")
Back to Journals » OncoTargets and Therapy » Volume 12
Evidence to date: talazoparib in the treatment of breast cancer
Authors Exman P, Barroso-Sousa R, Tolaney SM
Received 28 February 2019
Accepted for publication 3 June 2019
Published 2 July 2019 Volume 2019:12 Pages 5177—5187
DOI https://doi.org/10.2147/OTT.S184971
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jianmin Xu
Pedro Exman, Romualdo Barroso-Sousa, Sara M Tolaney
Breast Oncology Program, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
Abstract: Approximately 5–10% of all patients diagnosed with breast cancer have germline BRCA1/2 mutations, which make their disease more susceptible to DNA-damaging agents and a new class of drugs known as poly(ADP-ribose) polymerase (PARP) inhibitors. Talazoparib is a new PARP inhibitor that has been recently approved for use in patients with metastatic breast cancer with germline BRCA mutations after a phase III trial showed superior progression-free survival when compared to standard chemotherapy. In this review, we analyze the development of talazoparib as well as its safety profile and the potential role of the combination therapy with standard cytotoxic drugs and with novel therapies.
Keywords: talazoparib, breast cancer, BRCA mutation, homologous recombination deficiency
Introduction
Breast cancer is the leading cause of cancer death in women, and according to the American Cancer Society, there were an estimated 41,400 breast cancer–related deaths in the United States in 2018.1 The increased understanding of tumor biology, availability of better screening, and the development of more effective drugs have been associated with a reduction in breast cancer mortality.2 Approximately 50% of newly diagnosed breast cancer cases are associated with factors related to hyperestrogenic conditions, such as early age at menarche, older age at first live birth, older age at menopause, and obesity. Genetically, the majority of genomic alterations in breast cancer are sporadic and just 5–10% are germline mutations are classified as hereditary.3
The most common germline mutations related to breast cancer are in the breast and ovarian cancer susceptibility gene 1 (BRCA1) or BRCA2. Mutations in these genes, located on chromosome 17 and 13, respectively, are highly penetrant, classically more frequent in Ashkenazi Jews, and increase the lifetime risk of breast cancer up to 70% in patients with BRCA1 mutations and between 40% and 70% in patients with BRCA2 mutations.3
BRCA1 and BRCA2 proteins, which are at maximal expression during the DNA replication phases (S and G2 phases), present an essential role for cellular survival by integrating a complex mechanism of DNA double-strand damage restoration called homologous recombination repair.4 BRCA1 interacts with the MRE1/RAD50/Nbs1 protein complex and identifies, through histone phosphorylation, sites of DNA damage and subsequently resets and recreates the double-stranded DNA. Other indirect roles of BRCA1 DNA repair are degradation of proteins in the ubiquitin-mediated regulation and chromatin remodeling after double-strand damage.5,6 Similarly, BRCA2 is directly involved in homologous recombination repair by regulating an essential enzyme (RAD51), whose main function is to initiate the base pairing during the formation of the DNA double strand.7 The other two major pathways for double-strand damage repair are known as classical nonhomologous end joining (C-NHEJ) and alternative nonhomologous end joining (A-NHEJ). In C-NEHJ, BRCA1 interacts with Ku80 factor to stabilize Ku heterodimer complex binding to chromosomal breaks, promoting precisely the re-ligation of the broken DNA in the end of the double strand in the G1 phase. Additionally, A-NHEJ, also regulated by BRCA1, is active in all phases of the cell cycle and is completely independent of C-NHEJ, using an enzymatic complex that creates micro-homology to repair double-strand breaks.8,9
Dysfunctional BRCA1 or 2 proteins are responsible for the pathological accumulation of deficient chromosomal re-arrangement, such as translocations, deletions and broken chromosomes and consequent atypical cellular replication, leading to an increased risk of cancer. Breast cancers associated with BRCA1 mutations are mainly triple-negative (only ~10% are HER2-positive) and exhibit a higher mitotic index and increased lymphocytic infiltration compared to sporadic cancers. Alternatively, the majority of BRCA2 cancers are estrogen receptor-positive and have a lower mitotic index.10
Considering this specific disease biology, studies have investigated tailoring treatment for patients with BRCA1/2 mutations. Several preclinical and clinical studies have demonstrated that BRCA-associated cancers are associated with a higher sensitivity to chemotherapy, particularly platinum salts which cause covalent crosslinks in cell DNA and subsequently impair the ability of cells to repair this damage. High proliferation rates, as seen in tumors, and the absence of competent homologous recombination make cells more susceptible to these drugs.11
In addition, poly(ADP-ribose) polymerase-1/2 (PARP1/2) inhibitors have emerged as novel agents that act as a personalized targeted treatment in patients with BRCA1/2 mutations by inhibiting the single-strand DNA repair mechanism. In this context, two PARP inhibitors—olaparib (Lynparza®, AstraZeneca) and, more recently, talazoparib (Talzenna®, Pfizer)—have been approved by the United States Food and Drug Administration (FDA) for use in advanced breast cancer. Table 1 presents key data of the two FDA-approved PARP inhibitors.12–18
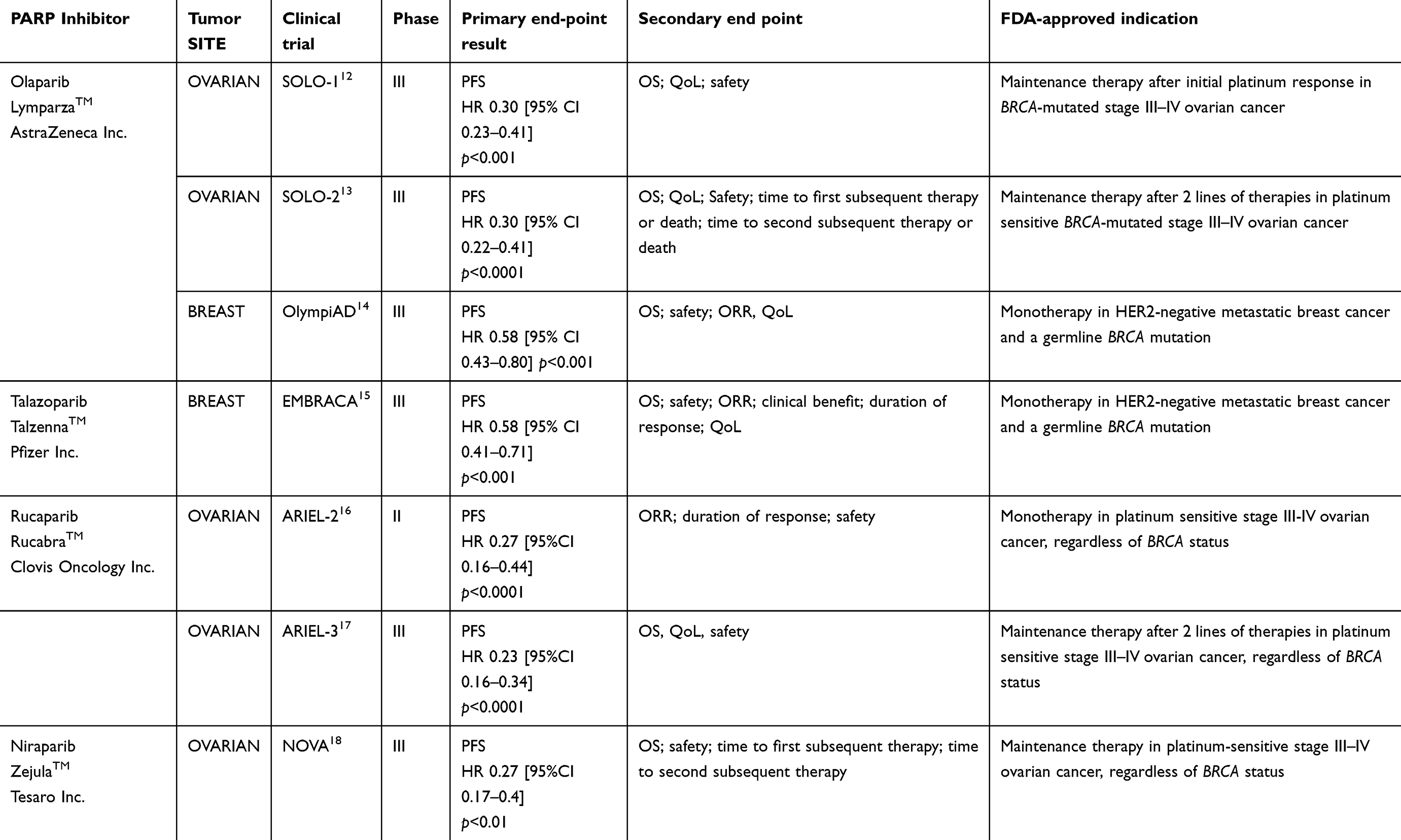 |
Table 1 FDA-approved PARP inhibitors |
This review will analyze talazoparib from its preclinical development to the recent randomized phase III trial, which led to the approval of this agent in the treatment of BRCA-mutated metastatic breast cancer.
Mechanism of action and synthetic lethality
Among all DNA damage repair mechanisms, PARP1 and PARP2 enzyme isoforms have a key role in base excision repair in response to single-stranded DNA breaks. Through recognition of DNA breaks, PARP1/2 are activated using ADP ribose and target histones to expose the chromatin to DNA repair by recruitment of restoration proteins. This process allows the stopped DNA replication and transcription processes to resume.19,20
PARP inhibition prevents the interaction between the enzyme itself and the ADP ribose, a process called PARylation, which impedes the DNA repair, maintaining single-strand breaks and a stalled replication fork, ultimately leading to a double-strand break. In physiologic conditions, proficient homologous recombination machinery would repair the double-strand damage and avoid cellular death. Conversely, in patients with BRCA mutation and other homologous recombination deficiencies, the DNA double-strand repair does not occur in the setting of PARP inhibition, causing apoptosis.21 This mechanism, which adds successive genetic repair defects leading to cellular death, is known as synthetic lethality and makes PARP inhibitors important drugs in the treatment of BRCA-mutated patients.22
An additional characteristic of PARP inhibitors is the ability to prevent the dissociation of PARP from DNA damage sites on chromatin, a process known as DNA trapping. This binding of PARP1/2 enzymes to the single-strand broken DNA is believed to be directly responsible for 2 different processes that cause cellular death: 1) the formation of cytotoxic PARP–DNA complex that directly causes cellular death and 2) trapped PARP inhibits the repair of single-stranded DNA more severely than the absence of PARP, which causes a higher level of double-strand breaks in DNA during replication and therefore cell death.21,22
Introduction to the drug
PARP inhibitors are molecular analogs of ADP ribose, inhibiting the interaction between PARP enzyme and the ADP ribose, and act as a PARP trapping that not just interferes with DNA repair, transcription and replication, but also causes direct lethal DNA double-strand breaks during S-phase by collapse of stalled replication forks. Currently, there are two FDA-approved PARP inhibitors for metastatic breast cancer: talazoparib (Talzenna, Pfizer Inc.) and olaparib (Lynparza, AstraZeneca Inc.). There are also 3 agents that are still in development: veliparib, niraparib and rucaparib.
Like other PARP inhibitors, talazoparib (formerly known as BMN673) demonstrated in-vitro antitumor activity only in tumors with defects in homologous recombination, mainly BRCA mutations. However, more potent antitumor responses at much lower concentrations were observed when compared to other PARP inhibitors. In a preclinical study, talazoparib demonstrated more potent antitumor activity with half maximal inhibitory concentration (IC50) of 0.57 nmol/L compared to veliparib (4.7 nmol/L), olaparib (2.0 nmol/L) and rucaparib (1.9 nmol/L). Although talazoparib enhanced antitumor effects of some cytotoxic agents (temozolomide, cisplatin, and carboplatin) in xenograft models, it showed superior PARP inhibition and activity as monotherapy.23,24 In addition to a higher catalytic inhibition of PARP enzymes, talazoparib has also demonstrated the highest efficiency (~100-fold more potent than olaparib) at trapping PARP–DNA complexes in preclinical studies.23–25
In this context, talazoparib emerged as novel potent PARP1/2 inhibitor with clinical potential for patients with germline BRCA mutations.
Phase I trial
The first-in-human phase I trial evaluating talazoparib was published by de Bono et al.25 This two-part study evaluated talazoparib in patients with advanced solid malignancies harboring germline BRCA1/2 mutations (Part 1, dose-escalation, and Part 2, expansion cohort). Initially, breast cancer cases were restricted to triple-negative disease and 8 patients were enrolled in part 1 followed by 12 (20.5%) enrolled in part 2, the expansion cohort (16.9%). A total of 110 patients received talazoparib and the study enrolled patients with other solid tumors including ovarian and peritoneal cancer (30.9%), prostate (3.6%), Ewing sarcoma (12.7%) and pancreatic cancers (11.8%).
In part 1, 2 of 6 patients experienced a dose-limiting toxicity (DLT) with grade 3 and 4 thrombocytopenia after being treated with 1.1 mg/day of talazoparib. The interim dose of 1.0mg/day was not associated with any DLTs and it was established as the maximum tolerated dose. In part 2, 71 patients received 1.0 mg/day continually with good tolerance. The most frequent any grade toxicities were hematological: anemia (35%), thrombocytopenia (21%) and neutropenia (15%). Nonhematological any grade side effects included nausea (32%), fatigue (37%) and alopecia (20%). Grade 3 and 4 adverse events were anemia (23%), thrombocytopenia (18%) and neutropenia (10%). Two patients (3%) experienced grade 3 or 4 fatigue. Pharmacokinetically, the median half-life was 2 days and plasma concentrations were maintained above 10 nmol/L. Constant PARP inhibition was seen in doses of ≥0.6 mg/day.25
Patients with breast cancer experienced higher objective response rates (ORR) compared to other tumor types. In 14 patients with BRCA-mutated breast cancer, the ORR was 50%. Five patients (35%) had stable disease and 1 (7%) had a complete response, with clinical benefit observed in 85% of the patients. In addition, progression-free survival (PFS) was 34.6 weeks in heavily pretreated patients.25
Phase II trial
With final results presented at the American Society of Clinical Oncology Annual Meeting in 2017 and recently published in Clinical Cancer Research, the ABRAZO trial was a 2-cohort, 2-stage phase II study that evaluated the use of talazoparib in patients with germline BRCA1/2- mutated metastatic breast cancer.26,27 Cohort 1 evaluated talazoparib in patients who received previous platinum treatment and cohort 2 consisted of platinum-naïve patients who had received at least 3 prior lines of therapy. A total of 49 patients were enrolled in cohort 1 and 35 in cohort 2; all received 1 mg of talazoparib once daily by mouth as continuous therapy until disease progression or unacceptable toxicity. Not surprisingly, cohort 1 had more triple-negative patients (59%) than cohort 2 (17%) and median of previous lines of therapy were 2 and 4, respectively. The primary end point was ORR and secondary end points included safety, PFS and overall survival (OS).
After a median follow-up of 13.7 months for each cohort, patients in cohort 1 had an ORR of 21% and patients in cohort 2 had an ORR of 37%. Remarkably, exploratory subgroup analyses suggested that a longer platinum-free interval following the last dose of platinum was associated with greater clinical activity. Although there was a small difference in ORR for patients with BRCA1 versus BRCA2 (23% versus 33%) mutations, no difference in ORR was observed when comparing patients with triple-negative (ORR=26%) versus hormone receptor-positive disease (ORR=29%).
PFS was 4.0 months (95% CI 2.8–5.4) in cohort 1 and 5.6 months in cohort 2 (95% CI 5.5–7.8). The median OS was 12.7 months (95% CI 9.6–15.8) in cohort 1 and 14.7 months (95% CI 11.0–24.4) in cohort 2. Common all grade adverse events observed were anemia (52%), fatigue (45%), nausea (42%), diarrhea (33%), thrombocytopenia (33%) and neutropenia (27%). Hematological grade 3 or 4 adverse events were anemia (35%), thrombocytopenia (19%), and neutropenia (15%); no nonhematological grade 3 or 4 events were observed, and only 4% of patients discontinued use of talazoparib due to adverse events.26,27
Phase III trial
The pivotal study which led to FDA approval of talazoparib was the EMBRACA trial.15 This phase III trial randomized 431 patients (2:1) and compared the efficacy and safety of talazoparib with standard chemotherapy of the physician’s choice for patients with metastatic breast cancer with a germline BRCA1/2 mutation, regardless of hormonal status. Patients may have received up to 3 previous lines of systemic chemotherapy and physician’s choice agents included capecitabine (44% of the patients), eribulin (40%), gemcitabine (10%) and vinorelbine (7%). There was no direct comparison with carboplatin or cisplatin, but patients were allowed to have previously received platinum agents in the neoadjuvant or adjuvant setting provided they had a disease-free interval of at least 6 months. Platinum was allowed in the metastatic setting, though patients were not allowed to have progressed on it. The primary end point was PFS and secondary end points included OS, response rate, safety and quality of life.
After a median follow-up of 11.2 months, there was a significant absolute improvement in PFS of 3 months in the talazoparib group compared with the physician’s choice group (8.6 months vs 5.6 months; HR 0.54 95% CI, 0.41 to 0.71; p<0.001). Of note, a subgroup analysis has demonstrated significant benefit of talazoparib over chemotherapy in all clinically relevant groups, but patients who received platinum agents previously did not reach a statistically significant gain (HR 0.76; 95% CI 0.40–1.45). In addition, the magnitude of benefit of talazoparib was similar among patients with BRCA1 versus BRCA2 mutations (HR 0.59 [95% CI 0.39–0.90] and 0.47 [95% CI 0.320.70], respectively); patients with triple-negative disease versus hormone receptor-positive disease (HR 0.60 [95% CI 0.41–0.87] and 0.47 [95% CI 0.32–0.71], respectively) and in patients who received talazoparib as first-line treatment versus those who received it as second or third/fourth line treatment (HR 0.57 [95% CI 0.34–0.95]; HR 0.51 [95% CI 0.33–0.80]; and HR 0.56 [95% CI 0.34–0.95], respectively).
As secondary end points, the ORR was 62.6% (95% CI, 55.8–69.0) in the talazoparib group versus 27.2% (95% CI, 19.3–36.3) among those who received standard therapy. Twelve patients who received talazoparib (5.5%) had a complete response. A subgroup analysis demonstrated similar response rates when comparing patients with BRCA1 versus BRCA2 mutations (64.1% and 62.3%, respectively); patients with triple-negative disease versus hormone receptor-positive disease (61.8 and 63.2, respectively) and patients with a history of brain metastases versus patients without brain disease (63.2 and 62.4, respectively). Patients who were previously exposed to platinum presented lower response rates when compared to platinum-naïve patients (50.0 and 65.2, respectively).
The interim OS analysis showed no significant differences between arms (22.3 months in the talazoparib group and 19.5 months in the standard therapy group [HR 0.76; 95% CI 0.55–1.06, p=0.11]), but the follow-up was short. Similar to the ABRAZO trial, common adverse effects were fatigue (any grade 50.3%), anemia (any grade 52.8%) and nausea (any grade 48.6%). Hematologic grade 3 or 4 adverse events occurred more frequently in the talazoparib arm (55% versus 38%) and non-hematologic grade 3 events were similar between arms (32% of patients in the talazoparib group and 38% of patients in the standard therapy group). Finally, a significant improvement in the quality of life was observed in the talazoparib group when compared to the standard treatment group.
Safety, tolerability and quality of life
As described earlier, talazoparib causes a strong inhibition of PARP compared to other drugs, and adverse events and patient’s quality of life should be carefully evaluated. Table 2 shows the most common adverse events reported in the main talazoparib trials.15,25,27
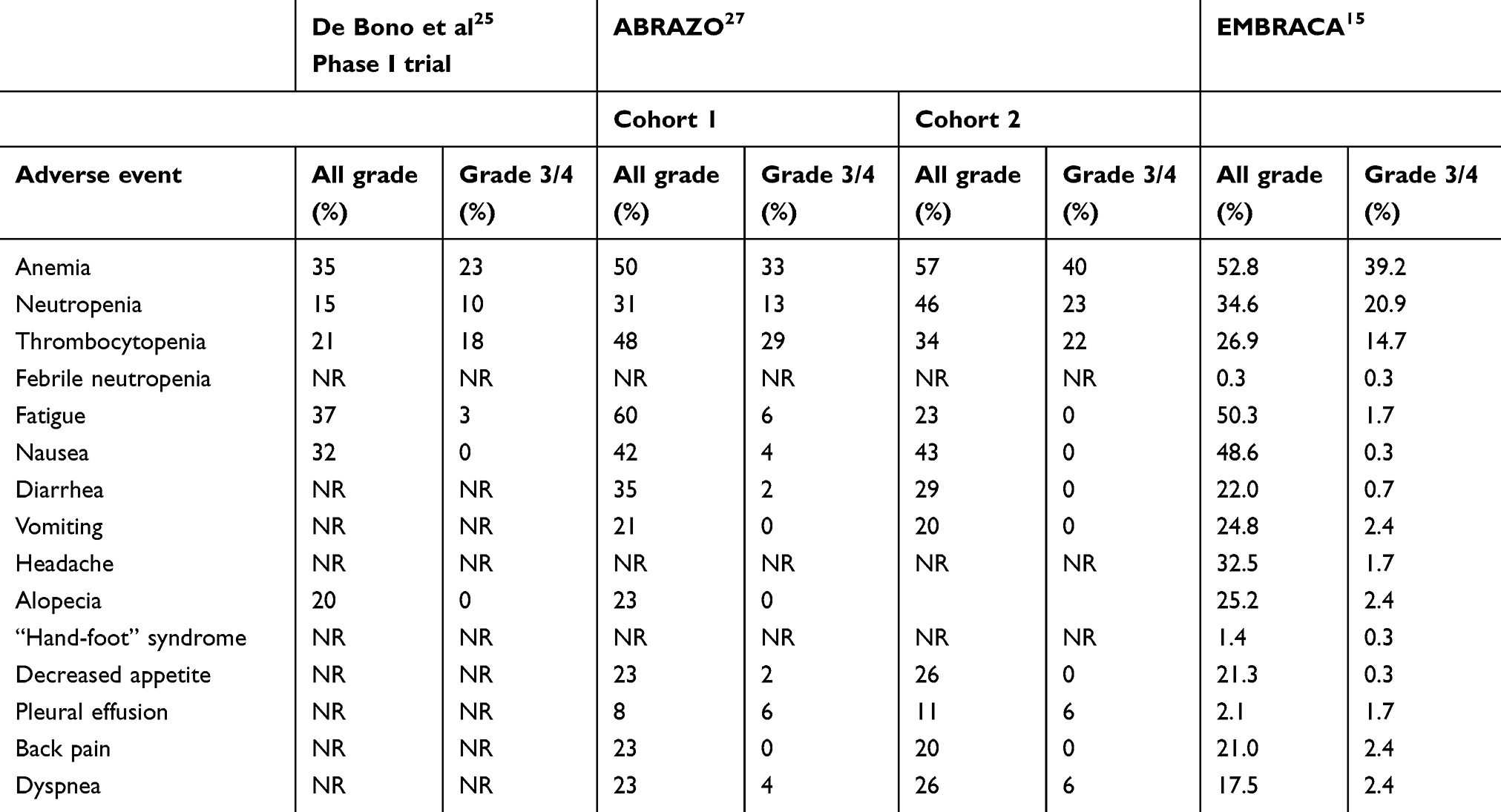 |
Table 2 Adverse events in published clinical trials of talazoparib |
In the ABRAZO trial,27 drug discontinuation due to toxicity was observed in only 3 patients (one patient with anemia and two with liver test abnormalities) and time to first dose reduction was 10.9 weeks in cohort 1 and 13.3 weeks in cohort 2. In addition, a high proportion (almost 30%) of patients needed blood transfusions due to symptomatic anemia during treatment. Grade 3 anemia was observed in 33% of the patients in cohort 1 and in 40% of the patients in cohort 2. No grade 4 anemia was observed. Relevant thrombocytopenia was also observed in both cohorts of the trial. Grades 3 and 4 thrombocytopenia were observed in 23% and 6%, respectively, in cohort 1, and in 11% (grades 3 and 4) in cohort 2. No acute myeloid leukemia or myelodysplastic syndrome related to talazoparib exposure was observed. Grade 3 or 4 nonhematological adverse events were not frequent: 6% of patients presented with grade 3 fatigue and 4% with grade 3 nausea. Interestingly, all grade fatigue was more frequent in patients who were previously exposed to platinum when compared to platinum-naïve patients (60% versus 23%).26,27
Similarly, the EMBRACA trial demonstrated a favorable safety profile for talazoparib, mainly when compared to physician’s choice chemotherapy.15 Only 5.9% of the patients had to discontinue the treatment due to severe side effects while most of the patients (50%) had a dose reduction after day 93 (13.3 weeks), and 25.5% presented grade 3 or 4 toxicity. Grade 3 anemia was observed in 38.5% of the patients, which was higher than seen in the ABRAZO trial. Also, grade 3 neutropenia was seen in 17.8% of the patients, but only one patient (0.3%) experienced febrile neutropenia (grade 4). Likewise, no cases of acute myeloid leukemia or myelodysplastic syndrome were reported in the talazoparib group, but one case was observed in a patient who received capecitabine. Nonhematologic grade 3 events were less frequent in the EMBRACA trial when compared to the ABRAZO study. Grade 3 or 4 fatigue was only seen in 1.7% of the patients and grade 3 or 4 vomiting was observed in 2.4% of the patients.15
Patient-reported quality of life was superior in the EMBRACA trial compared to treatment of physician’s choice.28,29 A statistically significant higher time to deterioration of quality of life was observed in the group of patients who received talazoparib over physician’s choice chemotherapy (median time 24.3 months versus 6.3, respectively; HR 0.38 95% CI 0.26–0.55, p=0.0001). Multiple treatment-specific and breast cancer-specific symptoms were also better in the talazoparib group.28,29
Table 3 shows the comparison of main adverse events between two FDA-approved PARP inhibitors in metastatic breast cancer.14,15
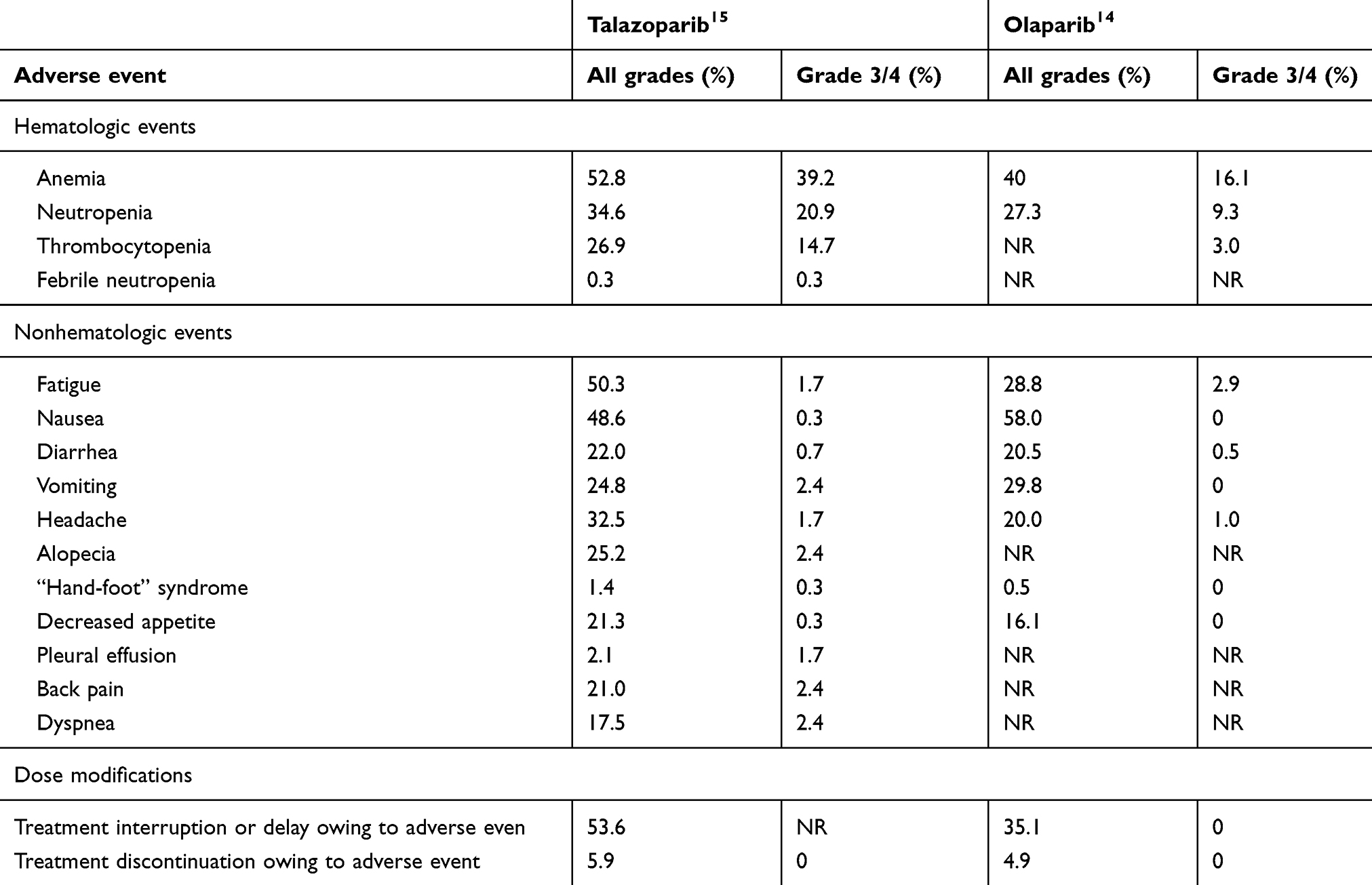 |
Table 3 Adverse events reported between the two approved PARP inhibitors, olaparib and talazoparib, in phase III trials in breast cancer |
Potential biomarkers
The development of reliable predictive biomarkers for PARP inhibitors aiming to determine which patients may have a greater benefit from receiving these drugs is still lacking. As mentioned earlier, BRCA1/2 mutation is the only established biomarker that confers sensitivity to the use of PARP inhibitors. Although the benefit was substantial in both BRCA1 or BRCA2 mutated patients, the subgroup analysis of the EMBRACA trial demonstrated that the magnitude of benefit seems to be higher in patients with a BRCA2 mutation (PFS HR 0.47; 95% CI 0.32–0.70) compared to those patients with a BRCA1 mutation (PFS HR 0.59, 95% CI 0.39–0.90).30
The previous use of platinum agents also seems to correlate with less benefit to talazoparib in patients with metastatic breast cancer. Similar to the EMBRACA trial in which previous platinum-treated patients (approximately 20% of the patients) were the only subgroup who did not demonstrate statistically significant benefit, the ABRAZO trial demonstrated a clear difference in ORR in patients who received previous platinum (cohort 1 – ORR 21%) compared to platinum-naïve patients (cohort 2 – ORR 37%). However, more specific studies are needed to address the use of PARP inhibitors in patients treated with prior platinum agents and to compare PARP inhibition to platinum treatment.15
No other clinicopathologic characteristics, such as disease subtype or the number of previous lines of therapy, demonstrated a predictive value to response of talazoparib.31
Studies evaluating molecular biomarkers are still incipient. The presence of additional homologous recombination deficiencies, assessment of levels of PARP1 in tumors and expression of SLFN11, which are involved in the replication fork, are all under investigation.31
Next steps and combination therapies
Combination with cytotoxic chemotherapy
Prospects of using talazoparib and other PARP inhibitors for other indications and in combination with other agents are promising (Table 4).32–35 First, the rationale of increasing chemotherapy sensitivity with the combination of PARP inhibitors and cytotoxic agents has been evaluated and phase I trials showed safety with certain combinations.19 The best results in terms of efficacy emerged from combination with cisplatin or carboplatin, as well as with topotecan and temozolomide, with response rates in BRCA-related breast cancer of up to 73%.19 The recently published phase III BrightNess trial demonstrated that the addition of veliparib to paclitaxel and platinum had equivalent ORR to paclitaxel and carboplatin (p=0.36).36 An ongoing Phase I trial (NCT02358200) is evaluating talazoparib in combination with carboplatin followed by paclitaxel in patients with BRCA-mutated or triple-negative metastatic breast cancer. However, there are still no phase III data to date that demonstrate the tangible clinical benefit of incorporating this drug combination into daily practice.
 |
Table 4 Most clinically relevant recruiting talazoparib trials |
Talazoparib as neoadjuvant treatment in early-stage disease
In the de-escalation era, the use of talazoparib monotherapy in the neoadjuvant setting is being studied and may become a real option. A small feasibility study of only 13 patients demonstrated that the use of single-agent talazoparib for 2 months before neoadjuvant therapy and definitive surgery achieved tumor volume decrease in 88% (range 30–98%) of patients, and all patients were able to receive standard chemotherapy regimens after talazoparib with no grade 4 toxicities seen.37 An expansion of this cohort was presented at the American Society of Clinical Oncology Annual Meeting in 2018 (NCT02282345), which evaluated the use of talazoparib before surgery for 6 months with 20 enrolled patients. Ten of the 19 patients who underwent surgery (53%) achieved pathologic complete response, only 1 patient experienced grade 4 toxicity, and 9 dose reductions were required.38 A larger nonrandomized, single-arm phase II trial evaluating talazoparib as a single agent for neoadjuvant treatment in early triple-negative patients with BRCA mutations (NCT03499353) is ongoing.
Non-BRCA homologous recombination deficiencies
With next-generation sequencing becoming more common, the diagnosis of somatic BRCA1/2 mutations and other homologous recombination deficiencies is increasing, and the use of PARP inhibitors in these clinical scenarios must be better investigated. An ongoing phase II trial of talazoparib is enrolling patients with pathologic mutations in a somatic or germline non-BRCA1/2 homologous recombination pathway gene, such as PTEN, PALB2, CHEK2, ATM, NBN, BARD1, BRIP1, RAD50, RAD51C, RAD51D, MRE11 and ATR (NCT02401347). Yet, data on the use of PARP inhibitors in non-BRCA homologous recombination deficiencies are still missing.
Talazoparib and immunotherapy
Given the significant advancements in cancer treatment caused by immune checkpoint inhibitors, the interplay between the immune system and tumor DNA damage has been explored more in recent years. The DNA damage response is directly linked to innate immunity, as cells have the capability of sensing damaged and foreign DNA.39 One of the mechanisms the cell can sense DNA is through the Stimulator of Interferon Genes (STING) pathway. This system is especially important for cells to recognize DNA viruses and can also be activated by DNA from microbials and is implicated in the pathogenesis of certain autoimmune disease. Also, interferon type I production by STING pathway is directly involved in tumor antigen-specific T-cell activation through the cross-presentation of antigen. In this context, patients with BRCA mutations present a decreased repair capacity, ultimately leading to increased mutation rates and the creation of neoantigens that can activate T-cell responses by the STING pathway.39,40
Recently, different studies have shown both in different cell lines and in preclinical models, that PARP inhibitors, through the activation of the STING pathway, elicit strong antitumor immunity response, with increased infiltration of proliferating CD8+ T cells in the tumor microenvironment.41,42 Moreover, these studies have also shown that the combination of PARP inhibitors and checkpoint inhibitors was synergistic, providing a rationale for clinical trials evaluating the efficacy of this combination.41,42
The MEDIOLA trial (NCT02734004) was aphase Ib/II trial designed to evaluate the efficacy and safety of the combination of olaparib and durvalumab in patients with HER2-negative metastatic breast cancer harboring germline BRCA mutations.43,44 Prior platinum therapy was allowed if its use was discontinued without disease progression. The primary end point was the 12-week disease control rate (DCR). Thirty-two patients were included in the efficacy analysis. The 12-week DCR was 81% (90% CI: 66%, 92%) and 28-week DCR was 47% (90% CI: 32%, 63%). The ORR for the overall cohort was 56% (95% CI: 38%, 74%), with 1 (3%) patient with complete response, 17 (53%) patients with partial responses, 8 (25%) patients with stable disease and 6 (19%) patients with progressive disease. The median PFS was 6.7 months (95% CI of 4.6, 11.7 months). The most common grade 3 or 4 adverse events reported were anemia (11.8%), neutropenia (8.8%), and pancreatitis (5.9%).
Another phase I/II study evaluating the efficacy and safety of a similar combination, niraparib plus pembrolizumab, in metastatic triple-negative breast cancer was the TOPACIO/KEYNOTE-162 trial.45 Differently from MEDIOLA, the TOPACIO trial included both patients with or without germline BRCA mutations. The primary outcome was ORR. Of 46 evaluable patients, 20 achieved durable clinical benefit (any complete response/partial response or stable disease ≥16 weeks), including 8 patients with tumor BRCA wild type, and 1 was BRCA unknown.45
Currently, the JAVELIN PARP MEDLEY (NCT03330405) is an ongoing phase I/II trial which is evaluating the combination of talazoparib plus avelumab in patients with solid tumors with a BRCA or ATM gene defect.35
Conclusion
Talazoparib is a new active PARP inhibitor which demonstrates high clinical efficacy with an acceptable adverse events profile. It has demonstrated activity in all disease subtypes, in patients with brain metastases and patients previously exposed to platinum. This new drug has been recently approved by the FDA for the use in patients with germline BRCA1/2 mutations and should be incorporated into clinical practice. Combinations of talazoparib with other agents, such as cytotoxic chemotherapy and anti-PD1 agents, appear promising and require further investigation.
Disclosure
RBS has served as an advisor/consultant to Eli Lilly and has received honoraria from Roche and Bristol-Myers Squibb (BMS) for participation in Speakers Bureau. RBS also reports personal fees from Roche and from Lilly, outside the submitted work. SMT receives institutional research funding from Novartis, Genentech, Eli Lilly, Pfizer, Merck, Exelixis, Eisai, Bristol Meyers Squibb, AstraZeneca, Cyclacel, Immunomedics, Odenate, and Nektar. SMT has served as an advisor/consultant to Novartis, Eli Lilly, Pfizer, Merck, AstraZeneca, Eisai, Puma, Genentech, Immunomedics, Nektar, Tesaro, and Nanostring. SMT also reports personal fees from Pfizer, AstraZeneca, Lilly, Merck, Nektar, Novartis, Genentech/Roche, Immunomedics, Eisai, Nanostring, Puma, Sanofi, Tesaro and Celldex, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Forouzanfar MH, Foreman KJ, Delossantos AM, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet. 2011;378(9801):1461–1484. doi:10.1016/S0140-6736(11)61351-2
3. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. Jama. 2017;317(23):2402–2416. doi:10.1001/jama.2017.7112
4. Pothuri B. BRCA1- and BRCA2-related mutations: therapeutic implications in ovarian cancer. Ann Oncol. 2013;24(Suppl 8):viii22–viii7. doi:10.1093/annonc/mdt307
5. Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci U S A. 2001;98(9):5134–5139. doi:10.1073/pnas.081068398
6. Bochar DA, Wang L, Beniya H, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102(2):257–265.
7. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182.
8. Saha J, Davis AJ. Unsolved mystery: the role of BRCA1 in DNA end-joining. J Radiat Res. 2016;57(Suppl 1):i18–i24. doi:10.1093/jrr/rrw032
9. Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol. 2018;2:313–336. doi:10.1146/annurev-cancerbio-030617-050502
10. Lakhani SR, Jacquemier J, Sloane JP, et al. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst. 1998;90(15):1138–1145. doi:10.1093/jnci/90.15.1138
11. Jin J, Zhang W, Ji W, Yang F, Guan X. Predictive biomarkers for triple negative breast cancer treated with platinum-based chemotherapy. Cancer Biol Ther. 2017;18(6):369–378. doi:10.1080/15384047.2017.1323582
12. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–2505. doi:10.1056/NEJMoa1810858
13. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. doi:10.1056/NEJMoa1105535
14. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–533. doi:10.1056/NEJMoa1706450
15. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–763. doi:10.1056/NEJMoa1802905
16. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. doi:10.1016/S1470-2045(16)30559-9
17. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
18. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
19. McCann KE, Hurvitz SA. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context. 2018;7:212540. doi:10.7573/dic.212540
20. Ame JC, Rolli V, Schreiber V, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274(25):17860–17868. doi:10.1074/jbc.274.25.17860
21. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi:10.1158/0008-5472.CAN-12-2753
22. Turk AA, Wisinski KB. PARP inhibitors in breast cancer: bringing synthetic lethality to the bedside. Cancer. 2018;124(12):2498–2506. doi:10.1002/cncr.31307
23. Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–5015. doi:10.1158/1078-0432.CCR-13-1391
24. Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–443. doi:10.1158/1535-7163.MCT-13-0803
25. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7(6):620–629. doi:10.1158/2159-8290.CD-16-1250
26. Turner NC, Telli ML, Rugo HS, et al. Final results of a phase 2 study of talazoparib (TALA) following platinum or multiple cytotoxic regimens in advanced breast cancer patients (pts) with germline BRCA1/2 mutations (ABRAZO) [abstract]. J Clin Oncol. 2017;35:1007. doi:10.1200/JCO.2017.35.15_suppl.1007
27. Turner NC, Telli ML, Rugo HS, et al. A phase II study of talazoparib after platinum or cytotoxic nonplatinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations (ABRAZO). Clin Cancer Res. 2019;25(9):2717–2724. doi:10.1158/1078-0432.CCR-18-1891
28. Hurvitz SA, Quek RGW, Turner NC, et al. Quality of life with talazoparib after platinum or multiple cytotoxic non-platinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations: patient-reported outcomes from the ABRAZO phase 2 trial. Eur J Cancer. 2018;104:160–168. doi:10.1016/j.ejca.2018.09.003
29. Ettl J, Quek RGW, Lee KH, et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol. 2018;29(9):1939–1947. doi:10.1093/annonc/mdy257
30. Eiermann W, Rugo HS, Diab S, et al. Analysis of germline BRCA1/2 mutated (gBRCAmut) hormone receptor-positive (HR+) and triple negative breast cancer (TNBC) treated with talazoparib (TALA) [abstract]. J Clin Oncol. 2018;36:1070. doi:10.1200/JCO.2018.36.15_suppl.1070
31. Thomas A, Murai J, Pommier Y. The evolving landscape of predictive biomarkers of response to PARP inhibitors. J Clin Invest. 2018;128(5):1727–1730. doi:10.1172/JCI120388
32. National Cancer Institute. Pilot trial of BMN 673, an oral PARP inhibitor, in patients with advanced solid tumors and deleterious BRCA mutations. Available from: https://clinicaltrials.gov/ct2/show/NCT01989546. NLM identifier: NCT01989546.
33. MD Anderson Cancer Center, Pfizer. Study of the PARP inhibitor BMN 673 in advanced cancer patients with somatic alterations in BRCA1/2, mutations/deletions in PTEN or PTEN loss, a homologous recombination defect, mutations/deletions in other BRCA pathway genes and germline mutation in BRCA1/2 (Not Breast or Ovarian Cancer). Available from: https://clinicaltrials.gov/ct2/show/NCT02286687. NLM identifier: NCT02286687.
34. National Cancer Institute. Phase II talazoparib in BRCA1+BRCA2 wild-type &triple-neg/HER2-negative breast cancer/solidtumors. Available from: https://clinicaltrials.gov/ct2/show/NCT02401347. NLM identifier: NCT02401347.
35. Pfizer. Javelin parp medley: avelumab plus talazoparib in locally advanced or metastatic solid tumors. Available from: https://clinicaltrials.gov/ct2/show/NCT03330405. NLM identifier: NCT03330405.
36. Loibl S, O’Shaughnessy J, Untch M, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19(4):497–509. doi:10.1016/S1470-2045(18)30111-6
37. Litton JK, Scoggins M, Ramirez DL, et al. A feasibility study of neoadjuvant talazoparib for operable breast cancer patients with a germline BRCA mutation demonstrates marked activity. NPJ Breast Cancer. 2017;3:49. doi:10.1038/s41523-017-0052-4
38. Litton JK, Scoggins M, Hess KR, et al. Neoadjuvant talazoparib (TALA) for operable breast cancer patients with a BRCA mutation (BRCA+) [abstract]. J Clin Oncol. 2018;36:508. doi:10.1200/JCO.2018.36.15_suppl.508
39. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–693. PMCPMC5659200. doi:10.1158/2159-8290.CD-17-0226
40. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi:10.1038/nri3921
41. Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79(2):311–319. doi:10.1158/0008-5472.CAN-18-1003
42. Ding L, Kim HJ, Wang Q, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–80 e5. doi:10.1016/j.celrep.2018.11.054
43. Domchek S, Bang YJ, Coukos G, et al. MEDIOLA: A phase I/II, open-label trial of olaparib in combination with durvalumab (MEDI4736) in patients (pts) with advanced solid tumours [abstract]. Ann Oncol. 2016;27:1103TiP. doi:10.1093/annonc/mdw141
44. Domchek S, Postel-Vinay S, Im SA, et al. Abstract PD5-04: an open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): updated results in patients with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Cancer Res. 2019;79:PD5–04.
45. Vinayak S, Tolaney SM, Schwartzberg L, et al. Abstract PD5-02: durability of clinical benefit with niraparib + pembrolizumab in patients with advanced triple-negative breast cancer beyond BRCA: (TOPACIO/Keynote-162). Cancer Res. 2019;79:PD5–02.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.