing PI3K/AKT and activating MAPK in GBM; these results indicate that Evo may be regarded as a new approach for GBM treatment.
Keywords: evodiamine, apoptosis, Akt, MAPK, glioma
Introduction
Gliomas are the most common primary malignant tumors of the central nervous system; glioma has a high incidence rate, and more than 50% of all gliomas are glioblastoma multiforme (GBM).1 The annual incidence of gliomas is increasing worldwide.2 Currently, the first line of treatment is surgical resection, and concomitant chemoradiation with temozolomide (TMZ) is used as the standard treatment; however, at least 50% of TMZ-treated patients do not respond to TMZ, and the prognosis for gliomas is generally poor.3 Despite significant advancements in therapeutic management, the median survival time for GBM remains less than 18 months, and the cumulative survival rate for 1 year remains lower than 30%.4 Therefore, developing more effective agents for treating GBM is important.
In the past few decades, many natural products have attracted great attention for their therapeutic effects against cancer.5 Evodiamine (Evo), a major alkaloidal compound extracted from the fruit of Evodia fructus, has been used traditionally in China.6,7 Previous studies have shown that evodiamine exhibits anti-inflammatory,8 anti-adipogenic,9 and anti-tumor effects on cancer. Evo inhibited cell proliferation through the PI3K/AKT/Caspase and Fas-L/NF-κB signaling pathways, which were augmented by ubiquitin-proteasome inhibition in human melanoma.10 Evo greatly reduced cell growth by inducing apoptosis, independent of p53 status via the intrinsic and extrinsic pathways in lung cancer.11 Evo induced apoptosis via activating JNK and PERK to disrupt mitochondrial membrane potential in ovarian cancer.12 Evo selectively killed cancer stem-like cells by changing the expression of cell cycle proteins via the p53-p21-Rb pathway in breast cancer.13 Evo decreased p-Akt and increased Bax/Bcl-2 and cleaved Caspase-3 levels to induce apoptosis to inhibit cell viability in hepatocellular carcinoma.14 Similarly, Liu et al15 reported that Evo induced dose- and time-dependent apoptosis through calcium signaling or JNK activation in glioma cells. Wu et al16 reported that Evo could induce G2/M arrest and disrupt mitochondrial membrane potential in glioma cells. In the present study, the anti-tumor effects and mechanism of Evo in GBM were investigated. Our results not only confirmed the previous findings of Liu et al15 and Wu et al,16 but also revealed that Evo could activate cellular apoptosis through suppressing PI3K/AKT and activating MAPK in cancer cells.
Materials and methods
Chemicals and reagents
Evo was purchased from Shanghai Aladdin Biochemical Technology Co, Ltd (Shanghai, China), and its purity was ≥99%. The powder was dissolved in dimethyl sulfoxide (DMSO), which was purchased from MP Biomedicals LLC (Santa Ana, CA, USA), to obtain a stock solution of 10 mM. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and 0.25% Trypsin-EDTA (1×) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Cell culture
The human GBM cell lines U251 and LN229 were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM supplemented with 10% FBS. The human normal glial cell line HEB and differentiated PC12 (the neuron-like cell line) were purchased from Nanjing Beierbo Co, Ltd (Nanjing, China). All cells were incubated at 37°C in a humidified atmosphere with 5% CO2. Cells used in the experiments were in the log phase of growth.
Cell proliferation assay
Cell proliferation was measured by MTT assays. MTT powder was purchased from Sigma Chemical Co. (St Louis, MO, USA). The cells were trypsinized and plated at 104 cells per well in a 96-well plate and then cultured overnight. The cells were treated with Evo at the indicated concentrations (0.2, 0.5, 1.0, 2.5, 5.0, 10, 20, and 40 μM) for 24 h and 48 h. Then, 20 μL of MTT reagent (5 mg/mL) was added to each well for 4 h at 37°C. The medium was removed and replaced with 150 μL of dimethyl sulfoxide for 10 min before measurement. The absorbance at 490 nm was assessed by using a microplate reader (Synergy2; BioTek, Winooski, VT, USA).
Hoechst 33258 staining
A Hoechst Staining Kit was purchased from Beyotime Institute of Biotechnology (C0003; Shanghai, China). The cells were seeded in a 6-well plate on coverslips and cultured overnight. After treatment with different concentrations of Evo for 24 h, the cells were fixed in stationary liquid for 10 min, washed with phosphate-buffered saline (PBS), stained with Hoechst 33258 for 10 min and then washed twice with PBS. Finally, the stained cells were visualized using a fluorescence microscope (IX71; Olympus Corporation, Japan).
Cell apoptosis assay
An Annexin V-FITC Apoptosis Detection Kit (c1063; Beyotime, Shanghai, China) was used to detect apoptosis in the cells. The cells were collected 24 h after treatment with the indicated concentrations of Evo, and then stained with Annexin V-FITC and propidium iodide (PI) for 15 min in the dark according to the manufacturer’s instructions. The stained cells were immediately analyzed by flow cytometry (FACSCanto II; BD Biosciences, San Jose, CA, USA).
Reactive oxygen species assay
Reactive oxygen species (ROS) production was detected using dichlorofluorescein diacetate (DCFH-DA) staining. An ROS assay kit was purchased from Beyotime Institute of Biotechnology (S0033; Shanghai, China). Cells treated with Evo for 24 h were incubated with 10 μM DCFH-DA for 20 min at 37°C in the dark, and then washed three times. The harvested cells were analyzed by flow cytometry.
Mitochondrial membrane potential measurement
A mitochondrial membrane potential (MMP) assay kit with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenaimidazolcarbocyanine iodide) was purchased from Beyotime Institute of Biotechnology (C2006; Shanghai, China). The changes in MMP were assessed after cells were treated with Evo for 24 h. After washing with PBS, the cells were incubated in freshly prepared JC-1 solution for 20 min at 37°C. Excess dye was removed by washing with PBS, and cell-associated fluorescence was analyzed on a flow cytometer using 488 nm excitation with 530 nm and 585 nm bandpass emission filters.
Western blot analysis
After treatment with Evo for 24 h, whole cell extracts and cytosol/mitochondria extracts were prepared by using a Whole Cell Lysis Assay Kit (Kgp2100; KeyGEN Biotec, Nanjing, China) and a Cell Mitochondria Isolation Kit (C3601; Beyotime, Shanghai, China), respectively. The protein concentration was measured using BCA (bicinchoninic acid) protein assays. Protein samples were boiled with 6× SDS loading buffer for 5 min, separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to hydrophobic polyvinylidene difluoride membranes. Membranes were blocked for 1 h and then incubated overnight at 4°C with the appropriate antibodies. Caspase-3 (9662), cleaved Caspase-3 (9661), Bax (2772), Bcl-2 (2870), Cytochrome c (4272), phosphoinositide 3-kinase (PI3K) (4249), phospho-PI3K (4228), phospho-Akt (4060), Akt (4691), phospho-SAPK/JNK (9251), SAPK/JNK (9252), phospho-p38 MAP kinase (9211), p38 MAPK (8690), phospho-p44/42 MAPK (Erk1/2) (9101), p44/42 MAPK (Erk1/2) (9102), and β-actin (4970) antibodies were purchased from Cell Signaling Technology (Danvers, CO, USA). Anti-PARP (32138) was from Abcam (Cambridge, UK). After washing three times, the membranes were incubated with horseradish peroxidase-conjugated secondary biotinylated antibodies (Jackson ImmunoResearch Laboratories, Inc, West Grove, PA, USA) for 1 h and washed again three times. The bound proteins were detected using an enhanced chemiluminescent substrate (32106; Thermo Scientific, Boston, MA, USA). The optical density of each protein was determined by using the NIH imaging program (ImageJ).
Statistical analysis
All data represent at least three independent experiments, and are presented as the mean ± SD. All statistical analyses were performed using GraphPad Prism 5.0 software, and comparisons were made using one-way ANOVA tests. P-values of less than 0.05 were considered statistically significant.
Results
Evo inhibited cell proliferation
To examine the effect of Evo on normal astrocytes and neuronal cultures, we first performed MTT assays on HEB and PC12 cells. As shown in Figure 1A, Evo could not induce any cytotoxicity from 0–40 μM. Next, we examined the anti-tumor effects of Evo on glioblastoma cell lines (U251 and LN229) at the indicated concentrations for 24 and 48 h. The proliferation rate was significantly lower in Evo-treated cells than the control cells; in addition, the proliferation rate was decreased in a concentration-dependent manner (Figure 1A). Based on these data, we chose 0.5, 2.5, and 10 μM for U251 cells and 1, 5, and 10 μM for LN229 cells as the working concentrations for the subsequent experiments. Moreover, we observed changes in cell number and cellular morphology by using an inverted microscope. Compared to the number of control cells, the number of Evo-treated cells was progressively reduced when the Evo concentration increased. Cell morphology was also dramatically changed (Figure 1B).
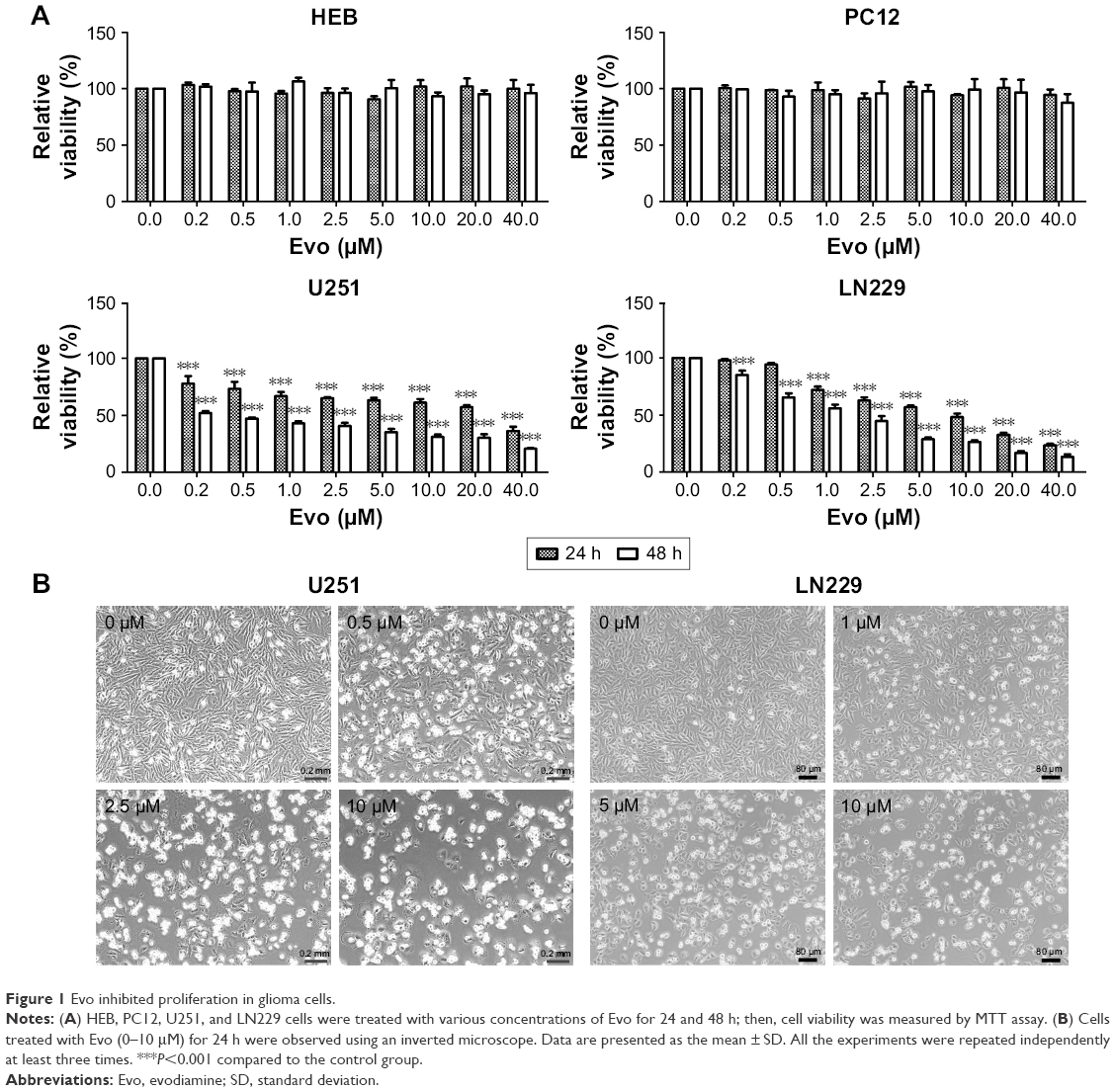 | Figure 1 Evo inhibited proliferation in glioma cells. |
Evo-induced cellular apoptosis, ROS production, and MMP dysfunction
To explore the underlying mechanism through which Evo inhibited cell proliferation in U251 and LN229 cells, we evaluated apoptosis using Hoechst 33258 staining and Annexin V-FITC/PI double staining assays. The images acquired by fluorescence microscopy revealed that nuclei changes, including chromatin condensation, nuclear fragmentation, and apoptotic bodies, were clearly observed in the cells treated with Evo for 24 h; these results indicated that the U251 and LN229 cells treated with Evo underwent morphological changes typical of apoptosis (Figure 2A). Flow cytometric analyses also showed that Evo-induced apoptosis; the percentages of early and late apoptotic cells were significantly increased in the cells after exposure to Evo for 24 h. The percentage of early apoptotic cells increased from 3.8% to 13.8% for U251, and from 2.5% to 20.5% for LN229 after treatment with Evo, whereas the percentage of late apoptotic cells increased from 2.9% to 16.2% for U251, and from 5.0% to 13.7% for LN229 when the Evo concentration increased from 0 μM to 10 μM (Figure 2B and C). Next, we examined the concentration of intracellular ROS using flow cytometry. Evo increased ROS production in a concentration-dependent manner compared to that of the control group (Figure 2D and E). Furthermore, we evaluated whether Evo-induced apoptotic cell death was involved in the mitochondrial pathway. Changes in MMP were next examined using JC-1 staining. Our data revealed that the ratio of red/green fluorescence intensity was lower in both U251 and LN229 cells treated with Evo than the control, indicating that Evo-induced apoptosis was associated with MMP depolarization (Figure 2F and G).
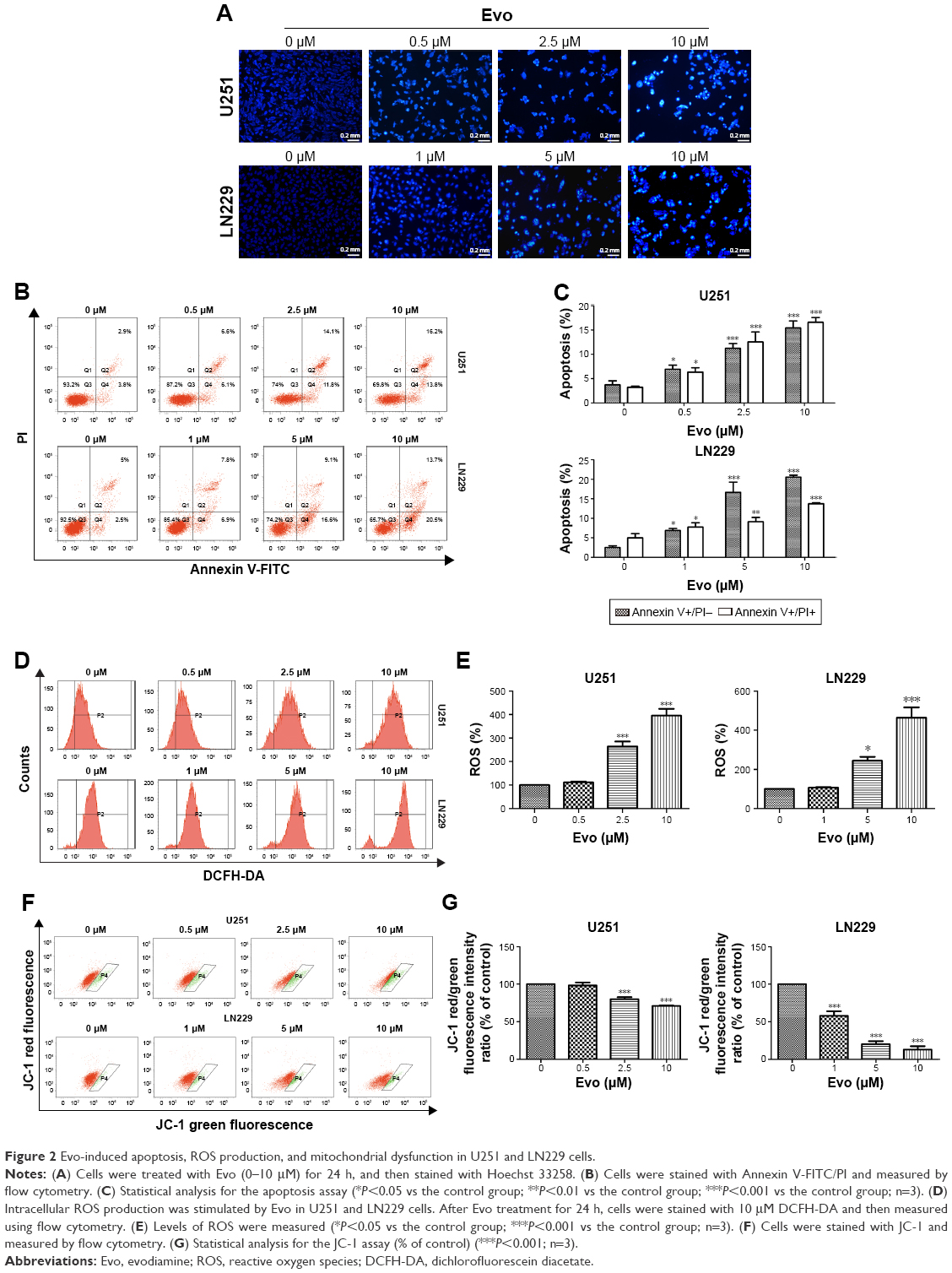 | Figure 2 Evo-induced apoptosis, ROS production, and mitochondrial dysfunction in U251 and LN229 cells. |
Evo suppressed the PI3K/AKT pathway
We next examined the effect of Evo on the PI3K/AKT pathway in U251 and LN229 cells. Although the expression levels of total PI3K were comparable, the phosphorylation levels of PI3K was markedly decreased after Evo treatment, compared to those in the control group in both U251 and LN229 cells (Figure 3A and B). Akt, the key downstream effector of PI3K, was changed similarly to PI3K. The expression levels of p-Akt were decreased in the cells treated with Evo, whereas the levels of total Akt were comparable in both U251 and LN229 cells (Figure 3A and C). Next, we analyzed the protein expression levels of Bax and Bcl-2, which are involved in the intrinsic cell death. With increasing concentrations of Evo, the expression levels of Bcl-2 were decreased. However, the levels of Bax were significantly lower in the Evo-treated group than in the control group, resulting in an obvious dose-dependent increase in the ratio of Bax to Bcl-2 in both U251 and LN229 cells (Figure 3A and D).
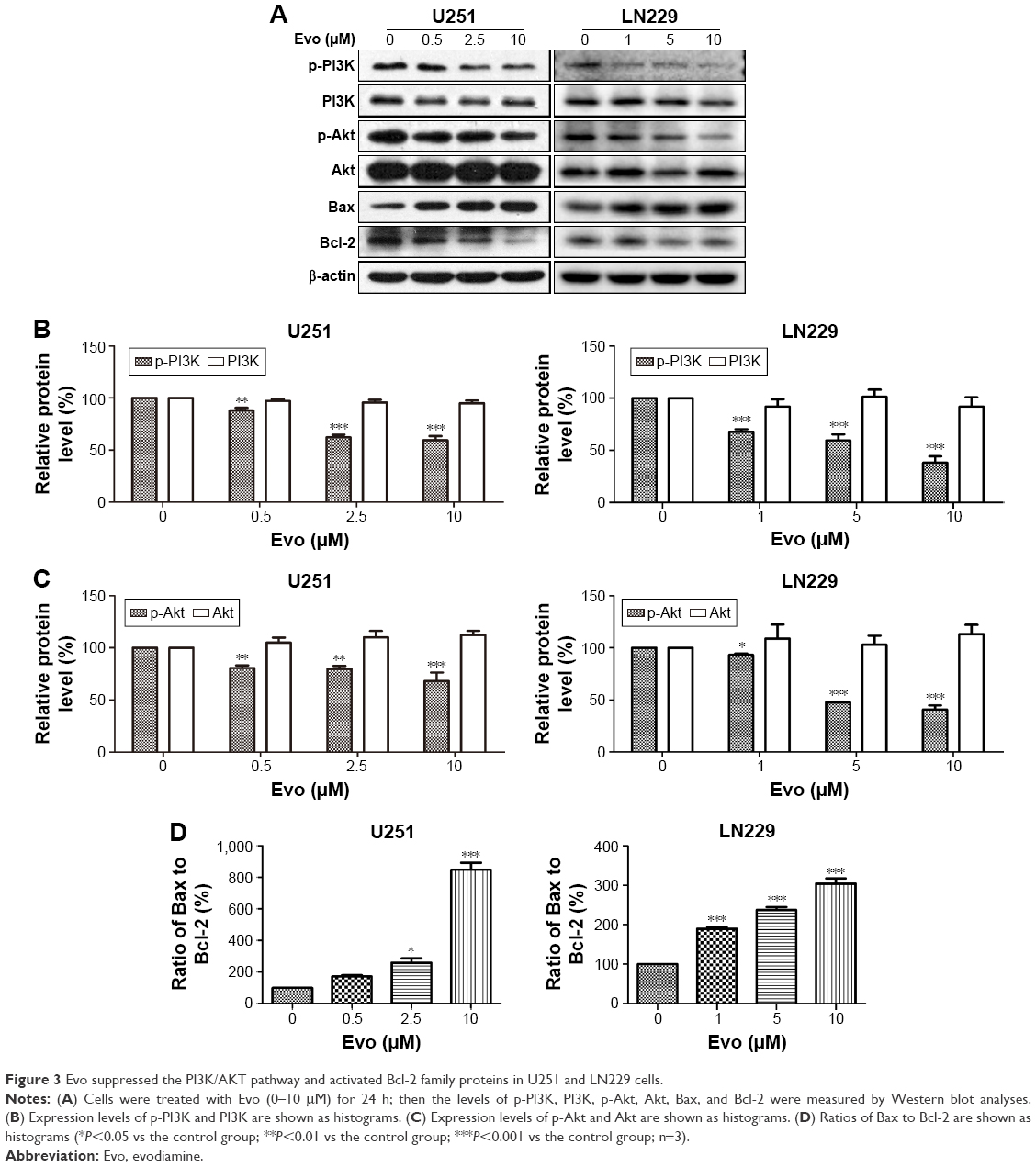 | Figure 3 Evo suppressed the PI3K/AKT pathway and activated Bcl-2 family proteins in U251 and LN229 cells. |
Evo activated the MAPK pathway
To further investigate the anti-tumor mechanism of Evo in U251 and LN229 cells, we next examined the MAPK signaling pathway, which is important for the action of chemotherapeutic drugs. According to Western blot assay results, p-p38 and p-JNK were upregulated in Evo-treated cells compared to the control group cells, but there were no changes in total p38, JNK, and ERK (Figure 4A–C). These data indicated that the activation of JNK and p38 MAPKs was mainly involved in Evo-induced apoptosis in glioma cells. We also measured Cytochrome c release from the mitochondria to the cytosol. In both U251 and LN229 cells, higher expression levels of Cytochrome c in the cytosol correlated with higher Evo concentrations (Figure 4A and D). Next, we measured the protein expression levels of cleaved Caspase-3 and poly (ADP ribose) polymerase (PARP). Cleaved Caspase-3 and cleaved PARP were expressed at higher levels in Evo-treated cells than control cells, whereas full-length Caspase-3 and pro-PARP levels were reduced in U251 and LN229 cells treated with Evo (Figure 4A, E and F). It was worthy to note that p-ERK was inhibited after Evo treatment in LN229 cells. However, we did not observe any differences in the phosphorylation levels of ERK between Evo-treated U251 cells and non-treated cells (Figure 4A and G). Taken together, these data suggest that evodiamine could suppress the PI3K/AKT pathway and activate the MAPK pathway to inhibit cell proliferation and induce cellular apoptosis in GBM (Figure 5).
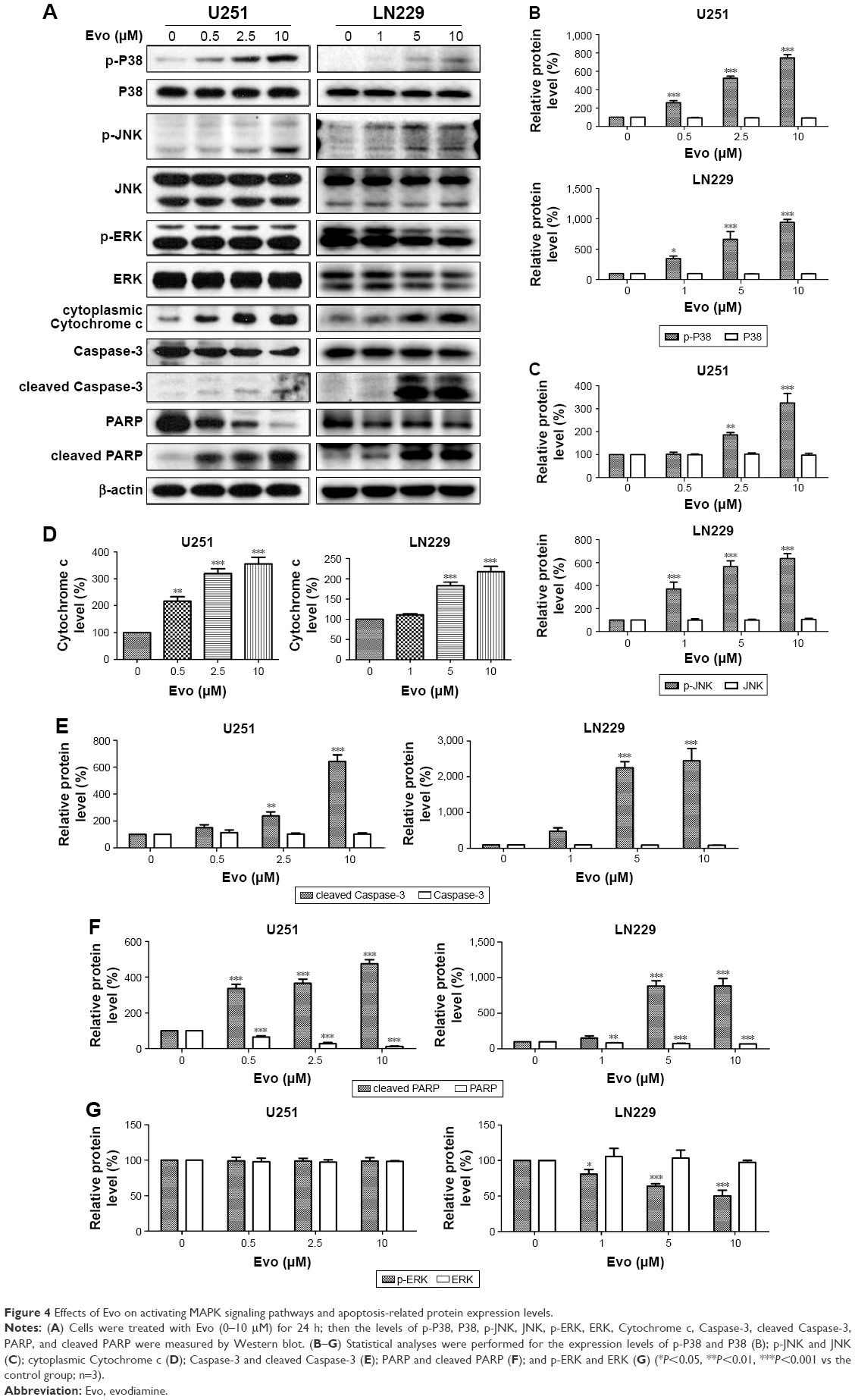 | Figure 4 Effects of Evo on activating MAPK signaling pathways and apoptosis-related protein expression levels. |
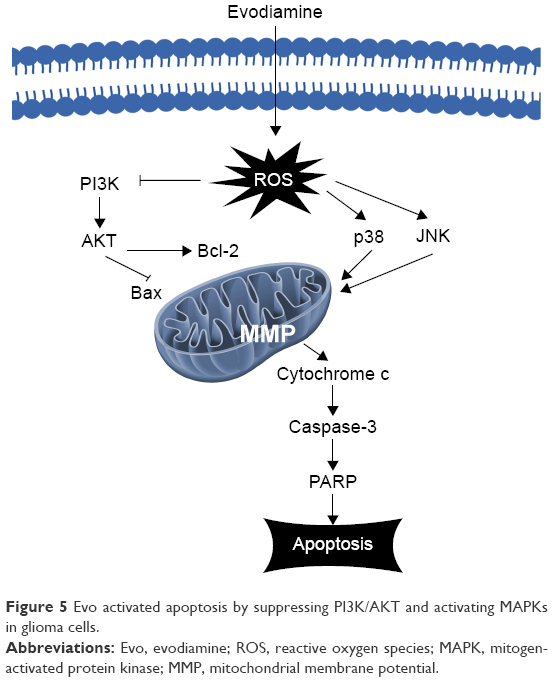 | Figure 5 Evo activated apoptosis by suppressing PI3K/AKT and activating MAPKs in glioma cells. |
Discussion
In recent years, natural Chinese compounds, especially plant-derived compounds, have received extensive attention as sources of new drugs for anti-cancer therapy.17 Evo, a quinolone alkaloid, is traditionally used for headache or stomach ache in China.18 However, its effects on brain tumors and its potential mechanisms are unclear. In the present study, Evo inhibited cell proliferation in a concentration- and time-dependent manner. Evo promoted cell apoptosis in a concentration-dependent manner by inducing chromosome condensation, nuclear fragmentation, and apoptotic bodies. Mechanistic studies revealed that Evo increased ROS production and caused MMP disruption. Furthermore, Evo induced apoptosis by suppressing PI3K/AKT signaling and inducing MAPK phosphorylation in GBM cancer cells.
Natural products typically exhibit their anti-cancer activities by triggering apoptosis.19 The apoptosis is initiated through increases in intracellular ROS.20 ROS function as an important intracellular signaling molecule and play an essential role in cell homeostasis, cell proliferation, and cell migration.21 When the production rate of ROS is greater than its clearance rate, cellular apoptosis is triggered.22 We observed that Evo induced ROS production in glioma cells, which indicated that the generation of ROS was a key event in mediating the apoptotic effects of Evo in glioma cells. Alterations to MMP during apoptosis have been reported in a number of studies; these data suggest that the depolarization of mitochondria is one of the first events occurring during apoptosis, which can induce cell death through caspase activation, chromatin condensation, and DNA fragmentation.23 We observed that Evo significantly induced the loss of the MMP in a dose-dependent manner, which was consistent with the morphology changes observed by microscopy.
Upon oxidative stress exposure, various signal transduction pathways may be activated in cells.24 PI3K/AKT signaling is frequently deregulated in various cancers and plays an important role in a variety of biological processes, including proliferation, migration, apoptosis, and differentiation.25,26 Western blot analyses indicated that Evo treatment considerably reduced PI3K/AKT pathway activation in GBM cells. The MAPK signaling pathway controls fundamental cellular processes in cancer, such as proliferation, apoptosis, and response to chemotherapeutic drugs.27,28 Currently, there are three main groups of mammalian MAPKs: ERK (p44/p42), JNK, and p38. JNK and p38 are involved in cell apoptosis, while ERK is associated with cell proliferation, migration, and senescence.29,30 Our findings revealed that treating GBM cells with Evo induced the activation and phosphorylation of p38 and JNK, but not ERK, indicating that p38 and JNK may play major roles in Evo-induced apoptosis, whereas ERK likely does not.
Apoptosis is a type of programmed cell death.31,32 There are two main pathways, the intrinsic and extrinsic cell death pathways; both of these result in caspase activation.33 The Bcl-2 family of proteins regulates intrinsic apoptosis through mitochondrial permeabilization.34 After permeabilization, the release of Cytochrome c from the mitochondrial intermembrane space to the cytoplasm promotes caspase activation and apoptosis.35 The extrinsic cell death pathway is activated by death receptors, which interact and recruit other adaptor molecules, such as FADD and caspase-8, and finally lead to apoptosis.36 In our study, Western blot assays showed that the expression levels of cleaved Caspase-3 and cleaved-PARP and the Bax/Bcl-2 ratio, as well as Cytochrome c release, were considerably higher in Evo-treated glioma cells than non-treated cells. These results suggest that Evo induces apoptosis in glioma cells through the intrinsic cell death pathways. In addition, we observed that Evo could not induce any cytotoxicity in normal astrocytes and neuronal cultures at a high concentration, but it could induce obvious cytotoxicity in glioma cells. Wu et al16 also reported that Evo was able to penetrate the blood–brain barrier, and could inhibit glioma in vivo. These results suggest that Evo may be a safe and therapeutic agent against glioma.
In summary, we have demonstrated that Evo exhibited potent anti-tumor effects against human GBM cells and promoted apoptosis through suppressing PI3K/Akt and activating MAPK. Based on these findings, Evo is a therapeutic agent worthy of further development as a clinical trial candidate for treating GBM.
Acknowledgments
This work was supported by Jiangsu Provincial Special Programme of Medical Science (BL2014035), Changzhou Science and Technology Support Program (CE20155060, CE20165048), Changzhou High-Level Medical Talents Training Project (2016CZBJ006), and Changzhou Municipal Commissions of Health and Family Planning Major Scientific and Technological Project (ZD201620).
Disclosure
The authors report no conflicts of interest in this work.
References
Reni M, Mazza E, Zanon S, Gatta G, Vecht CJ. Central nervous system gliomas. Crit Rev Oncol Hematol. 2017;113:213–234. | ||
Chen R, Cohen AL, Colman H. Targeted therapeutics in patients with high-grade gliomas: past, present, and future. Curr Treat Options Oncol. 2016;17(8):42. | ||
Jiang H, Cui Y, Wang J, Lin S. Impact of epidemiological characteristics of supratentorial gliomas in adults brought about by the 2016 World Health Organization classification of tumors of the central nervous system. Oncotarget. 2017;8(12):20354–20361. | ||
Grier JT, Batchelor T. Low-grade gliomas in adults. Oncologist. 2006;11(6):681–693. | ||
Luo R, Wang J, Zhao L, et al. Synthesis and biological evaluation of baicalein derivatives as potent antitumor agents. Bioorg Med Chem Lett. 2014;24(5):1334–1338. | ||
Liao JF, Chiou WF, Shen YC, Wang GJ, Chen CF. Anti-inflammatory and anti-infectious effects of Evodia rutaecarpa (Wuzhuyu) and its major bioactive components. Chin Med. 2011;6(1):6. | ||
Yu H, Jin H, Gong W, Wang Z, Liang H. Pharmacological actions of multi-target-directed evodiamine. Molecules. 2013;18(2):1826–1843. | ||
Lv Q, Xue Y, Li G, et al. Beneficial effects of evodiamine on P2X(4)-mediated inflammatory injury of human umbilical vein endothelial cells due to high glucose. Int Immunopharmacol. 2015;28(2):1044–1049. | ||
Wang T, Wang Y, Yamashita H. Evodiamine inhibits adipogenesis via the EGFR-PKCalpha-ERK signaling pathway. FEBS Lett. 2009;583(22):3655–3659. | ||
Wang C, Li S, Wang MW. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-kappaB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol In Vitro. 2010;24(3):898–904. | ||
Mohan V, Agarwal R, Singh RP. A novel alkaloid, evodiamine causes nuclear localization of cytochrome-c and induces apoptosis independent of p53 in human lung cancer cells. Biochem Biophys Res Commun. 2016;477(4):1065–1071. | ||
Chen TC, Chien CC, Wu MS, Chen YC. Evodiamine from Evodia rutaecarpa induces apoptosis via activation of JNK and PERK in human ovarian cancer cells. Phytomedicine. 2016;23(1):68–78. | ||
Han S, Woo JK, Jung Y, et al. Evodiamine selectively targets cancer stem-like cells through the p53-p21-Rb pathway. Biochem Biophys Res Commun. 2016;469(4):1153–1158. | ||
Yang F, Shi L, Liang T, et al. Anti-tumor effect of evodiamine by inducing Akt-mediated apoptosis in hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;485(1):54–61. | ||
Liu AJ, Wang SH, Chen KC, et al. Evodiamine, a plant alkaloid, induces calcium/JNK-mediated autophagy and calcium/mitochondria-mediated apoptosis in human glioblastoma cells. Chem Biol Interact. 2013;205(1):20–28. | ||
Wu WS, Chien CC, Liu KH, Chen YC, Chiu WT. Evodiamine prevents glioma growth, induces glioblastoma cell apoptosis and cell cycle arrest through JNK activation. Am J Chin Med. 2017;45(4):879–899. | ||
Zhou XJ, Liu M, Yan JJ, Cao Y, Liu P. Antidepressant-like effect of the extracted of Kai Xin San, a traditional Chinese herbal prescription, is explained by modulation of the central monoaminergic neurotransmitter system in mouse. Jf Ethnopharmacol. 2012;139(2):422–428. | ||
Gavaraskar K, Dhulap S, Hirwani RR. Therapeutic and cosmetic applications of Evodiamine and its derivatives – A patent review. Fitoterapia. 2015;106:22–35. | ||
Tan W, Lu J, Huang M, et al. Anti-cancer natural products isolated from Chinese medicinal herbs. Chin Med. 2011;6(1):27. | ||
Gniadecki R, Thorn T, Vicanova J, Petersen A, Wulf HC. Role of mitochondria in ultraviolet-induced oxidative stress. J Cell Biochem. 2000;80(2):216–222. | ||
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. | ||
Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005;70(2):229–241. | ||
Smaili SS, Hsu YT, Youle RJ, Russell JT. Mitochondria in Ca2+ signaling and apoptosis. J Bioenerg Biomembr. 2000;32(1):35–46. | ||
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. | ||
Carnero A, Paramio JM. The PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front Oncol. 2014;4:252. | ||
Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. | ||
Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis. 2002;23(11):1831–1838. | ||
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. | ||
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | ||
Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60(3):261–310. | ||
Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–219. | ||
Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7(7):532–542. | ||
Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 2015;16(2):2942–2955. | ||
Hata AN, Engelman JA, Faber AC. The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015;5(5):475–487. | ||
Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai). 2005;37(11):719–727. | ||
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.