")
Back to Journals » Cancer Management and Research » Volume 10
Evidence-based complementary treatment of pancreatic cancer: a review of adjunct therapies including paricalcitol, hydroxychloroquine, intravenous vitamin C, statins, metformin, curcumin, and aspirin
Authors Bigelsen S
Received 7 January 2018
Accepted for publication 30 April 2018
Published 13 July 2018 Volume 2018:10 Pages 2003—2018
DOI https://doi.org/10.2147/CMAR.S161824
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Stephen Bigelsen
Department of Allergy, Asthma and Immunology, Rutgers New Jersey Medical School, Newark, NJ, USA
Abstract: Despite new and exciting research and renewed optimism about future therapy, current statistics of survival from pancreatic cancer remains dismal. Patients seeking alternative or complementary treatments should be warned to avoid the hype and instead look to real science. A variety of relatively safe and inexpensive treatment options that have shown success in preclinical models and/or retrospective studies are currently available. Patients require their physicians to provide therapeutic guidance and assistance in obtaining and administrating these various therapies. Paricalcitol, an analog of vitamin D, has been shown by researchers at the Salk Institute for Biological Studies to break though the protective stroma surrounding tumor cells. Hydroxychloroquine has been shown to inhibit autophagy, a process by which dying cells recycle injured organelles and internal toxins to generate needed energy for survival and reproduction. Intravenous vitamin C creates a toxic accumulation of hydrogen peroxide within cancer cells, hastening their death. Metformin inhibits mitochondrial oxidative metabolism utilized by cancer stem cells. Statins inhibit not only cholesterol but also other factors in the same pathway that affect cancer cell growth, protein synthesis, and cell cycle progression. A novel formulation of curcumin may prevent resistance to chemotherapy and inhibit pancreatic cancer cell proliferation. Aspirin therapy has been shown to prevent pancreatic cancer and may be useful to prevent recurrence. These therapies are all currently available and are reviewed in this paper with emphasis on the most recent laboratory research and clinical studies.
Keywords: vitamin D, autophagy, stroma, T cells, integrative medicine, supplements, stellate cell
Background
Despite new, exciting research and renewed optimism about future therapy, current statistics of survival from pancreatic cancer remains dismal. Certainly, patients should be encouraged to join clinical trials where opportunities for better outcomes exist, while supporting the critical cause of advancing the state of cancer treatment for all. Unfortunately, for many patients, clinical trials remain unavailable or impractical, and in fact, only 4% of all pancreatic cancer patients are enrolled in trials. Patients should be given the opportunity to design their own trial with currently available experimental treatments, particularly those that have shown promise in preclinical trials, many of which have already advanced to early-phase human trials.
Patients seeking “alternative” treatments should be warned to avoid hype and instead look to real science. These treatments should never be used as replacement for recommended treatments such as surgery or chemotherapy, but, rather, to supplement them. Certainly, physicians should provide patients with all the proper warnings in regard to using off-label treatments which lack clearly proven results but would be remiss in not availing patients to treatments offering real hope of improving their odds of survival.
On a personal note, the author is a physician specializing in allergy and asthma who became interested in this subject after being diagnosed with stage 4 pancreatic cancer in July 2016. At the time of diagnosis, the author had tumors in the head and the tail with scattered peritoneal metastases and a CA19-9 of 11,575 U/mL. Working with physicians from Weill-Cornell and Johns Hopkins universities, the author began treatment with chemotherapy, plus intravenous (IV) paricalcitol (25 μg 3×/week) and oral hydroxychloroquine (600 μg BID). The author has now enjoyed a complete response with the latest CA19-9 of 15 U/mL and no evidence of active disease on the most recent CT scan. Although it is only a study of one, this response occurs no >1% of the time with chemotherapy alone. In a large-scale study of 340 stage 4 pancreatic cancer patients comparing gemcitabine to FOLFIRINOX, only one patient achieved a complete response.1 Whether or not the author’s response was indeed due to the use of these off-label, complementary treatments, he understands the desire of patients to improve their odds, especially with therapies that have shown results in laboratory studies, retrospective studies, and animal models. Box 1 provides a list of human studies looking at the off-label agents in the treatment of pancreatic cancer.
 | Box 1 Human studies of agents for treatment of pancreatic cancer Abbreviations: Vit D, vitamin D; HCQ, hydroxychloroquine; chemo, chemotherapy; pre-op SCRT, preoperative short-course chemoradiation; Gem, gemcitabine; Vit C, vitamin C; IV, intravenous; OS, overall survival; Retro, retrospective study; Mt, metformin; DM, diabetes mellitus; PEXG, cisplatin, epirubicin, capecitabine, and gemcitabine; PC, pancreatic cancer. |
Methods
The relevant medical and scientific English literature was reviewed using PubMed, Google Scholar, and ClinicalTrials.gov. To be included in this review, treatments were required to meet the following criteria:
- Have shown positive results in multiple studies using pancreatic cancer cell lines and animal studies.
- Have completed at least Phase I trials in humans and are advancing to Phase II trials and/or have large retrospective studies supporting their use.
- Are available to the general public who are willing to utilize off-label treatments if prescribed by a physician, without enrolling in a clinical trial.
Vitamin D
Vitamin D deficiency and cancer
Vitamin D deficiency appears common in most cancer patients. One study found that 75% of cancer patients had low vitamin D levels. In this study, low serum vitamin D levels predicted advanced-stage disease. In fact, in patients with levels under 24 ng/mL, the risk of stage 3 disease was almost triple that of those with higher vitamin D levels.2
In another study, cancer patients had a significantly lower mean serum vitamin D level (24.9 ng/mL) relative to a cohort of noncancer primary care patients (30.6 ng/mL, P < 0.001).3
In regard to pancreatic cancer, in a study looking at 2 large US cohorts totaling 122,198 people of whom 365 developed pancreatic cancer, higher dietary intake of foods containing vitamin D was associated with a lower risk for pancreatic cancer.4
In a pooled analysis of 5 prospective cohorts with 451 cases and 1,167 controls, higher plasma levels of vitamin D were associated with a lower risk for pancreatic cancer (P = 0.005).5
Paricalcitol, a synthetic analog of vitamin D
Paricalcitol is a modified form of vitamin D that acts as a vitamin D receptor agonist and is not associated with systemic toxicity of vitamin D resulting in conditions such as hypercalcemia. It is currently available intravenously or orally to treat or prevent hyperparathyroidism in dialysis patients.
Recently, investigators at the Salk Institute for Biological Studies have found that paricalcitol helps break though the pancreatic tumor’s stroma, which acts as a protective shield, incasing the tumor. The stroma is part of an extracellular matrix obstructing the tumor’s vasculature and inhibiting chemotherapy delivery to the tumor site. Specifically, the pancreatic stellate cells (those surrounding the tumor cells) are particularly activated in pancreatic cancer, driving the production of the stroma, as shown in Figure 1. These stellate cells have high levels of vitamin D receptors, and the blocking of these receptors by paricalcitol inactivates the stromal production.6 These stellate cells also produce cytokines and growth factors that enhance local tumor growth, contribute to angiogenesis, and enable metastasis. Furthermore, stellate cells metastasize along with the cancer cells assisting in their seeding, survival, and proliferation.7
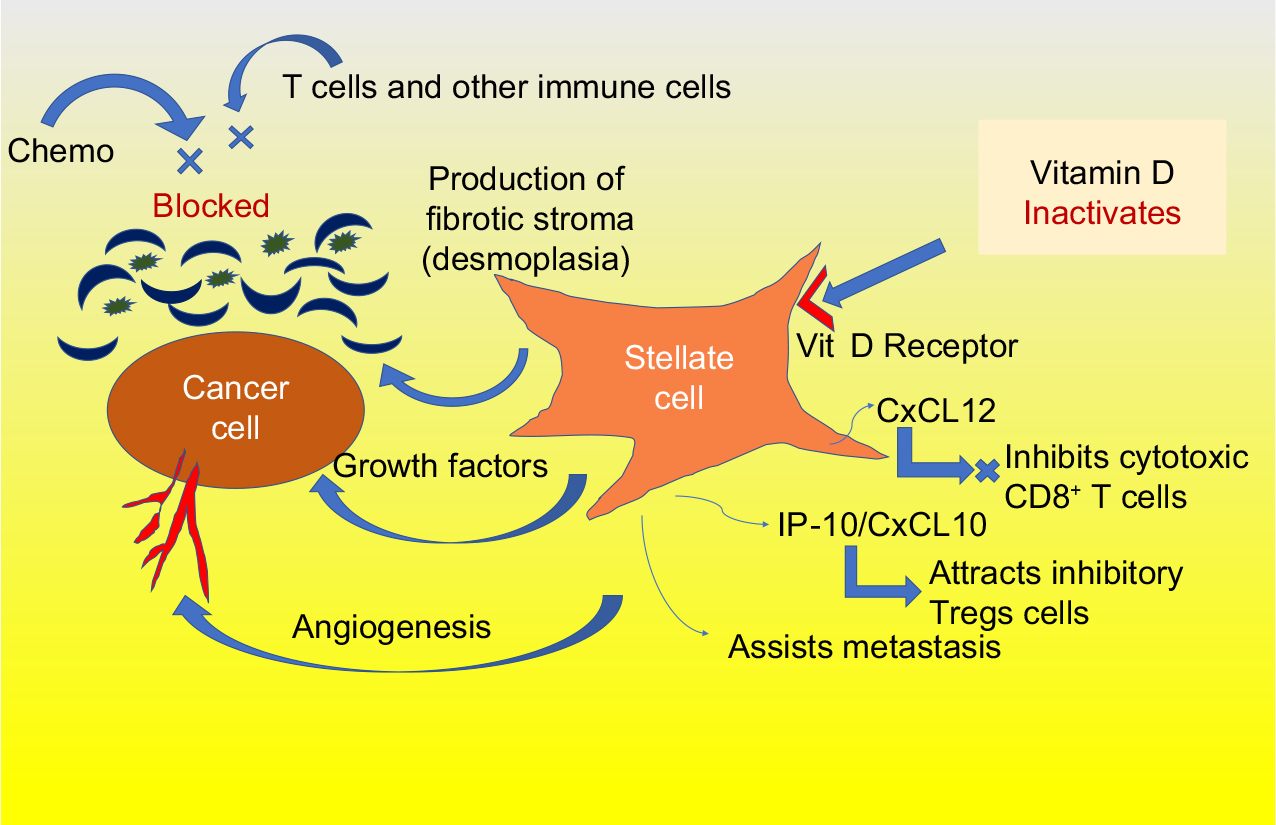 | Figure 1 Stellate cells are overactive in pancreatic cancer and are inactivated by vitamin D. Abbreviation: Vit D, vitamin D. |
In mice, when paricalcitol was given along with gemcitabine, stromal activation and tumor size were both significantly reduced, resulting in a 57% prolongation of survival.7
In addition to stromal inactivation, vitamin D has been shown to exert antiproliferative effects, secondary to the upregulation of the cell cycle inhibitors, especially p21 and p27, which control cell proliferation, differentiation, and division.8 Studies have shown a reduction of several pancreatic tumor lines in mice treated with paricalcitol correlating with the degree of cell cycle kinase inhibition.8
Lastly, paricalcitol has been shown to increase T cell penetration into the tumor. In a small Phase I study in patients treated with paricalcitol for 1 month prior to tumor resection, a 10- to 100-fold increase in the number of T cells was observed in and around the tumor.9 The hope that vitamin D affects the tumor’s immune environment has inspired the start of a Phase II study combining paricalcitol with immunotherapy and chemotherapy.10
Vitamin D may have many other anticancer effects, as well, not limited to pancreatic cancer. Evidence suggests that vitamin D promotes apoptosis leading to quicker cancer cell death.11 This has been evaluated in other cancers such as retinoblastoma.12 Vitamin D has been shown to inhibit angiogenesis within tumors.13 Tumors cannot grow larger than a few millimeters or metastasize unless they are well vascularized.
Safety of paricalcitol
In terms of safety, as stated, paricalcitol is less likely to produce hypercalcemia, hyperphosphatemia, or elevations in calcium and phosphorus levels compared to other forms of vitamin D, primarily due to its decreased effect on intestinal absorption of calcium and phosphorus.14
In a Phase I dose-escalating trial of IV paricalcitol in men with advanced prostate cancer, patients received as much as 25 μg 3×/week intravenously. Significant hypercalcemia was rare, and the maximally tolerated dose of paricalcitol was not reached in that study, indicating that even higher doses may be free of significant side effects.15 Paricalcitol has also been shown to be well tolerated in mice at relatively high levels.
In summary, paricalcitol given intravenously at a dose of 25 μg, 3×/week, appears to be well tolerated with little risk of serious adverse side effects in humans. It has worked well in vitro and in vivo (mouse studies) indicating possible benefit in combination with chemotherapy in human pancreatic cancer. Large-scale studies in humans are just beginning.
Hydroxychloroquine
Hydroxychloroquine is a relatively inexpensive drug currently available for the treatment of malaria, lupus, and rheumatoid arthritis. It is currently in clinical trials, combined with chemotherapy, for the treatment of pancreatic and other cancers.
Hydroxychloroquine has been shown to inhibit autophagy. Autophagy is a process of self-cannibalization in which injured cancer cells ingest pieces of themselves, such as organelles and macromolecules, to conserve energy, and, therefore, thrive. Additionally, autophagy helps rid the cancer cells of toxic substances and free radicals, such as hydrogen peroxide and superoxide. When combining chemotherapy with autophagy inhibition, damaged cancer cells are unable to conserve the needed energy to survive.
How autophagy works
First, the structures within the cells including toxic substances, free radicals, and damaged organelles that are no longer needed are targeted for removal. They are engulfed by a double-membrane structure that elongates and wraps around them to form an autophagosome. The autophagosome then fuses with a lysosome, leading to the degradation and removal of the enveloped structures. This process, as shown in Figure 2, creates energy to replenish other critical cell functions necessary for cancer cell survival.16 In addition to creating energy, autophagy acts to remove toxic substances that may be damaging to the cell.17 Drugs such as chloroquine, and the less toxic, hydroxychloroquine, inhibit the last step in the process, preventing the lysosome from clearing the undesirable substances contained in the autophagosome.18
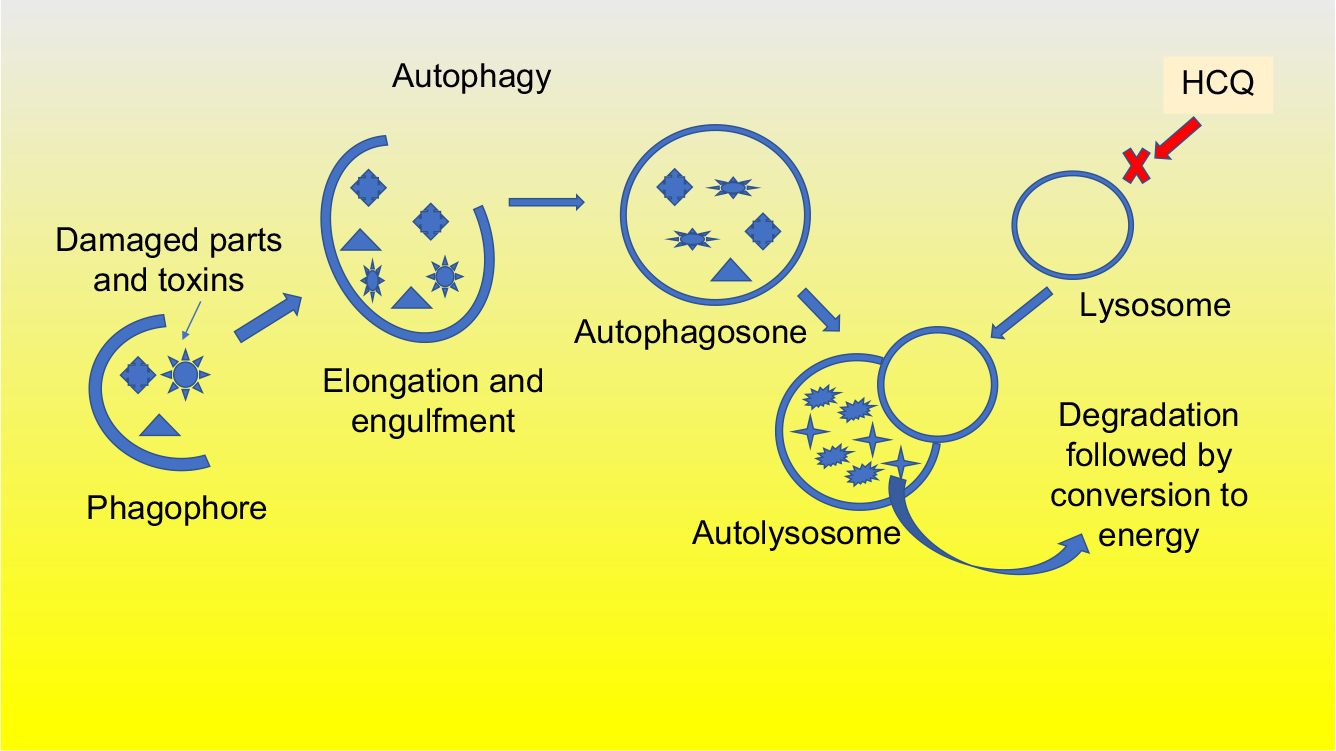 | Figure 2 Autophagy is activated by KRAS mutation. The process of autophagy is highly active in pancreatic cancer cells and clears the damaged cancer cells of toxins and dying organelles to create the needed energy to survive and divide. Hydroxychloroquine prevents this autophagy process. Abbreviation: HCQ, hydroxychloroquine. |
Why is autophagy important in pancreatic cancer?
The KRAS genetic mutation, found in over 90% of pancreatic tumors, appears to upregulate the process of autophagy which may be responsible for the extreme resilience of pancreatic cancer cells.19 When the KRAS oncogene was introduced into mice, it enhanced autophagy, which lead to faster growing, more aggressive tumors.20 Because of this transformation, pancreatic cancer, more so than other cancers, appears to have a distinct dependence on autophagy, with studies showing increased autophagic activity occurring within these cancer cells.21 The rapidly dividing cells within tumors require more energy than normal cells to reproduce. When chemotherapy agents attack the pancreatic cancer cells, their ability to conserve energy, through autophagy, becomes especially critical.
Studies of pancreatic cancer cells in laboratories have shown that inhibition of autophagy makes survival of cancer cells more difficult by such processes as increasing reactive oxygen toxins, elevating DNA damage, and causing a metabolic defect leading to decreased mitochondrial oxidative phosphorylation.21
In mice studies, decreased autophagy has led to robust tumor regression and prolonged survival of the mice. In a 16-mouse xenograft study, the response to chloroquine was dramatic. Of the 8 mice treated with chloroquine, 7 (88%) survived over 180 days, compared to all 8 untreated mice dying within 140 days.21 Additional studies in mice with genetic pancreatic tumors also showed promising results.
Human studies
One human study using hydroxychloroquine alone (without chemotherapy) produced disappointing results with only 2 of 20 patients without progressive disease.22 In another negative study of 50 patients on hydroxychloroquine 800 mg daily plus preoperative short-course chemoradiation and gemcitabine, disease-free survival did not significantly improve.23 On the bright side, an interim report of an ongoing Phase II study showed encouraging results. This study which analyzed 54 patients with resectable or borderline resectable pancreatic cancer receiving hydroxychloroquine 1,200 mg daily, in addition to chemotherapy, showed that the percentage of tumor destroyed was better in the hydroxychloroquine group (P = 0.004). Additionally, the CA19-9 tumor marker in patients receiving hydroxychloroquine decreased by 20%, as compared to 10% in the chemotherapy-alone group (P = 0.014), and at the time of surgery, the ratio of positive lymph nodes to total number of lymph nodes was lower in the hydroxychloroquine group vs. the control group (0.03 vs. 0.05; P = 0.02). The hydroxychloroquine group had greater apoptosis in their tumors, less stromal activation, and greater infiltration of CD4 and CD8 T cells (P = 0.016 and P = 0.046, respectively), and greater tumor expression of PD-L1. No adverse effects were noted in this study.24
Risks associated with hydroxychloroquine
The major risk associated with hydroxychloroquine is retinopathy, potentially leading to blindness. At the typical dose of 200 mg 2×/day (for autoimmune diseases), the risk is exceedingly small with <2% of patients developing retinopathy after 20 years.25 The higher doses being tested to prevent autophagy (800–1,200 mg daily dosages are currently in clinical trials) carry a higher risk. Two small studies have shown some degree of retinal damage occurring in under 2 years.26 Therefore, at high doses, screening by an ophthalmologist is recommended every 6 months, as early detection is the only method to prevent serious retinal damage.
IV vitamin C
Evidence that vitamin C (ascorbic acid) is beneficial in prolonging life in cancer patients dates back to the 1970s.27,28 However, while some studies on oral vitamin C have shown success in breast cancer,29,30 most studies in other cancers failed to show any success with oral administration.31,32 Evidence suggests the high blood concentrations required to induce cytotoxicity can only be achieved with IV administration.33 IV vitamin C should be avoided in patients with G6PD deficiency due to a risk of hemolysis, but otherwise appears extremely well tolerated in almost all patients.
When vitamin C is given orally, plasma levels peak at 100 μM. With greater oral doses, absorption decreases while urine excretion increases, so that blood levels cannot rise. In contrast, when vitamin C is administered intravenously, plasma concentrations of 1 mM or higher can be achieved without toxicity.33–36
Vitamin C is believed to work because it breaks down into hydrogen peroxide, which is especially toxic to catalase-deficient cancer cells. Healthy noncancerous cells produce enough catalase to protect themselves from the toxic effects of hydrogen peroxide, resulting in no adverse effects to them.37–39 For example, in one study, there was an increase in measured hydrogen peroxide production that correlated with concentrations of vitamin C. Cell death by vitamin C was reversed when scavengers of hydrogen peroxide were added to cell lines.38 In another study, when 11 human cancer cell lines were exposed to serial dilutions of vitamin C, a correlation between catalase activity and the susceptibility to ascorbic acid was observed.40
Another theory on the effect of vitamin C in the treatment of cancer suggests that its toxicity is due to an increased uptake of its oxidized form, dehydroascorbate (DHA), via the GLUT1 glucose transporter that is upregulated in KRAS- and BRAF-mutated cells. Increased DHA uptake is believed to cause an oxidative stress by depleting glutathione and inactivating glyceraldehyde phosphate dehydrogenase, resulting in an energetic crisis and cell death. This was studied in colorectal cancer cells with these mutations, and hopefully, also applies to pancreatic cancer cells, of which >90% contain the same KRAS mutation. In that study, vitamin C treatment inhibited KRAS- and BRAF-mutant cell growth and colony formation much greater than in their nonmutant counterparts. In the same study, vitamin C treatment significantly reduced tumor growth compared to vehicle control treatment in mice bearing established cancer xenografts.41
It is likely that both theories are correct. As one study showed, adding catalase, which neutralizes hydrogen peroxide, to cell lines treated with vitamin C reversed 75% of the effect but not all of it.39 Additionally, studies have shown that both cell lines with low catalase activity37,40,42 and cell lines expressing the KRAS mutation38,41,43 are much more sensitive to vitamin C.
IV vitamin C may also cause a metabolic defect in the difficult-to-kill pancreatic cancer stem cells (CSCs). These are the cells most resistant to chemotherapy and are responsible for recurrence of cancer even after the metastatic cells have been destroyed. These stem cells rely on oxidative phosphorylation (OXPHOS) as their primary energy source, as opposed to glycolysis. A recent study showed that vitamin C can be used to target the stem cell population, as it is an inhibitor of energy metabolism that feeds into the mitochondrial tricarboxylic acid cycle and OXPHOS.44
Studies in cancer cell lines
In laboratory studies, vitamin C has proven to be potently cytotoxic to a wide variety of cancer cell lines45–53 including pancreatic cancer.38,47 Additionally, it has been shown to boost the cytotoxicity of several common chemotherapy drugs.37,41,54
Mice studies
In vitro studies have been further confirmed in animal studies, where IV vitamin C decreased the growth rates of liver, ovarian, pancreatic, and glioblastoma tumors with dosages easily achievable in humans.55
In a study looking at 7 different pancreatic cell lines, gemcitabine–vitamin C combinations administered to mice bearing pancreatic tumor xenografts consistently enhanced inhibition of growth compared to gemcitabine alone. Growth inhibition of 50% more than gemcitabine alone was seen, and vitamin C administration demonstrated a gemcitabine dose-sparing effect.39
In mice treated with vitamin C, a slower rate of growth in pancreatic tumors was observed in comparison to the control group of animals that received NaCl. On day 21 of the experiment, the control group had a mean tumor volume of 472 mm3, while the vitamin C group had a mean tumor volume of 138 mm3. Additionally, mice that received vitamin C had increased survival compared to controls (68 days vs. 78 days; P < 0.0001).38
Human studies
The primary goal of Phase I studies is to evaluate safety and determine dosing, although some small insight into efficacy can be ascertained.
A small Phase I clinical trial in the USA has just shown that adding IV vitamin C to gemcitabine for pancreatic cancer extended patients’ average survival time to 12 months, compared to historical survival times of 5.65 months for such patients.56
In another Phase I study of IV vitamin C in combination with gemcitabine and erlotinib, 7 of 9 subjects had stable disease while only 2 had progressive disease.57
Neither of these studies showed any significant toxicity. Larger Phase II studies are just beginning.
Combinations of off-label treatments with vitamin C
Almost no research in mice or humans has been done combining different off-label treatments. Theoretically, some combinations may be synergistic. For example, IV vitamin C increases the free radical hydrogen peroxide within cancer cells. This triggers autophagy,38,58 presumably to detoxify the cells. Adding hydroxychloroquine that inhibits autophagy would make it more difficult to clear the hydrogen peroxide, leading to quicker cell death.
Lastly, metformin and vitamin C may be synergistic as they both have been shown to block OXPHOS in the pancreatic stem cells.
Metformin
The diabetic drug, metformin, seems to have an effect of inhibiting pancreatic CSCs, but not metastatic cancer cells. Therefore, it may be useful in cancer prevention, in early-stage disease, and in prevention of recurrence after remission, although it is likely not helpful in metastatic disease.
How it works
Pancreatic CSCs are dependent on mitochondrial oxidative metabolism for their energy requirements, whereas metastatic cells rely on glycolysis.59 Glycolysis breaks down glucose and forms pyruvate with the production of 2 molecules of ATP. Alternatively, in mitochondrial oxidative metabolism, glucose plus oxygen leads to CO2 production and a plethora of ATP. Metformin inhibits the mitochondria, and thereby, shuts down oxidative metabolism in the stem cells resulting in an energy crisis leading to apoptosis. Because metformin decreases mitochondrial respiration, cells treated with metformin become energetically inefficient. Inhibition of mitochondrial oxidative metabolism in CSCs has been shown to significantly decrease their survival.60 Unfortunately, in at least one study, the stem cells eventually adapted by changing their metabolic process and became metformin resistant.57
A second effect of metformin is indirect inhibition of mammalian target of rapamycin. In pancreatic cancer, the mTOR pathway functions downstream of RAS, and therefore, is in part activated by the KRAS mutation. Activation of this pathway correlates significantly with a poor prognosis.61 MTORC1, the primary regulator of the mTOR pathway, stimulates ribosome biogenesis and transcription of genes, leading to cell growth, division, and differentiation primarily within stem cells.
A third effect metformin has is to reduce desmoplasia, similar to that seen with vitamin D. This occurs by inhibiting the activation of the pancreatic stellate cells that produce the extracellular matrix and by reprogramming immune cells to reduce inflammation. For metformin, this effect is primarily seen in diabetic and obese patients.62,63
Lastly, evidence suggests that metformin inhibits proliferation, migration, and invasion of drug-resistant pancreatic cancer cells by attenuating CSC function by deregulation of certain microRNAs (miRNAs). miRNAs are small noncoding RNAs involved in the modulation of several biological activities ranging from invasion to metastases development, as well as drug resistance in pancreatic cancer.64
Mice studies
In a xenograft mouse model, low doses of metformin inhibited cellular transformation and selectively killed CSCs in 4 genetically different types of breast cancer.65
In a study on the effects of metformin on pancreatic intraepithelial neoplasia (PanIN) and its progression to pancreatic cancer in mice, 2 doses of metformin decreased pancreatic tumor weights by 34% and 49%, respectively (P < 0.03–0.001). The drug treatment caused suppression of PanIN3 (carcinoma in situ) lesions by 28% and 39%, respectively (P < 0.002), and significant inhibition of carcinoma spread in the pancreas. The CSC markers were significantly decreased (P < 0.04–0.0002) in the pancreatic tissue. This study implied that the biologic effects of metformin are mediated through decreased CSC markers CD44 and CD133, CSC markers and modulation of the mTOR signaling pathway.66
In another study of hamsters fed a high-fat diet, 50% of the hamsters not given metformin developed malignant lesions, compared to none in the metformin group (P < 0.05). The non-metformin group also developed significantly more hyperplastic and premalignant lesions, most of which were found within the islets, (8.6 lesions/hamster) than in the metformin group (1.8 lesions/hamster).67
In a third study, metformin given orally to mice inhibited MaiPaca-2 implanted xenografts by 67% and markedly reduced the growth of preestablished PANC-1 xenografts in a dose-dependent manner. A decrease in MTORC1 and extracellular-signal-regulated kinase signaling in the metformin-treated xenografts was also demonstrated.68
In another study comparing metformin and rapamycin, both significantly reduced tumor burden compared with vehicle, although the effect of rapamycin was more dramatic. Additionally, both metformin and rapamycin significantly decreased tumoral mTOR activity.69
Prevention of pancreatic cancer in humans
In a retrospective cohort study of 62,809 diabetics treated in the UK, metformin monotherapy carried the lowest risk of cancer. Metformin use was associated with lower risk of cancer of the colon or pancreas, although it did not affect the risk of breast or prostate cancer.70
In another hospital-based case–control study at MD Anderson Cancer Center performed over 4 years, diabetic patients who had taken metformin had a significantly lower risk of pancreatic cancer compared with those who had not taken metformin (P = 0.001). In contrast, diabetic patients who had taken insulin or insulin secretagogues had a significantly higher risk of pancreatic cancer compared with diabetic patients who had not taken these drugs. This study demonstrates that metformin use was associated with reduced risk and insulin or insulin secretagogue use was associated with increased risk of pancreatic cancer in diabetic patients.71
Positive human studies in pancreatic cancer
Two recent small studies showed improved survival in early-stage disease. A recent small Phase II study of 44 patients showed trend toward improved survival with the use of metformin in diabetic patients with resectable pancreatic cancer. The median overall survival of 10.4 months was longer in those who took metformin than in those who did not. Furthermore, the long-term survival was higher in the metformin group than in the control group, with 5-year survival rates of 34% and 14%, respectively. Due to the small size of this study, the results did not reach statistical significance.72
In another retrospective study of 302 diabetic patients with pancreatic cancer, the 2-year survival rate was 30.1% for the metformin group and 15.4% for the non-metformin group (P = 0.004). The median overall survival time was 15.2 months for the metformin group, and 11.1 months for the non-metformin group (P = 0.004). The beneficial effect of metformin was seen in all disease stages but reached statistical significance only in patients with nonmetastatic disease.73
Negative studies in humans with metastatic disease
Unfortunately, metformin has not been shown to be effective in 2 Phase II studies of 60 and 121 patients with metastatic disease.74,75
In a retrospective study of 980 diabetic patients on metformin with pancreatic cancer, the findings failed to show any benefit in metastatic disease and showed only a small protective effect in patients with locally advanced disease.76
A Surveillance, Epidemiology, and End Results (SEER) data analysis of 12,572 Medicare patients with pancreatic cancer, exposed to statins but not metformin, showed that use of statins alone was significantly associated with reduced overall mortality and the combination of the 2 was not superior to statins use alone.77
Statins
Several large-scale retrospective studies of pancreatic patients taking statins show impressive results in reducing mortality, especially in early-stage disease. Evidence also shows statins help prevent pancreatic and various other types of cancers. Hydrophobic/lipophilic statins (atorvastatin, simvastatin, lovastatin, fluvastatin) are likely more effective than hydrophilic statins (pravastatin and rosuvastatin) in cancer treatment since they are able to cross biological membranes, and therefore, have greater intracellular access.78
Antitumor effects by statins
Cancer cells require increased lipid biosynthesis to meet their metabolic needs and supply cholesterol to the cell membrane.78 The inhibition of cholesterol production, however, plays only a small role in the effect statins have on cancer cells. More importantly, statins, through inhibition of HMG-CoA reductase, inhibit not only cholesterol formation but also the entire mevalonate pathway. In addition to cholesterol, this pathway also leads to the production of isoprenoids, dolichol, ubiquinone, and isopentenyl adenine. Several members of this pathway have been shown to be essential for the survival of several cancer cell lines. Inhibiting the production of these factors leads to a decrease in cancer cell growth, protein synthesis, and cell cycle progression and to an increase in apoptosis in many cancer types. These effects appear to be independent of cholesterol, and in fact, studies have not shown a correlation between cholesterol levels and cancer progression. Two other products of the mevalonate pathway, farnesyl pyrophosphate and geranylgeranyl pyrophosphate, are required to activate the RAS protein, and their inhibition has been shown to increase cell apoptosis.79
Studies with pitavastatin in pancreatic cell lines revealed dose-dependent growth inhibition. At the molecular level, pitavastatin induced expression of the cyclin-dependent kinase inhibitor p21 in a cholesterol-independent manner.80
A recent study revealed a significant increase in survival of mice with pancreatic cancer fed atorvastatin (171.9 ± 6.2 days) compared to the control mice (144.9 ± 8.4 days; P < 0.05). Atorvastatin treatment resulted in a significant reduction in tumor volume and cell proliferation. Atorvastatin also inhibited several key proteins, including KRAS protein, and their activities.81
Statins in prevention of pancreatic cancer
In a meta-analysis of 18 studies that included 1,799,157 patients, the incidence of pancreatic cancer was 0.28% in the statin therapy group vs. 0.54% in the group without statin therapy.82
In a study of over 500,000 veterans, statin use of >6 months was associated with a risk reduction of pancreatic cancer of 67%. An impressive 80% risk reduction was found with use of a statin for >4 years.83
While several studies showed a decreased risk of pancreatic cancer among statin users, several studies looking at all cancers showed an increased risk of cancer, especially in the elderly. This pro-cancer effect is thought to be due to a stimulatory effect on Treg cells.84
Retrospective studies in pancreatic cancer patients
A study of 2,427 pancreatic cancer patients, of whom 680 were taking simvastatin and 149 were taking atorvastatin, demonstrated a 31% decrease in mortality in the group taking Zocor (simvastatin) and a 39% decrease in the group taking Lipitor (atorvastatin).85
In another study among the 1,761 pancreatic cancer patients of whom 118 had used statins, the 5-year overall survival was 16.6% for statin users and 8.9% for nonusers (P = 0.012). Simvastatin showed the greatest benefit.79
Among 226 patients undergoing resection for pancreatic cancer, 71 (31.4%) had prior simvastatin use and 27 (11.9%) had prior lovastatin use. Active use of moderate- to high-dose simvastatin at baseline was associated with improved overall and disease-free survival.86
A study of 7,813 elderly patients with pancreatic cancer showed statin treatment after cancer diagnosis was associated with enhanced survival in patients with low-grade, resectable pancreatic cancer.87
A 14-year study showed that statin use at the time of any cancer diagnosis was associated with 15% reduced cancer-related mortality in Danish patients.88
As previously mentioned, a SEER data analysis of 12,572 Medicare patients with pancreatic cancer, exposed to statins but not metformin, showed that statin use was significantly associated with reduced overall mortality, especially in post-diagnosis statin users.77
Mixed studies in metastatic disease
Unfortunately, a double-blind prospective study of 114 stage 4 patients failed to show any benefit from simvastatin 40 mg.89
Another study with 180 patients treated with erlotinib–gemcitabine for unresectable pancreatic cancer showed that a history of statin treatment resulted in improved overall survival (P = 0.026).90
Curcumin
Curcumin is the most studied of the nutraceuticals that are considered anticancer agents found in natural plants. Agents such as epigallocatechin-3-gallate from green tea have been shown in vitro to induce apoptosis and inhibit tumor progression by modulating different signaling pathways in pancreatic cancer.91,92 Others, such as isoflavone from soybeans, resveratrol from grapes, lycopene which is the red pigment in tomatoes, and garcinol from the rind of the fruit, have shown promise in the laboratories but may be limiting due to lack of absorption and bioavailability. Nutraceuticals are also believed to work by affecting the expression of certain miRNAs which modulate cellular signaling networks leading to the inhibition of pancreatic cancer cell growth and pancreatic CSC self-renewal.93,94
Curcumin, from the plant Curcuma longa and a component of turmeric, has exhibited multiple anticancer effects in numerous studies in pancreatic cell lines and mice studies.95–114 When used in combination, curcumin has also been shown to potentiate the effects of other cytotoxic agents, including gemcitabine, cisplatin, oxaliplatin, and 5-fluorouracil, in preclinical models of a variety of cancers.96,105 Most importantly, it seems to prevent chemoresistance especially to gemcitabine.102,104–109
A low incidence of cancer has been documented in countries that incorporate high consumption of turmeric root, of which curcumin is believed to be the active ingredient.110,111 After testing >1,000 different potential agents for cancer prevention, the National Cancer Institute has chosen only 40, of which curcumin was included, to be moved to clinical trials.112 In several mice studies, curcumin has been shown to prevent cancer including mammary adenocarcinoma, esophageal cancer, and familial adenomatous polyposis.113–115
Disappointingly, its low bioavailability limits its effectiveness. To improve the bioavailability of curcumin, numerous approaches have been undertaken, including the formation of liposomes, micelles, and phospholipid complexes, as well as attempting different routes of administration such as subcutaneous dose of microparticles, intraperitoneal delivery, and IV administration.116
Fortunately, new formulations including the nanoparticles of curcumin have been recently investigated. One such product, with the brand name Theracurmin®, is currently available and appears to provide significantly greater blood levels and greater hopes of efficacy. Other preparations are also under development.
How curcumin works
Curcumin has demonstrated a plethora of functions affecting various cell signaling pathways at multiple levels, as shown in Figure 3. Studies have identified numerous factors inhibited by curcumin related to cancer cell survival, proliferation, invasion, angiogenesis, and metastasis, suppression of apoptosis, and chemoresistance. Curcumin has been shown to inhibit a variety of factors including STAT3,100 COX-2,117 survivin,100 miR-200, and miR-21,111 the hedgehog pathway,118 and IAP proteins.119 Additionally, it has been demonstrated to activate the cell cycle inhibitors, p27 and p27,94 and upregulate the p53 modulator of apoptosis.112 Curcumin has been shown to inhibit pancreatic tumor growth and angiogenesis in mouse models.120
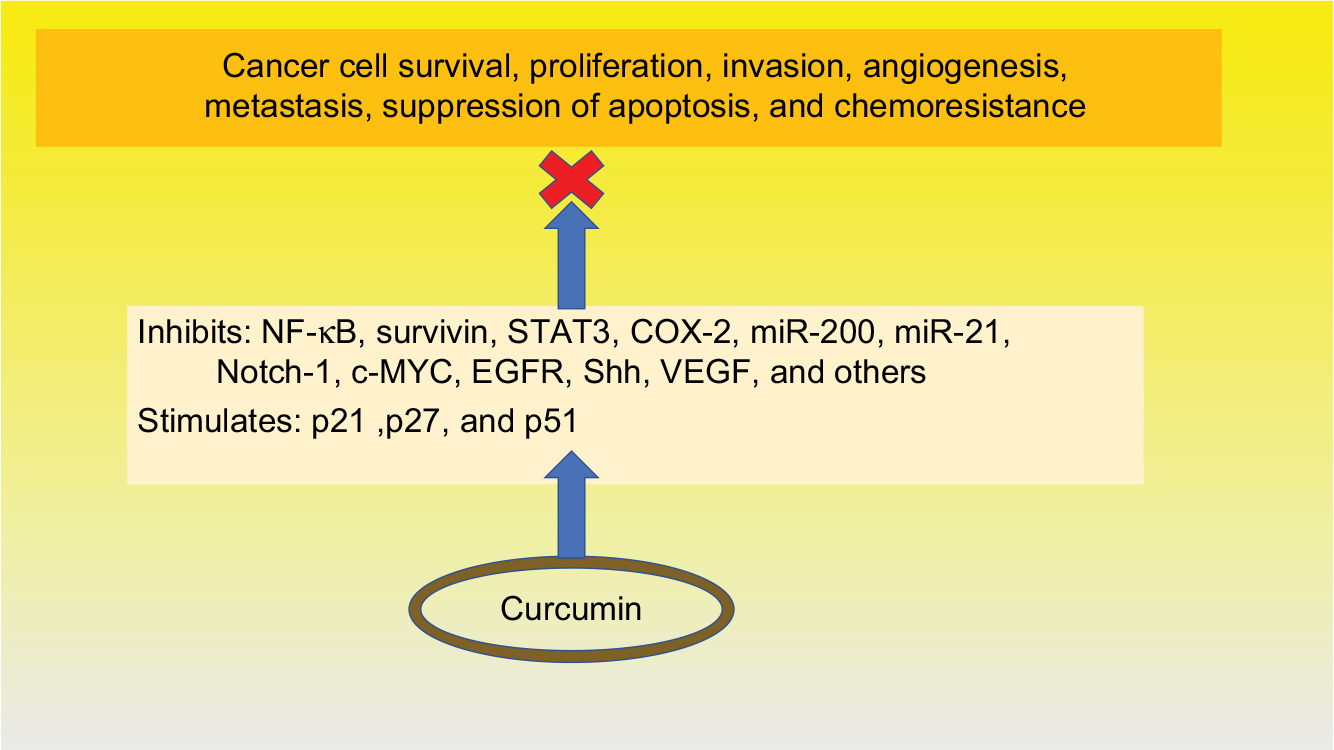 | Figure 3 Functions of curcumin. Abbreviations: NF-κB, nuclear factor kappa enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3; COX-2, cyclooxygenase 2; miR, microRNAs; Notch-1, neurogenic locus notch homolog protein-1; c-MYC, c-mycproto-oncogene; EGFR, epidermal growth factor receptor; shh, sonic hedgehog; VEGF, vascular endothelial growth factor; P21, p27 and p51, cyclin-dependent kinase inhibitors. |
Curcumin’s most important effect seems to stem from inhibition of the transcription factor NF-κB and all its downstream products.96,97,110,117,118 Many lines of evidence suggest that NF-κB plays a major role in growth, proliferation, angiogenesis, and most importantly, chemoresistance.104–106,117,121 Becoming resistant to chemotherapy is the main cause of death in most pancreatic patients. Curcumin seems to block this resistance.122 For example, one study showed that resistance to gemcitabine is induced by NF-κB activity and that curcumin inhibits this process.107 A recent study demonstrated curcumin restores sensitivity in gemcitabine-resistant cancer cells and confirmed this finding in a xenograft mouse model.108
Nanoparticles
Unfortunately, the bioavailability of curcumin is very low. The most likely explanation of its low plasma and tissue levels appears to be poor absorption, rapid metabolism, and rapid systemic elimination.123 Multiple studies report plasma levels of curcumin rarely exceed 40 ng/mL even with extremely high oral intake including amounts over 8 g.124–126 Therefore, Phase I and II human studies with curcumin have demonstrated good tolerability, albeit with only limited effectiveness.127,128
In order to increase bioavailability, there have been several attempts to create different formulations, the most successful of which have been nanoparticles.95,122 Nanoparticles of curcumin are formed by encapsulation in polymeric micelles, liposomes, or hydrogels, all of which make these particles water-soluble, and therefore, easily absorbable. One such nanoparticle formulation has shown a much greater bioavailability in several studies129,130 including a >40-fold increase in area under the blood concentration–time curve compared with conventional curcumin in rat models and a 27-fold increase in a human trial.131 In another study, the maximal plasma curcumin concentration of Theracurmin was 10.7–5.6 times higher than 2 other curcumin preparations also claiming a novel drug-delivery system.132 Small human studies have shown an excellent safety profile with good tolerability,129,130 although there are only limited data on efficacy. Larger-scale studies have yet to be performed.
Aspirin
Aspirin may be useful in pancreatic cancer prevention as indicated in several large retrospective studies. Many pancreatic cancer patients are greatly concerned about family members as pancreatic cancer appears to have a genetic tendency, even when no known oncogenes such as BRCA are detected. We have already reviewed statins, metformin, and curcumin, all of which have shown some evidence in terms of cancer prevention.
Additionally, many patients who have undergone surgery or complete chemotherapy and are now cancer-free are not receiving any medications or treatment to prevent recurrence despite a high recurrence rate in these patients. Prevention of recurrence after surgery is an intriguing possibility, although it has yet to be studied in pancreatic cancer. Two studies, however, one in colon cancer and one in breast cancer, have shown prevention of recurrence with the use of aspirin.133,134
Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) have shown promise in prevention of a variety of other cancers.135 The greatest evidence has been seen with colorectal cancer, both in observational epidemiological studies136–139 and in prospective clinical trials.139,140 There is also evidence for a prophylactic effect for several other types of cancers, including stomach cancer,140 esophageal cancer,141 leukemia,142 breast cancer,143,144 ovarian cancer,145 endometrial cancer,146 and prostate cancer.147,148
Four studies have shown aspirin likely helps in pancreatic cancer prevention. The first is a population-based study performed during 2006–2011 in Shanghai, People’s Republic of China, with 761 pancreatic cancer patients and 794 control subjects who were matched on sex and age. The results were rather impressive, demonstrating that regular use of aspirin reduced the risk of pancreatic cancer by almost half.149
A population-based Connecticut study, conducted from January 2005 to August 2009, of 362 pancreatic cancer cases matched to 690 randomly sampled controls, showed that subjects who regularly used aspirin had a lower risk of pancreatic cancer. The more years of aspirin use, the greater the benefit.150
In another prospective study from 1992 through 1999, among 28,283 postmenopausal women who lived in Iowa, of whom 80 developed pancreatic cancer, there was a trend of decreasing risk of pancreatic cancer with aspirin use, but not with nonsteroidal anti-inflammatory medications.151
Lastly, the Mayo Clinic performed a clinic-based case–control study from April 2004 to September 2010, evaluating the association between aspirin, NSAID, and acetaminophen use with pancreatic cancer risk using a sample of 904 patients with pancreatic cancer, and 1,224 age- and sex-matched healthy controls. They found that aspirin use, but not NSAID or acetaminophen use, was associated with a lowered risk of developing pancreatic cancer.152
Unfortunately, not all studies show a reduced risk. In a study of 408 pancreatic cancer patients and 816 matched controls, overall statin use, but not aspirin use, was associated with a reduced pancreatic cancer risk. The authors of the study suggest that prior positive results for aspirin use may have resulted from concomitant statin use as many cardiovascular patients take both.153
Studies of aspirin for treatment of pancreatic cancer have not yet been performed. A recent systematic review and meta-analysis of 58 mostly observational studies of various cancers, but not including pancreatic cancer, showed reductions in metastatic spread and a decrease in overall mortality by about 15%.154
Although the mechanism by which aspirin prevents cancer is unknown, studies indicate it may be due to aspirin’s ability to inhibit platelet upregulation of c-MYC which stimulates cancer cell proliferation. This has been demonstrated in both colon and pancreatic cancer cell lines.155,156 Evidence suggests the anticancer effect of aspirin relates to its ability to reduce metastasis possibly though its effect on platelets.157 It has also been suggested that aspirin may work by inhibiting survivin, a protein which inhibits apoptosis and is overly expressed in pancreatic cancer.158
Final thoughts
Given the dismal 7% overall survival rate in pancreatic cancer, with only 1% for stage 4, almost all patients are desperately seeking alternative options. Patients seeking the above-mentioned treatments should go to their oncologists armed with this paper and other medical publications rather than resorting to alternative or holistic providers who may not practice evidence-based medicine. Oncologists, therefore, must be prepared to assist patients in finding the most scientifically sound therapeutic options, lest they turn to the extremes of unconventional therapies, or even worse, to the counsel of charlatans.
Acknowledgments
The author would like to thank Allyson Ocean, MD, for encouraging and guiding him in finding treatment options which gave him hope at a near-hopeless time, Jorge Monge, MD, for his help with reference work, and lastly his son, Ryan Bigelsen, for his technical assistance and proofreading.
Disclosure
The author reports no conflicts of interest in this work.
References
Conroy T, Desseigne F, Ychou M, et al; Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. | ||
Churilla TM, Lesko SL, Brereton HD, Klem M, Donnelly PE, Peters CA. Serum vitamin D levels among patients in a clinical oncology practice compared to primary care patients in the same community: a case-control study. BMJ Open. 2011;1(2):e000397. | ||
Churilla TM, Brereton HD, Klem M, Peters CA. Vitamin D deficiency is widespread in cancer patients and correlates with advanced stage disease: a community oncology experience. Nutr Cancer. 2012;64(4):521–525. | ||
Skinner HG, Michaud DS, Giovannucci E, Willett WC, Colditz GA, Fuchs CS. Vitamin D intake and the risk for pancreatic cancer in two cohort studies. Cancer Epidemiol Biomarkers Prev. 2006;15(9):1688–1695. | ||
Wolpin BM, Ng K, Bao Y, et al. Plasma 25-hydroxyvitamin D and risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2012;21(1):82–91. | ||
Apte MV, Wilson JS. Dangerous liaisons: pancreatic stellate cells and pancreatic cancer cells. J Gastroenterol Hepatol. 2012;27 Suppl 2:69–74. | ||
Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93. | ||
Schwartz GG, Eads D, Naczki C, Northrup S, Chen T, Koumenis C. 19-nor-1 alpha,25-dihydroxyvitamin D2 (paricalcitol) inhibits the proliferation of human pancreatic cancer cells in vitro and in vivo. Cancer Biol Ther. 2008;7(3):430–436. | ||
O’Dwyer PJ, Sherman M, Johnston K, et al. A randomized pilot/pharmacodynamic/genomic study of neoadjuvant paricalcitol to target the microenvironment in resectable pancreatic cancer: a SU2C trial. In: American Society of Clinical Oncology Annual Conference; June 2nd 2015; Chicago, IL. | ||
Borazanci EH, Jameson GS, Borad MJ, et al. A phase II pilot trial of nivolumab + albumin bound paclitaxel + paricalcitol + cisplatin + gemcitabine (NAPPCG) in patients (pts) with previously untreated metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. 2017;35(Suppl 4):TPS511. | ||
Chakraborti CK. Vitamin D as a promising anticancer agent. Indian J Pharmacol. 2011;43(2):113–120. | ||
Audo I, Darjatmoko SR, Schlamp CL, et al. Vitamin D analogues increase p53, p21, and apoptosis in a xenograft model of human retinoblastoma. Invest Ophthalmol Vis Sci. 2003;44(10):4192–4199. | ||
Kalkunte S, Brard L, Granai CO, Swamy N. Inhibition of angiogenesis by vitamin D-binding protein: characterization of anti-endothelial activity of DBP-maf. Angiogenesis. 2005;8(4):349–360. | ||
Sprague SM, Llach F, Amdahl M, Taccetta C, Batlle D. Paricalcitol versus calcitriol in the treatment of secondary hyperparathyroidism. Kidney Int. 2003;63(4):1483–1490. | ||
Schwartz GG, Hall MC, Stindt D, Patton S, Lovato J, Torti FM. Phase I/II study of 19-nor-1alpha-25-dihydroxyvitamin D2 (paricalcitol) in advanced, androgen-insensitive prostate cancer. Clin Cancer Res. 2005;11(24 Pt 1):8680–8685. | ||
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. | ||
White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. | ||
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–312. | ||
Perera RM, Bardeesy N. Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov. 2015;5(12):1247–1261. | ||
White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. | ||
Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. | ||
Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19(6):637–638. | ||
Hong TS, Wo JY, Jiang W, et al. Phase II study of autophagy inhibition with hydroxychloroquine (HCQ) and preoperative (preop) short course chemoradiation (SCRT) followed by early surgery for resectable ductal adenocarcinoma of the head of pancreas (PDAC). J Clin Oncol. 2017;35(Suppl 15):4118. | ||
Miller-Ocuin JL, Bahary NS, Singhi AD, et al. Inhibition of autophagy improves pathologic and biomarker response to preoperative gemcitabine/Nab-paclitaxel in potentially resectable pancreatic cancer: a phase II randomized controlled trial. Ann Surg Oncol. 2017;24(Suppl 1):s6. | ||
Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123(6):1386–1394. | ||
Leung LS, Neal JW, Wakelee HA, Sequist LV, Marmor MF. Rapid onset of retinal toxicity from high-dose hydroxychloroquine given for cancer therapy. Am J Ophthalmol. 2015;160(4):799–805.e1. | ||
Cameron E, Campbell A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Biol Interact. 1974;9(4):285–315. | ||
Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1978;75(9):4538–4542. | ||
Nechuta S, Lu W, Chen Z, et al. Vitamin supplement use during breast cancer treatment and survival: a prospective cohort study. Cancer Epidemiol Biomarkers Prev. 2011;20(2):262–271. | ||
Harris HR, Orsini N, Wolk A. Vitamin C and survival among women with breast cancer: a meta-analysis. Eur J Cancer. 2014;50(7):1223–1231. | ||
Creagan ET, Moertel CG, O’Fallon JR, et al. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N Engl J Med. 1979;301(13):687–690. | ||
Moertel CG, Fleming TR, Creagan ET, Rubin J, O’Connell MJ, Ames MM. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N Engl J Med. 1985;312(3):137–141. | ||
Padayatty SJ, Sun H, Wang Y, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140(7):533–537. | ||
Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93(8):3704–3709. | ||
Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR Jr, Wang Y, Levine M. Phamacokinetic model for ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14(9):1133–1139. | ||
Hoffer LJ, Levine M, Assouline S, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19(11):1969–1974. | ||
Doskey CM, Buranasudja V, Wagner BA, et al. Tumor cells have decreased ability to metabolize H2O2: implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016;10:274–284. | ||
Du J, Martin SM, Levine M, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16(2):509–520. | ||
Espey MG, Chen P, Chalmers B, et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med. 2011;50(11):1610–1619. | ||
Du J, Doskey CM, Wilkes JG, Buettner GR, Cullen JJ. Catalase as a potential biomarker of pharmacological ascorbate cancer therapy. Free Radic Biol Med. 2016;100:S121. | ||
Yun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–1396. | ||
Klingelhoeffer C, Kammerer U, Koospal M, et al. Natural resistance to ascorbic acid induced oxidative stress is mainly mediated by catalase activity in human cancer cells and catalase-silencing sensitizes to oxidative stress. BMC Complement Altern Med. 2012;12:61. | ||
Aguilera O, Munoz-Sagastibelza M, Torrejon B, et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget. 2016;7(30):47954–47965. | ||
Bonuccelli G, De Francesco EM, de Boer R, Tanowitz HB, Lisanti MP. NADH autofluorescence, a new metabolic biomarker for cancer stem cells: identification of vitamin C and CAPE as natural products targeting “stemness”. Oncotarget. 2017;8(13):20667–20678. | ||
Chen P, Stone J, Sullivan G, Drisko JA, Chen Q. Anti-cancer effect of pharmacologic ascorbate and its interaction with supplementary parenteral glutathione in preclinical cancer models. Free Radic Biol Med. 2011;51(3):681–687. | ||
Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102(38):13604–13609. | ||
Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med. 2009;47(1):32–40. | ||
Frömberg A, Gutsch D, Schulze D, et al. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemother Pharmacol. 2011;67(5):1157–1166. | ||
Chen P, Yu J, Chalmers B, et al. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs. 2012;23(4):437–444. | ||
Lin ZY, Chuang WL. Pharmacologic concentrations of ascorbic acid cause diverse influence on differential expressions of angiogenic chemokine genes in different hepatocellular carcinoma cell lines. Biomed Pharmacother. 2010;64(5):348–351. | ||
Pathi SS, Lei P, Sreevalsan S, Chadalapaka G, Jutooru I, Safe S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr Cancer. 2011;63(7):1133–1142. | ||
Takemura Y, Satoh M, Satoh K, Hamada H, Sekido Y, Kubota S. High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem Biophys Res Commun. 2010;394(2):249–253. | ||
Hardaway CM, Badisa RB, Soliman KF. Effect of ascorbic acid and hydrogen peroxide on mouse neuroblastoma cells. Mol Med Rep. 2012;5(6):1449–1452. | ||
Peng LJ, Lu DX, Qi RB, Zhang T, Wang Z, Sun Y. [Therapeutic effect of intravenous high-dose vitamin C on implanted hepatoma in rats]. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(2):264–266. Chinese [with English abstract]. | ||
Chen Q, Espey MG, Sun AY, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105(32):11105–11109. | ||
Welsh JL, Wagner BA, van’t Erve TJ, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol. 2013;71(3):765–775. | ||
Monti DA, Mitchell E, Bazzan AJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One. 2012;7(1):e29794. | ||
Cullen JJ. Ascorbate induces autophagy in pancreatic cancer. Autophagy. 2010;6(3):421–422. | ||
Sancho P, Burgos-Ramos E, Tavera A, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22(4):590–605. | ||
Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014; 2:12. | ||
Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013;3:3230. | ||
Incio J, Suboj P, Chin SM, et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS One. 2015;10(12):e0141392. | ||
Duan W, Chen K, Jiang Z, et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017;385:225–233. | ||
Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila). 2012;5(3):355–364. | ||
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69(19):7507–7511. | ||
Mohammed A, Janakiram NB, Brewer M, et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol. 2013;6(6):649–659. | ||
Schneider MB, Matsuzaki H, Haorah J, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120(5):1263–1270. | ||
Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42(5):781–785. | ||
Cifarelli V, Lashinger LM, Devlin KL, et al. Metformin and rapamycin reduce pancreatic cancer growth in obese prediabetic mice by distinct microRNA-regulated mechanisms. Diabetes. 2015;64(5):1632–1642. | ||
Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. | ||
Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137(2):482–488. | ||
Ambe CM, Mahipal A, Fulp J, Chen L, Malafa MP. Effect of metformin use on survival in resectable pancreatic cancer: a single-institution experience and review of the literature. PLoS One. 2016;11(3):e0151632. | ||
Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res. 2012;18(10):2905–2912. | ||
Kordes S, Pollak MN, Zwinderman AH, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16(7):839–847. | ||
Reni M, Dugnani E, Cereda S, et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: an open-label, randomized Phase II trial. Clin Cancer Res. 2016;22(5):1076–1085. | ||
Chaiteerakij R, Petersen GM, Bamlet WR, et al. Metformin use and survival of patients with pancreatic cancer: a cautionary lesson. J Clin Oncol. 2016;34(16):1898–1904. | ||
E J-Y, Lu S-E, Lin Y, et al. Effects of concomitant metformin and statins on overall survival of pancreatic cancer patients: results from SEER-Medicare data analyses. Cancer Res. 2017;77(13):abstr 3288. | ||
Bathaie SZ, Ashrafi M, Azizian M, Tamanoi F. Mevalonate pathway and human cancers. Curr Mol Pharmacol. 2017;10(2):77–85. | ||
Lee HS, Lee SH, Lee HJ, et al. Statin use and its impact on survival in pancreatic cancer patients. Medicine (Baltimore). 2016;95(19):e3607. | ||
Villarino N, Signaevskaia L, van Niekerk J, et al. A screen for inducers of bHLH activity identifies pitavastatin as a regulator of p21, Rb phosphorylation and E2F target gene expression in pancreatic cancer. Oncotarget. 2017;8(32):53154–53167. | ||
Liao J, Chung YT, Yang AL, et al. Atorvastatin inhibits pancreatic carcinogenesis and increases survival in LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre mice. Mol Carcinog. 2013;52(9):739–750. | ||
Cheema A, Hassan H, Qureshi W, Shakeel M, Saif W. Effect of statins on risk of pancreatic cancer: a meta-analysis of randomized controlled trials (RCTs) and observational studies (OS). J Clin Oncol. 2016;34:abstr e15751. | ||
Khurana V, Sheth A, Caldito G, Barkin JS. Statins reduce the risk of pancreatic cancer in humans: a case-control study of half a million veterans. Pancreas. 2007;34(2):260–265. | ||
Goldstein MR, Mascitelli L, Pezzetta F. Do statins prevent or promote cancer? Curr Oncol. 2008;15(2):76–77. | ||
Huang BZ, Chang JI, Li E, Xiang AH, Wu BU. Influence of statins and cholesterol on mortality among patients with pancreatic cancer. J Natl Cancer Inst. 2017;109(5):djw275. | ||
Wu BU, Chang J, Jeon CY, et al. Impact of statin use on survival in patients undergoing resection for early-stage pancreatic cancer. Am J Gastroenterol. 2015;110(8):1233–1239. | ||
Jeon CY, Pandol SJ, Wu B, et al. The association of statin use after cancer diagnosis with survival in pancreatic cancer patients: a SEER-medicare analysis. PLoS One. 2015;10(4):e0121783. | ||
Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367(19):1792–1802. | ||
Hong JY, Nam EM, Lee J, et al. Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemother Pharmacol. 2014;73(1):125–130. | ||
Moon do C, Lee HS, Lee YI, et al. Concomitant statin use has a favorable effect on gemcitabine-erlotinib combination chemotherapy for advanced pancreatic cancer. Yonsei Med J. 2016;57(5):1124–1130. | ||
Bimonte S, Cascella M, Leongito M, et al. An overview of pre-clinical studies on the effects of (−)-epigallocatechin-3-gallate, a catechin found in green tea, in treatment of pancreatic cancer. Recenti Prog Med. 2017;108(6):282–287. | ||
Bimonte S, Leongito M, Barbieri A, et al. Inhibitory effect of (−)-epigallocatechin-3-gallate and bleomycin on human pancreatic cancer MiaPaca-2 cell growth. Infect Agents Cancer. 2015;10(1):22. | ||
Bimonte S, Barbieri A, Leongito M, et al. Curcumin anticancer studies in pancreatic cancer. Nutrients. 2016;8(7):E433. | ||
Bimonte S, Barbieri A, Leongito M, et al. The role of miRNAs in the regulation of pancreatic cancer stem cells. Stem Cells Int. 2016;2016:8352684. | ||
Li Y, Go VL, Sarkar FH. The role of nutraceuticals in pancreatic cancer prevention and therapy: targeting cellular signaling, miRNAs and epigenome. Pancreas. 2015;44(1):1–10. | ||
Kanai M. Therapeutic applications of curcumin for patients with pancreatic cancer. World J Gastroenterol. 2014;20(28):9384–9391. | ||
Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004;101(10):2351–2362. | ||
Kunnumakkara AB, Diagaradjane P, Guha S, et al. Curcumin sensitizes human colorectal cancer xenografts in nude mice to gamma-radiation by targeting nuclear factor-kappaB-regulated gene products. Clin Cancer Res. 2008;14(7):2128–2136. | ||
Sahu RP, Batra S, Srivastava SK. Activation of ATM/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. Br J Cancer. 2009;100(9):1425–1433. | ||
Glienke W, Maute L, Wicht J, Bergmann L. Curcumin inhibits constitutive STAT3 phosphorylation in human pancreatic cancer cell lines and downregulation of survivin/BIRC5 gene expression. Cancer Invest. 2010;28(2):166–171. | ||
Jutooru I, Chadalapaka G, Lei P, Safe S. Inhibition of NFkappaB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J Biol Chem. 2010;285(33):25332–25344. | ||
Ali S, Ahmad A, Banerjee S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70(9):3606–3617. | ||
Youns M, Fathy GM. Upregulation of extrinsic apoptotic pathway in curcumin-mediated antiproliferative effect on human pancreatic carcinogenesis. J Cell Biochem. 2013;114(12):2654–2665. | ||
Li Y, Revalde JL, Reid G, Paxton JW. Modulatory effects of curcumin on multi-drug resistance-associated protein 5 in pancreatic cancer cells. Cancer Chemother Pharmacol. 2011;68(3):603–610. | ||
Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007;67(8):3853–3861. | ||
Fujioka S, Son K, Onda S, et al. Desensitization of NFkappaB for overcoming chemoresistance of pancreatic cancer cells to TNF-alpha or paclitaxel. Anticancer Res. 2012;32(11):4813–4821. | ||
Uwagawa T, Yanaga K. Effect of NF-kappaB inhibition on chemoresistance in biliary-pancreatic cancer. Surg Today. 2015;45(12):1481–1488. | ||
Arlt A, Gehrz A, Muerkoster S, et al. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22(21):3243–3251. | ||
Yoshida K, Toden S, Ravindranathan P, Han H, Goel A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis. 2017;38(10):1036–1046. | ||
Gupta SC, Sung B, Kim JH, Prasad S, Li S, Aggarwal BB. Multitargeting by turmeric, the golden spice: from kitchen to clinic. Mol Nutr Food Res. 2013;57(9):1510–1528. | ||
Ferrucci LM, Daniel CR, Kapur K, et al. Measurement of spices and seasonings in India: opportunities for cancer epidemiology and prevention. Asian Pac J Cancer Prev. 2010;11(6):1621–1629. | ||
Park W, Amin AR, Chen ZG, Shin DM. New perspectives of curcumin in cancer prevention. Cancer Prev Res (Phila). 2013;6(5):387–400. | ||
Singletary K, MacDonald C, Wallig M, Fisher C. Inhibition of 7,12-dimethylbenz[a]anthracene (DMBA)-induced mammary tumorigenesis and DMBA-DNA adduct formation by curcumin. Cancer Lett. 1996;103(2):137–141. | ||
Ushida J, Sugie S, Kawabata K, et al. Chemopreventive effect of curcumin on N-nitrosomethylbenzylamine-induced esophageal carcinogenesis in rats. Jpn J Cancer Res. 2000;91(9):893–898. | ||
Perkins S, Verschoyle RD, Hill K, et al. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2002;11(6):535–540. | ||
Prasad S, Tyagi AK, Aggarwal BB. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: the golden pigment from golden spice. Cancer Res Treat. 2014;46(1):2–18. | ||
Surh YJ, Chun KS, Cha HH, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat Res. 2001;480–481:243–268. | ||
Sun XD, Liu XE, Huang DS. Curcumin reverses the epithelial-mesenchymal transition of pancreatic cancer cells by inhibiting the Hedgehog signaling pathway. Oncol Rep. 2013;29(6):2401–2407. | ||
Díaz Osterman CJ, Gonda A, Stiff T, et al. Curcumin induces pancreatic adenocarcinoma cell death via reduction of the inhibitors of apoptosis. Pancreas. 2016;45(1):101–109. | ||
Bimonte S, Barbieri A, Palma G, Luciano A, Rea D, Arra C. Curcumin inhibits tumor growth and angiogenesis in an orthotopic mouse model of human pancreatic cancer. Biomed Res Int. 2013;2013:810423. | ||
Arlt A, Schafer H, Kalthoff H. The ‘N-factors’ in pancreatic cancer: functional relevance of NF-kappaB, NFAT and Nrf2 in pancreatic cancer. Oncogenesis. 2012;1:e35. | ||
Diaz Osterman CJ, Wall NR. Curcumin and pancreatic cancer: a research and clinical update. J Nat Sci. 2015;1(6):e124. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Lao CD, Ruffin MT 4th, Normolle D, et al. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med. 2006;6:10. | ||
Vareed SK, Kakarala M, Ruffin MT, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol Biomarkers Prev. 2008;17(6):1411–1417. | ||
Garcea G, Berry DP, Jones DJ, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev. 2005;14(1):120–125. | ||
Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–4499. | ||
Kanai M, Yoshimura K, Asada M, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68(1):157–164. | ||
Kanai M, Imaizumi A, Otsuka Y, et al. Dose-escalation and pharmacokinetic study of nanoparticle curcumin, a potential anticancer agent with improved bioavailability, in healthy human volunteers. Cancer Chemother Pharmacol. 2012;69(1):65–70. | ||
Kanai M, Otsuka Y, Otsuka K, et al. A phase I study investigating the safety and pharmacokinetics of highly bioavailable curcumin (Theracurmin) in cancer patients. Cancer Chemother Pharmacol. 2013;71(6):1521–1530. | ||
Sasaki H, Sunagawa Y, Takahashi K, et al. Innovative preparation of curcumin for improved oral bioavailability. Biol Pharm Bull. 2011;34(5):660–665. | ||
Sunagawa Y, Hirano S, Katanasaka Y, et al. Colloidal submicron-particle curcumin exhibits high absorption efficiency-a double-blind, 3-way crossover study. J Nutr Sci Vitaminol (Tokyo). 2015;61(1):37–44. | ||
Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2017;302(6):649–658. | ||
Holmes MD, Chen WY, Li L, Hertzmark E, Spiegelman D, Hankinson SE. Aspirin intake and survival after breast cancer. J Clin Oncol. 2010;28(9):1467–1472. | ||
Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6(2):130–140. | ||
Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Curhan GC, Fuchs CS. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA. 2005;294(8):914–923. | ||
Mahipal A, Anderson KE, Limburg PJ, Folsom AR. Nonsteroidal anti-inflammatory drugs and subsite-specific colorectal cancer incidence in the Iowa women’s health study. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1785–1790. | ||
Tan XL, Nieters A, Hoffmeister M, Beckmann L, Brenner H, Chang-Claude J. Genetic polymorphisms in TP53, nonsteroidal anti-inflammatory drugs and the risk of colorectal cancer: evidence for gene-environment interaction? Pharmacogenet Genomics. 2007;17(8):639–645. | ||
Collet JP, Sharpe C, Belzile E, Boivin JF, Hanley J, Abenhaim L. Colorectal cancer prevention by non-steroidal anti-inflammatory drugs: effects of dosage and timing. Br J Cancer. 1999;81(1):62–68. | ||
Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348(10):883–890. | ||
González-Pérez A, García Rodríguez LA, López-Ridaura R. Effects of non-steroidal anti-inflammatory drugs on cancer sites other than the colon and rectum: a meta-analysis. BMC Cancer. 2003;3:28 | ||
Kasum CM, Blair CK, Folsom AR, Ross JA. Non-steroidal anti-inflammatory drug use and risk of adult leukemia. Cancer Epidemiol Biomarkers Prev. 2003;12(6):534–537. | ||
Harris RE, Chlebowski RT, Jackson RD, et al. Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women’s Health Initiative. Cancer Res. 2003;63:6096–6101. | ||
Marshall SF, Bernstein L, Anton-Culver H, et al. Nonsteroidal anti-inflammatory drug use and breast cancer risk by stage and hormone receptor status. J Natl Cancer Inst. 2005;97:805–812. | ||
Prizment AE, Folsom AR, Anderson KE. Nonsteroidal anti-inflammatory drugs and risk for ovarian and endometrial cancers in the Iowa Women’s Health Study. Cancer Epidemiol Biomarkers Prev. 2010;19(2):435–442. | ||
Viswanathan AN, Feskanich D, Schernhammer ES, Hankinson SE. Aspirin, NSAID, and acetaminophen use and the risk of endometrial cancer. Cancer Res. 2008;68(7):2507–2513. | ||
Jacobs EJ, Rodriguez C, Mondul AM, et al. A large cohort study of aspirin and other nonsteroidal anti-inflammatory drugs and prostate cancer incidence. J Natl Cancer Inst. 2005;97(13):975–980. | ||
Salinas CA, Kwon EM, FitzGerald LM, et al. Use of aspirin and other nonsteroidal antiinflammatory medications in relation to prostate cancer risk. Am J Epidemiol. 2010;172(5):578–590. | ||
Risch HA, Lu L, Streicher SA, et al. Aspirin use and reduced risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2017;26(1):68–74. | ||
Streicher SA, Yu H, Lu L, Kidd MS, Risch HA. Case-control study of aspirin use and risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(7):1254–1263. | ||
Anderson KE, Johnson TW, Lazovich D, Folsom AR. Association between nonsteroidal anti-inflammatory drug use and the incidence of pancreatic cancer. J Natl Cancer Inst. 2002;94(15):1168–1171. | ||
Tan XL, Reid Lombardo KM, Bamlet WR, et al. Aspirin, nonsteroidal anti-inflammatory drugs, acetaminophen, and pancreatic cancer risk: a clinic-based case-control study. Cancer Prev Res (Phila). 2011;4(11):1835–1841. | ||
Archibugi L, Piciucchi M, Stigliano S, et al. Exclusive and combined use of statins and aspirin and the risk of pancreatic cancer: a case-control study. Sci Rep. 2017;7(1):13024. | ||
Elwood PC, Morgan G, Pickering JE, et al. Aspirin in the treatment of cancer: reductions in metastatic spread and in mortality: a systematic review and meta-analyses of published studies. PLoS One. 2016;11(4):e0152402. | ||
Mitrugno A, Sylman JL, Ngo AT, et al. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: implications for the oncoprotein c-MYC. Am J Physiol Cell Physiol. 2017;312(2):C176–C189. | ||
Ai G, Dachineni R, Muley P, Tummala H, Bhat GJ. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: a potential role in chemoprevention. Tumour Biol. 2016;37(2):1727–1738. | ||
Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomized controlled trials. Lancet. 2012;379(9826):1591–1601. | ||
Yang L, Zhu H, Liu D, et al. Aspirin suppresses growth of human gastric carcinoma cell by inhibiting survivin expression. J Biomed Res. 2011;25(4):246–253. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.