")
Back to Journals » International Journal of General Medicine » Volume 16
Events in CNS Tumor Pathology Post-2016 WHO CNS: cIMPACT-NOW Updates and Other Advancements: A Comprehensive Review Plus a Summary of the Salient Features of 2021 WHO CNS 5
Authors Ahmad Z, Rahim S, Abdul-Ghafar J , Chundriger Q, Ud Din N
Received 26 October 2022
Accepted for publication 29 December 2022
Published 7 January 2023 Volume 2023:16 Pages 107—127
DOI https://doi.org/10.2147/IJGM.S394872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Zubair Ahmad,1 Shabina Rahim,1 Jamshid Abdul-Ghafar,2 Qurratulain Chundriger,1 Nasir Ud Din1
1Department of Pathology and Laboratory Medicine, Aga Khan University Hospital, Karachi, Pakistan; 2Department of Pathology and Clinical Laboratory, French Medical Institute for Mothers and Children (FMIC), Kabul, Afghanistan
Correspondence: Jamshid Abdul-Ghafar, Department of Pathology and Clinical Laboratory, French Medical Institute for Mothers and Children (FMIC), Kabul, Afghanistan, Tel +93 792 827 287, Email [email protected]
Introduction: The 2016 World Health Organization Classification (WHO) of Tumors of the Central Nervous System (CNS) represented a major change. It recommended an “integrated diagnosis” comprising histologic and molecular information facilitating a more precise diagnosis of specific CNS tumors. Its goal was to provide greater diagnostic precision and reproducibility resulting in more clinical relevance and predictive value, ultimately leading to better patient care. Advances in molecular classification, mostly resulting from DNA methylation array profiling of CNS tumors, were occurring at a very rapid pace and required more rapid integration into clinical practice.
Methods: cIMPACT-NOW updates and other recent papers plus salient features of 2021 WHO CNS5 in this comprehensive write-up were reviewed.
Results: CNS tumor classification needs to be updated at a rapid pace and mechanisms put into place to guide diagnosticians and clinicians in the interim period if major changes in the classification of tumor types came to light. Recognizing the need to integrate these into clinical practice more rapidly and without inordinate delay, the International Society of Neuropathology (ISN) 2016 sponsored an initiative called cIMPACT-NOW.
Discussion and/or Conclusion: Goal of cIMPACT-NOW was to provide clarification regarding contentious issues arising in the wake of the 2016 WHO CNS update as well as report new advancements in molecular classification of CNS tumors and new tumor entities emerging as a result of these advancements. cIMPACT-NOW updates: It thus laid the foundation for the 5th edition of the WHO Classification of CNS tumors (2021 WHO CNS 5). We have discussed cIMPACT updates in detail in this review. In addition, molecular diagnostics including DNA methylation-based classification of CNS tumors and the practical use of molecular classification in the prognostication and treatment of CNS tumors is discussed. Finally, the salient features of the new CNS tumor classification are summarized.
Keywords: cIMPACT-NOW, DNA-methylation profiling, molecular diagnostics, integrated reporting, hypermutated gliomas
Introduction
The revised 4th edition of the World Health Organization Classification (WHO) Classification of Central Nervous System (CNS) tumors (2016 WHO CNS) was published as an update of the 4th edition (2007) in response to the rapid advancements and breakthroughs in our understanding of the molecular basis of CNS tumors which were associated with greater diagnostic precision and reproducibility of CNS tumor entities and necessitated early integration into clinical practice for better predictive response and care of patients suffering from CNS tumors in the intervening period between the 4th (2016 WHO CNS) and 5th (2021 WHO CNS 5) editions. 2016 WHO CNS represented a major change in the way CNS tumors were classified. It defined tumor entities by combining histological and molecular information and recommended an “integrated diagnosis” in a layered format comprising histologic classification, WHO grade, and molecular information thus facilitating a more definite diagnosis and risk stratification of CNS tumors.1–4 For example, the integration of traditional histological features and molecular information is essential to achieve maximum risk stratification of adult IDH-mutant diffuse gliomas.5 Since advancements in molecular typing of CNS tumors continued rapidly even after 2016, integration of these into routine clinical practice before the publication of 2021 WHO CNS 5 was essential for optimum patient care. For example, large scale genomic DNA analysis demonstrated differences between tumors with similar pathological features.6 In addition, several contentious issues such as marked variation in the use of the term NOS (not otherwise specified); confusion regarding diagnosis of diffuse middle gliomas; need to distinguish IDH wild-type diffuse astrocytic glioma from IDH mutant types came to light in the wake of 2016 WHO CNS, based on experiences of pathologists and clinicians diagnosing and treating CNS tumors which required clarification and needed to be resolved to achieve the goal of optimum patient care. Also, newer entities were continually being recognized and definitive information continued to emerge regarding entities already included in the 2016 WHO CNS as a result of new studies which was significantly different and needed to be integrated into clinical practice without inadvertent delay. Recognizing this, the International Society of Neuropathology (ISN) sponsored an initiative named cIMPACT-NOW, the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy Not Official WHO. Its goal was to provide clarifications regarding contentious issues arising in wake of the 2016 Update and to define newly recognized entities.7–10 Members of cIMPACT-NOW Steering Committee were principal authors of 2016 WHO CNS. A Clinical Advisory Panel was also constituted which provided feedback on the clinical implications of the recommendations proposed by cIMPACT-NOW. Three Working Committees were assigned specific topics of interest and published four updates in Round 1 following which the committees were reconstituted and published three updates in Round 2. Each update was published in Acta Neuropathologica and an accompanying commentary that focused on the practical use of the proposed recommendations in clinical practice was published in Brain Pathology. While some updates were meant to “clarify” confusing issues, others reported the advancements in molecular classification of CNS tumors, new entities as well as changes in grading, prognostication, and treatment. Thus, these updates laid an effective foundation for the 5th edition of CNS tumors (WHO CNS5). In this review, we will discuss the salient features of the seven cIMPACT-NOW updates and review the advancements in molecular classification of CNS tumors including DNA methylation profiling of CNS tumors. Hypermutated gliomas will be discussed as examples of CNS tumors where treatment benefits from the integration of molecular information in diagnosis and results in targeted (personalized) therapy. We will also briefly discuss the templates for integrated, layered CNS tumor reporting and summarize the salient features of the new 2021 WHO CNS5.
Main Text
cIMPACT-NOW update 1 clarified the use of the term “Not Otherwise Specified (NOS)” and proposed the additional term “Not Elsewhere Classified (NEC)”.11 The designation NOS was introduced by the 2016 WHO CNS to designate tumors about which sufficient information was not available. It included tumors on which molecular testing had not been performed as well as those on which it was performed but did not show specific molecular features.1
However, feedback showed marked variation in the use of this term. Update 1 clarified that use of NOS was not appropriate when molecular testing was performed but specific features were not found. It emphasized that it is difficult to treat tumors designated NOS and this term should only be used in situations when additional diagnostic testing was necessary in order to reach a specific diagnosis but was not done or was performed but failed. Examples include determination of IDH status in diffuse glioma or determination of IDH and 1p/19q status in oligodendroglioma, etc. The update recommended that this term not be used when additional diagnostic testing was not required to establish a specific WHO diagnosis. Examples include testing for EGFR or MGMT methylation status in glioblastomas. It emphasized that the report should clearly state the reason for using NOS and strongly recommended that if tissue was available, it should be sent to a larger center for workup.
A new term NEC (Not Elsewhere Classified) was defined for cases in which necessary testing was performed but failed to achieve a definite diagnosis. It was recommended that NEC be used when a tumor cannot be assigned to a specific entity. For example, when the histological, IHC, and/or genetic features do not match or when new entities appear to have emerged. NEC diagnosis can be given when molecular genetic findings are not required for diagnosis but are important for adequate classification, for example, BRAF V600E mutations in Pilocytic Astrocytoma (PA) or Pleomorphic Xanthoastrocytoma (PXA).
It was recommended that the NEC diagnosis should be in form of a layered format and include histological features and genetic information facilitating the interpretation of the report. It is hoped that NEC designation will need to be used much less often in the future as more and more tumors are specifically classified.9,12–14 For example, any CNS embryonal tumor which cannot be assigned a specific diagnosis even after adequate testing is currently diagnosed as CNS embryonal tumor, NOS. Update 1 recommended that such cases be diagnosed as CNS embryonal tumor, NEC, while those cases in which additional testing could not be performed be diagnosed as CNS embryonal tumor, NOS. For atypical teratoid/rhabdoid tumor (AT/RT), the descriptive diagnosis of CNS embryonal tumor with rhabdoid features, NOS should be given if testing is not performed. If testing is done but is negative, the diagnosis of CNS embryonal tumor with rhabdoid features, NEC should be given.
The main features of Update 1 are summarized below:
- Meaning of “Not otherwise specified” (NOS) designation in diagnoses refined.
- New designation “Not elsewhere classified” (NEC) introduced.
- NOS to be used when diagnostic information (histologic or molecular) required for a specific WHO diagnosis is not available because necessary molecular testing is not performed, either because it is not available or is performed but does not work (does not yield adequate results).
- NEC to be used when molecular testing is performed and test results are valid and adequate but do not lead to a precise WHO diagnosis. In other words, available diagnostic (molecular) information is not currently sufficient to allow the diagnosis of a specific WHO entity.
cIMPACT-NOW Update 2 clarified and updated the criteria for diagnosing diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH mutant.15
Diffuse midline glioma mostly occurs in children in a midline location (brain stem, thalamus, spinal cord) diffusely infiltrates adjacent brain tissue, is usually astrocytic, and demonstrates H3 K27M mutation. However, many other tumors have now been shown to harbor the same mutation. These include PA, pediatric diffuse astrocytoma, ganglioglioma, ependymoma, etc. Their clinicopathological features vary, and they do not constitute a distinct entity. The prognostic significance of H3 K27M mutation in these tumors is not fully known, and some of these tumors are associated with a long survival. In contrast, Diffuse midline glioma, H3 k27M mutation has a poor prognosis and the two-year survival rate is less than 10%.16–23
The 2016 WHO CNS stated that H3 K27M mutations only occur in diffuse midline glioma, H3 K27M mutant. This has now been proved to be incorrect. It was stressed that since H3 K27M mutation is seen in other tumors as well, it cannot by itself be used for the diagnosis of a diffuse midline glioma, H3 K27M mutant.
The second clarification was regarding diffuse astrocytoma, IDH mutant, and anaplastic astrocytoma, IDH mutant. There was confusion about whether all diffuse gliomas needed to be analyzed for 1p/19q codeletion. The 2016 WHO CNS stated that IDH-mutant phenotypically oligodendrocytic tumors with astrocytic areas should be classified as oligodendroglioma if they demonstrate 1p/19q codeletion. This implied that 1p/19q codeletion analysis was only required for oligodendrogliomas and mixed oligoastrocytomas and not for pure astrocytomas. However, a review article in Acta Neuropathologica declared that an IDH-mutant phenotypically astrocytic tumor which demonstrated 1p/19q codeletion should be diagnosed as oligodendroglioma. This implied that 1p/19 q analysis was required in all diffuse gliomas, IDH mutant including pure astrocytomas.
It was clarified that a diffuse astrocytic glioma, WHO grade II or III, can be diagnosed as diffuse astrocytoma, IDH mutant or anaplastic astrocytoma, IDH mutant without 1p/19q testing if the tumor shows IDH mutation plus loss of ATRX nuclear expression and/or diffuse strong p53 immunopositivity.
Thus, Update 2 provided relief as it stated that the use of surrogate IHC markers may yield a full WHO diagnosis in a histological diffuse astrocytic glioma that is positive for IDH R132 H.
The main features of Update 2 are summarized below:
- It was clarified that the term “diffuse midline glioma, H3 K27M-mutant” should be reserved for those H3 K27M-mutant tumors which satisfy all the following attributes: diffuse infiltrative growth in the CNS parenchyma, affect midline structures (brain stem, thalamus, spinal cord), are gliomas on histological examination and are H3 K27M-mutant.
- It should not be applied to those tumors with H3 K27M-mutations that are not diffuse midline gliomas and do not behave as high-grade tumors. Examples include ependymoma, pilocytic astrocytoma, and ganglioglioma.
- It was clarified that every IDH-mutant WHO grade II or III diffuse astrocytic gliomas do not require 1p 19q testing and can be designated as diffuse astrocytoma, IDH mutant or anaplastic astrocytoma, IDH mutant in the absence of 1P 19q testing if the tumor immunohistochemically shows strong and diffuse nuclear staining for p53 and/or clear loss of nuclear expression of ATRX.
- In other words, a diagnosis of IDH-mutant diffuse astrocytoma can be made if surrogate IHC markers are suggestive of the diagnosis without the requirement of testing for 1p 19q codeletion.
cIMPACT-NOW Update 3 distinguished IDH wild-type diffuse astrocytic gliomas from IDH-mutant astrocytic gliomas. H3 G34-mutant diffuse glioma was not recognized as a specific entity in 2016 WHO CNS. It is now known that the detection of an H3 G34 mutation in a diffuse glioma of any histological grade is an indicator of aggressiveness and survival is only slightly longer than survival times in IDH wild-type glioblastomas. These tumors occur mainly in children and adolescents and are distinct from the IDH wild-type astrocytic gliomas.12,13,24
Studies that showed that a substantial subset of adult diffuse astrocytoma, IDH wild type, WHO grade II and anaplastic astrocytoma, IDH wild type, WHO grade III had a more aggressive clinical course and demonstrated survival times similarly or only slightly better than Glioblastoma, IDH wild type, WHO grade IV were discussed.25–28 The literature was searched for molecular criteria reliably identifying these aggressive IDH wild type grade II and III astrocytomas. Only those molecular features which are associated with a bad prognosis as clinical management of these tumors could be substantially altered by such markers were considered. Based on this search, three diagnostic molecular criteria were recommended: (1) High-level EGFR amplification OR (2) Combined whole chromosome 7 gain and whole chromosome 10 loss (+7/-10) OR (3) TERT promoter mutation.
Multiple studies had shown that IDH wild type grade II and grade III diffuse astrocytic tumors with the above genetic alterations behaved aggressively with significantly shorter survival times similar to patients with IDH wild type glioblastoma. Many such tumors correspond histologically to anaplastic astrocytoma, WHO grade III.26–28
EGFR amplification was shown to have excellent specificity for aggressive gliomas and was not present in more indolent gliomas. Only high-level copy number gains are considered consistent with EGFR amplification. Combined +7/-10 were shown to have excellent specificity for these aggressive IDH wild-type astrocytic gliomas, grade II and III, and were linked with poor survival. TERT promoter mutations were quite common in glioblastoma grade IV, and astrocytoma grade II & III IDH-wildtype and usually occurred in those tumors which also showed +7/-10 and EGFR amplification. TERT promoter mutations, even when they occurred in the absence of +7/-10 or EGFR amplification, were still associated with aggressive clinical behavior and can serve as markers for WHO grade IV behavior in IDH wild type grade II and grade III astrocytic tumors. It was emphasized that almost all oligodendrogliomas harbor TERT mutations, while some less aggressive gliomas including PXA, ganglioglioma, ependymoma, etc. may occasionally harbor TERT mutations. The possibility of oligodendroglioma should be ruled out in a tumor with TERT promoter mutations by IDH and 1p/19q testing.26–28
The presence of the molecular changes discussed above makes it possible to predict poor outcomes similar to glioblastoma in tumors which do not fulfill the histological criteria of glioblastoma.24,29 Patients with these tumors will be recommended combined chemo and radiotherapy if they demonstrate EGFR amplification or +7/-10 or TERT promoter mutations. A layered format for diagnosis was recommended to clearly show the histological and molecular features.
These tumors also harbor homozygous CDKN 2A/B deletions in a high frequency of cases. However, other gliomas also harbor these mutations. A combination of BRAF mutation and CDKN 2A/B deletion is often seen in PXA, but outcomes are substantially better. An additional subset of IDH wildtype astrocytic gliomas with frequent deletions of CDKN2A/B was recognized recently, and ATRX mutation or loss of nuclear expression and BRAF pathway alterations were also typically present in these tumors. These tumors designated as “anaplastic astrocytoma with piloid features” also have a better prognosis. The presence of CDKN 2A/B deletion was not found to be indicative of aggressive behavior in these tumors. It was noted that these tumors clustered tightly with the usual IDH wildtype glioblastomas on DNA methylation profiling, which is a powerful new method for precisely segregating similar CNS tumors. Gliomas with indolent behavior do not cluster with these tumors. DNA methylation profiling was suggested as an alternative method to identify these aggressive tumors.9,12,13,24
The main features of Update 3 are summarized below:
- A large subset of histologic grade II and III IDH-wild type diffuse astrocytomas which lack necrosis and/or florid microvascular proliferation but behave aggressively similar to glioblastomas were studied.
- It was concluded that the presence of the following 3 molecular criteria or any combination of whole chromosome 7 gain and whole chromosome 10 loss, high-level EGFR amplification, and TERT promoter mutations even in histologically grade II or III IDH-wild type diffuse astrocytomas results in aggressive behavior similar to glioblastoma and upgrading of the tumor to WHO grade IV.
- It was strongly recommended that molecular analysis of grade II and III IDH wild-type diffuse astrocytomas be performed in adult patients.
It was recommended that such tumors be designated as “diffuse astrocytic glioma, IDH-wild type, with molecular features of glioblastoma, WHO grade IV”.
cIMPACT-NOW update 4: In the 2016 WHO CNS, IDH wildtype and H3 wildtype pediatric diffuse gliomas were assigned to the NOS category. However, later, it became apparent that these pediatric glial tumors harbored distinct molecular alterations with distinct prognostic and predictive outcomes. Although patients may suffer significant morbidity during the course of their disease, these tumors are associated with good overall survival.30–33
H3 K27M and H3 G34 mutant diffuse gliomas of children and young adults were found to have genetic features and prognosis distinct from IDH wildtype glioblastoma, WHO grade IV. Others harbor activating BRAF V600E mutations and distinct gene expression and methylation profiles. Their prognosis is better, and they may respond to targeted drugs. BRAF V600E mutations can be targeted with BRAF inhibitors such as Dabrafenib.34
Another small group of tumors that occurs mostly in children harbors MYB/MYBL rearrangements and has a better prognosis. It is important to recognize such distinct molecular signatures for targeted therapy.30 Owing to this genetic heterogeneity, the histologic grade is now considered less useful in predicting prognostic and predictive behavior in such tumors. These tumors are often associated with epilepsy, are astrocytic or oligodendroglial, histologic grade II with infiltration of adjacent CNS parenchyma and absent or low mitotic activity. Microvascular endothelial proliferation and necrosis are absent.
These entities include:
Diffuse glioma, MYB-altered
Diffuse glioma, MYBL1-altered
Diffuse glioma, FGFR1 TKD-duplicated
Diffuse glioma, FGFR1-mutant
Diffuse glioma, BRAF V600E-mutant
Diffuse glioma, other MAPK pathway alteration.
It was recommended that until more data regarding the prognosis of these tumors were available, they should not be assigned a WHO grade.
It was recommended that surrogate IHC markers (for BRAF V600E) and FISH (for detection of MYB or MYBL1 rearrangements) which are commonly available be used. It was suggested that if molecular analysis was performed but failed to discover the genetic alterations, such tumors be diagnosed as diffuse astrocytoma, IDH wild type, NEC or oligodendroglioma, IDH wild type, NEC. If molecular testing is not available, a diagnosis of diffuse astrocytoma, IDH wild type, NOS or oligodendroglioma, IDH wild type, NOS should be rendered.
It was noted that since these pediatric diffuse gliomas carried distinct molecular alterations despite the histological similarity to adult gliomas, it was essential to perform appropriate molecular testing for accurate classification and identification of potential therapeutic targets.12,13,30
The main features of Update 4 are summarized below:
- In general, IDH-wild-type diffuse astrocytic gliomas, WHO Grade II carry a worse prognosis compared to IDH-mutant tumors. However, some IDH-wild type and H3 wild-type (lacking H3 K27M mutation) diffuse astrocytomas, WHO Grade II in children and some young adults appear to have a better prognosis. Thus, when this diagnosis is made in this age group, it is important to characterize the tumor further at the molecular level to determine if it has a specific genetic alteration that signifies a favorable prognosis and can be targeted therapeutically.
- Such tumors, when they demonstrate BRAF V600 E mutation (but without CDKN2A/B deletion) or EGFR1 alterations (mutation or duplication) or a MYB or MYBL1 rearrangement or any other MAPK pathway alteration, should be diagnosed by combining the histologic and specific genetic features (integrated diagnosis) in a layered format.
- This refined categorization of pediatric type diffuse gliomas has great potential for targeted therapies.
- These tumors are thus distinct entities with prognostic and potentially therapeutic implications.
cIMPACT NOW Update 5 recommended grading criteria and terminologies for IDH-mutant diffuse astrocytoma, WHO grade II; anaplastic astrocytoma, WHO grade III and glioblastoma, WHO grade IV.35 These tumors are much more aggressive compared to IDH mutant astrocytomas and are recognized as distinct clinical and genetic entities. It was noted that while molecular classifications represented a major step forward for these tumors, the grading schemes had not been modified and the same grading criteria are applied to both IDH mutant and wild-type diffuse astrocytic gliomas. Many recent studies showed that the current grading system based on morphology (mitotic activity, nuclear atypia, microvascular proliferation, and/or necrosis) was not optimal.36–38 However, some other recent studies showed that traditional histology-based grading schemes are still useful for stratifying risk in these tumors.39–41 Several newer studies which investigated potential morphologic, proliferative, or molecular markers which correlate with aggressive clinical behavior and can improve risk stratification were discussed.26,37,39,42–46
It considered thresholds of proliferative activity based on mitotic figures or Ki-67 indices and any morphologic features typical of a high grade which could stratify risk better than current histologic criteria.
Multiple studies which showed that homozygous deletion of CDKN2A/B was an independent marker of poor prognosis in all grades of (II, III, IV) IDH mutant diffuse astrocytic gliomas and had a strong association with shorter survival were cited. The association was greatest for WHO grades III and IV. CDKN2A/B homozygous deletions were least frequent in grade II and most frequent in grade IV (16 to 34%).38,43,46
There was conflicting evidence for the role of CDK4 amplification with some studies showing that it was associated with poor prognosis and others showing that it was not. The same was true for homozygous deletion of RB1 with some studies demonstrating its strong association with poor prognosis and others not finding any such association. Altered RB1 pathway genes (CDKN 2A/B homozygous deletion, CDK4 amplification, or RB1 mutation) when present together were found to have a strong and statistically significant association with poor prognosis in these tumors. On multivariate analysis, PIK3R1 mutations were found to be independent markers of poor prognosis in these tumors.37,39,42,47
Multiple studies showed that PDGFRA amplification was associated with shorter survival in IDH mutant astrocytomas. This was confirmed on multivariate analysis. MYCN amplification was associated with shorter overall survival on univariate analysis.37,39,42,46,48
The presence of genomic instability, presence of both somatic mutations, and high levels of copy number variations (CNVs) were associated with shorter overall survival in these tumors. The prognosis was worse for tumors with high CNVs compared to tumors with low CNVs.39,42,49
Patients with G-CIMP-low (globally reduced levels of DNA methylation) IDH mutant diffuse astrocytomas had shorter overall survival compared to those with G-CIMP-high tumors. There was a significant association between G-CIMP-low tumors and those with alterations in RB pathway genes (CDKN2A/B, CDK4) leading to shorter overall survival in grades II, III, and IV astrocytomas.26,50
We have evaluated and discussed the evidence from multiple retrospective studies in detail. It was concluded that homozygous deletion of CDKN2A/B is a marker for grade IV behavior in IDH-mutant astrocytomas grade III and IV. It is agreed that alterations in genes encoding members of the RB pathway as well as amplification and mutations in PIK3R1 and PIK3CA and MYCN amplification all appeared to be associated with aggressive behavior. However, more evidence was required. The Committee’s observations were the same for genomic instability and G-CIMP-low DNA methylation. Evidence was not sufficient to establish a new threshold of mitotic activity to distinguish between grade II and grade III IDH mutant astrocytomas.
Thus, IDH wild type and IDH mutant diffuse astrocytic gliomas represent distinct clinical and genetic entities. However, until recently, similar terms were used for their classification. It was desirable to have terminologies that more clearly distinguished between these distinct tumor entities.
It was proposed that IDH wild-type astrocytomas with histologic or genetic features predictive of a highly aggressive clinical behavior be termed “glioblastoma” and this term be reserved for such tumors. For IDH mutant diffuse astrocytic gliomas, the Committee recommended that these be graded based upon the histologic and genetic features that correspond to WHO grades II, III, and IV. It was proposed that the term “glioblastoma” no longer be used for IDH mutant diffuse astrocytic gliomas.
It was recommended that Arabic (2,3,4) rather than Roman numerals (II, III, IV) be used to harmonize with WHO grading schemes of non-CNS tumors. It was noted that the distinction of 2 vs 3 was less susceptible to error in a report than II vs III and there was less possibility of typographic and interpretive errors. The following terminology, classification, and grading criteria for IDH mutant astrocytomas were proposed:
Astrocytoma, IDH mutant, grade 2: A diffusely infiltrative astrocytic glioma with an IDH1 or IDH2 mutation that is well differentiated and lacks histologic features of anaplasia. Mitotic activity is not detected or low. Microvascular proliferation, necrosis, and CDKN2A/B homozygous deletion are absent.
Astrocytoma, IDH mutant, grade 3: A diffusely infiltrative astrocytic glioma with an IDH 1 or IDH 2 mutation that exhibits focal or dispersed anaplasia and displays significant mitotic activity. Microvascular proliferation, necrosis, and CDKN2A/B homozygous deletions are absent.
Astrocytoma, IDH mutant grade 4: A diffusely infiltrative astrocytic glioma with an IDH1 or IDH 2 mutation that exhibits microvascular proliferation or necrosis or CDKN2A/B homozygous deletion or any combination of these features.
It was noted that no validated published criteria exist for mitotic count cut-off values for grading IDH mutant astrocytomas and “significant” mitotic activity remains the criterion to distinguish grade 3 from grade 2 tumors.
The main features of Update 5 are summarized below:
- IDH mutant diffuse astrocytomas generally have a more favorable prognosis than IDH-wild-type diffuse astrocytomas.
- However, the presence of a homozygous deletion of CDKN2A/B in IDH-mutant tumors is associated with a worse prognosis and such tumors behave aggressively as high-grade tumors.
- Update 5 recommended that grade II or III IDH-mutant diffuse astrocytic tumors should be graded as WHO grade IV even in the absence of necrosis and/or microvascular proliferation if they show the presence of homozygous CDKN2A/B deletion.
It was recommended that molecular testing for CDKN2A/B deletions be performed in all IDH-mutant grade II and III diffuse astrocytic tumors.
For cIMPACT-NOW update 6, Working Committee 3 convened in Utrecht, The Netherlands, for a meeting designed to review emerging tumor types for the new CNS tumor classification.
A substantial number of newly recognized tumor types and subtypes were recommended for inclusion in the new CNS tumor classification. A set of principles related to classification categories, approaches to classification, nomenclature, and grading was also endorsed.51
In preparation for the meeting, a list of 40 potential tumor types was compiled. Eight groups evaluated histopathologically related novel tumor types and addressed the following issues for each of these: (i) review literature on the particular tumor type and determine whether the literature was strong enough for classification of the entity as a distinct tumor type, (ii) decide if it was a type or a subtype, (iii) generate a new definition or alter an existing definition and (iv) suggest where it should be incorporated in the new WHO CNS tumor classification.
It then gave its recommendations on these issues. Some entities were left sub judice ie, lacking sufficient published evidence to make a decision. The categories of CNS tumors recommended by Update 6 along with examples of tumor entities in each category are shown in Table 1.
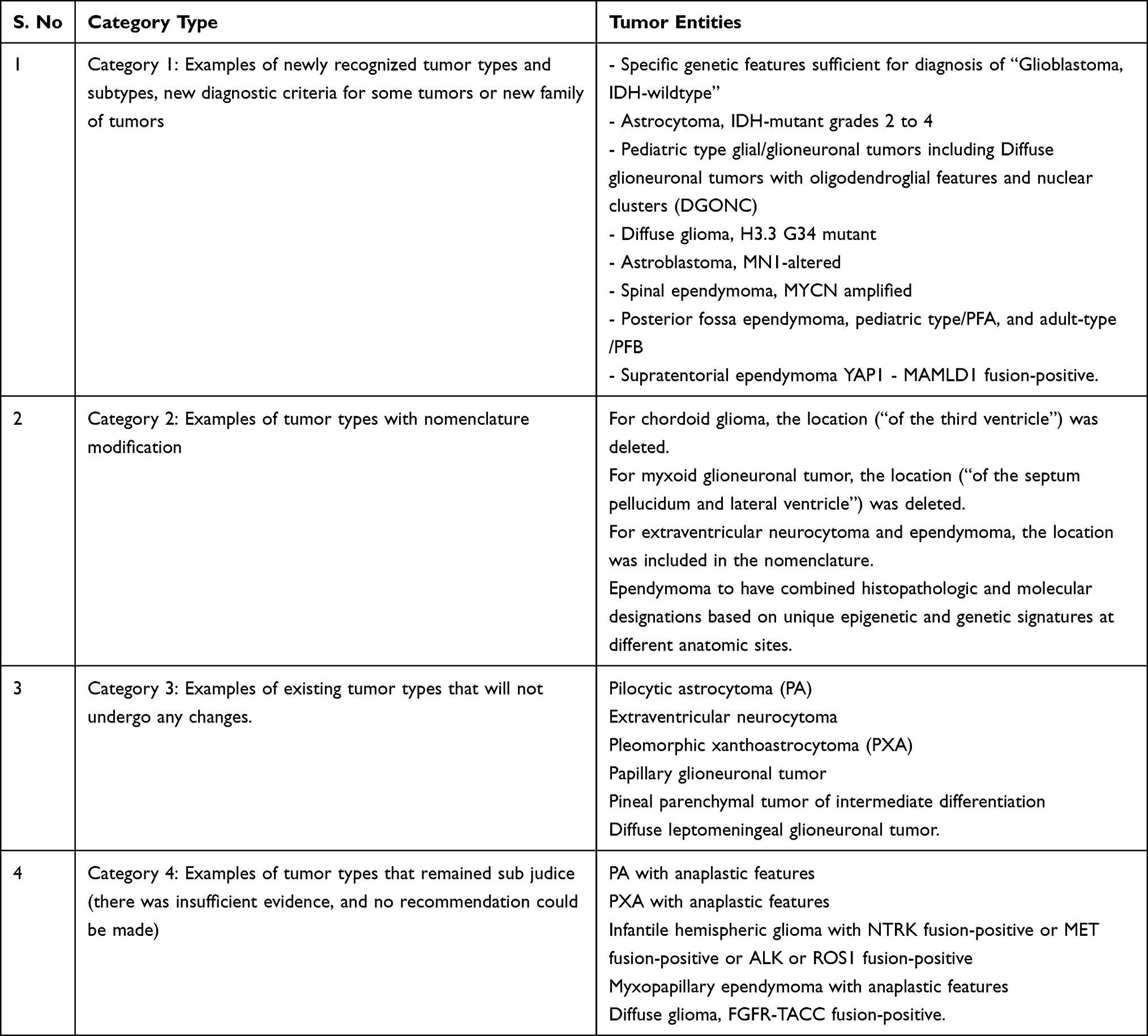 |
Table 1 Categories of CNS Tumors Recommended by Update 6 with Examples of Each |
The terms “types” and “subtypes” in place of the terms “entities” and “variants” were recognized to conform with the terminology used across organ systems.
Type: A neoplasm in which multiple parameters (for example, clinical, anatomic, histopathologic, and/or molecular) differ from other types, for example, myxoid glioneuronal tumor.
Subtype: A variant of a type in which a single or couple of parameters (for example, clinical, anatomic, histopathologic, and/or molecular) suggest it differs from other subtypes and needs to be recognized as a separate lesion, for example, MC-1 and MC-2 subtypes of diffuse leptomeningeal glioneuronal tumor.
The clinical utility of a “two list” or “mix-match” approach similar to the one used for the classification of medulloblastomas into histologically defined and genetically defined tumors was strongly endorsed, since such an approach afforded greater flexibility and conveyed key diagnostic information in a succinct layered format. The group of pediatric diffuse gliomas IDH-wildtype and H3 wildtype were used as models. Examples of “two list” or “mix-match” approach for clarification of CNS tumors are shown in Table 2.
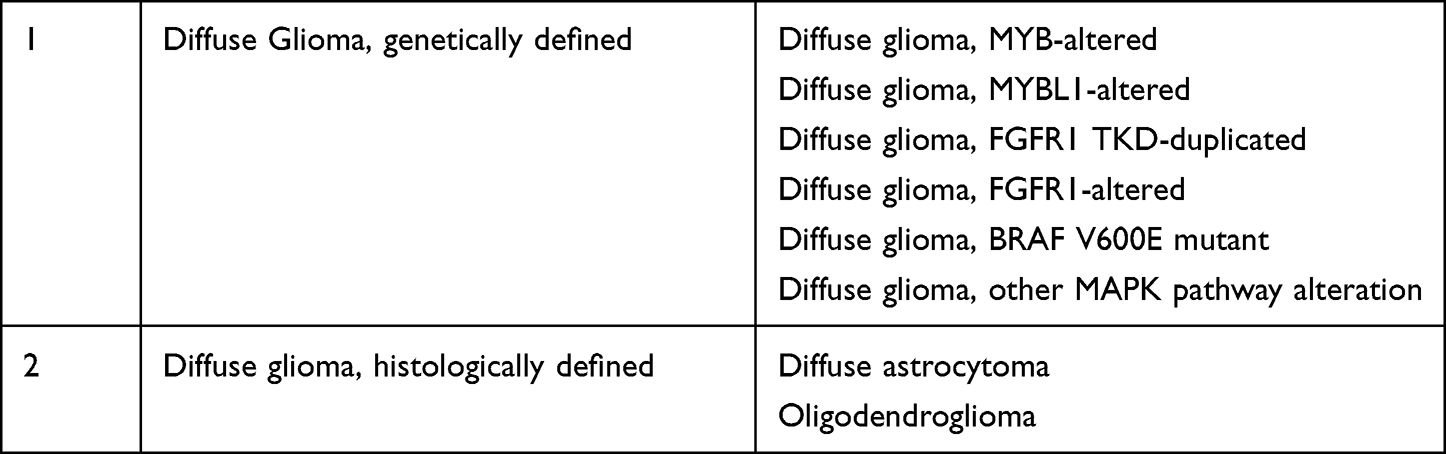 |
Table 2 Examples of Mix Match Approach Recommended by Update 6 for the Classification of Tumors |
The presence of homozygous CDKN2A/B deletion was approved as a marker for the highest malignancy grade (grade 4) in the group of diffuse, IDH-mutant astrocytoma, and the term “Astrocytoma, IDH-mutant grade 4” (rather than Glioblastoma IDH-mutant) was proposed for this subgroup. This makes it easier to discriminate IDH-mutant astrocytic tumors grade 4 from the much more common and aggressive IDH-wildtype glioblastomas. It was recommended that the nomenclature of the “Glioblastoma, IDH wildtype” including the cumbersome designation of “Diffuse astrocytic glioma, IDH wildtype with molecular features of glioblastoma, WHO grade IV” for histologic grade 2 and 3 IDH wildtype astrocytic gliomas be simplified. A diagnosis of “Glioblastoma, IDH wildtype, WHO grade 4” be rendered if there is microvascular proliferation or necrosis (or both) and even if these histologic features are absent if one (or more) of the following three genetic alterations ie, TERT promoter mutation, EGFR gene high amplification and +7/-10 copy number changes are present.13,51
Diffuse Glioma H3.3 G34 mutant was defined as follows:
A diffuse IDH-wild type glioma of the cerebral hemispheres with a missense mutation exchanging glycine for arginine or valine at position 34 of the mature histone H3.3 proteins.
This tumor mainly occurs in children and young adults. Histologically, it shows features of a diffusely infiltrating astrocytic neoplasm with anaplasia, mitoses, microvascular proliferation, and/or necrosis. The H3.3 protein is encoded by two replication-independent genes, H3F3A at 1q 42.12 and H3F3B at 17q 25.1. In addition to DNA sequencing, surrogate immunohistochemical markers of H3.3 G34 mutant proteins that are H3.3 G34R or H3.3 G34V are available and provide an alternative diagnostic method. Loss of ATRX nuclear expression and P53 overexpression is seen in all cases, and lack of OLIG2 expression is present in up to 90% of cases. These tumors correspond to WHO grade 4. The prognosis is slightly better than glioblastoma, IDH-wildtype but much worse than Astrocytoma, IDH-mutant, WHO grade 4.25,52,53
High-grade astrocytoma with piloid features was defined as follows:
An astrocytoma displaying histological features of anaplasia alongside a piloid cytology and frequent MAPK pathway gene alteration (most often affecting NF1 followed by BRAF and FGFR1) combined with homozygous deletion of CDKN 2A/B and loss of nuclear ATRX expression (indicative of mutation) as well as a distinct DNA methylation pattern.
It is mostly located in the cerebellum and has an unfavorable prognosis which however is better compared to IDH-wildtype glioblastoma.54,55
“Astroblastoma, MN1 altered” was defined as follows: “A circumscribed glial neoplasm with MN1 alteration (usually a fusion between MN1 and BEND2) and usually with cuboidal or columnar cells with variable pseudopapillary or perivascular growth, perivascular anuclear zones and vascular and pericellular hyalinization and focal IHC expression of EMA and podoplanin”. These tumors arise most commonly in the cerebral hemispheres of young patients. While EMA and podoplanin positivity is seen in all cases, expression of GFAP, S100 protein, and OLIG 2 is variable.56 It is recommended that tumors with classic astroblastoma histology which do not show MN1 alterations on testing should be designated “Astroblastoma, NEC”, while tumors with astroblastoma histology in which testing for MN1 was not or could not be performed be designated as “Astroblastoma, NOS”.
Definitions were proposed and characteristic features were described for other new, rare tumor types including Diffuse Leptomeningeal glioneuronal tumor, Myxoid glioneuronal tumor, PLNTY, and CNS tumor with BCOR internal tandem duplication (CNS-BCOR ITD).
Definitions were proposed, characteristic features were described, and additional comments were given for specific ependymoma subtypes.57–63 Supratentorial ependymoma, YAP1-MAMLD1 fusion-positive was defined as follows: “an ependymoma that arises in the supratentorial compartment and has fusion between YAP1 and MAMLD1”. These tumors mostly occur in young children, are common in females, often present as large intra/paraventricular lesions with prominent cyst formation and comprise approximately 4% of supratentorial ependymomas. They have a characteristic methylation profile. Posterior fossa ependymoma, pediatric type/PFA was defined as follows: “a posterior fossa ependymoma that has a PFA DNA methylation profile and/or immunohistochemical loss of nuclear H3 K27me3 expression”. Posterior fossa ependymoma, adult-type/PFB was defined as follows: “a posterior fossa ependymoma that has a PFB DNA methylation profile and/or immunohistochemical retention of nuclear H3 K27me3 expression”.
The presence of chromosome 1 gain was recognized as an adverse prognostic factor across all PF ependymomas. Some PF ependymomas, pediatric type/PFA even without the presence of chromosome 1 gain, have a relatively poor prognosis.
Spinal Ependymoma, MYCN amplified was defined as follows “an ependymoma in the spinal cord with MYCN amplification”. Most of these tumors occur in the thoracic or cervical cord and have anaplastic histologic features and a distinct DNA methylation profile. These tumors frequently disseminate and have a poor prognosis. Definitive classification requires the demonstration of MYCN gene amplification.
It was noted that among ependymomas, overlapping histopathological features can be associated with markedly different clinical behaviors stemming in part from the biologic heterogeneity demonstrated by ependymomas across the principal anatomic compartments of the CNS, which in turn results from their distinct genetic and epigenetic signatures. It was proposed that site be incorporated into ependymoma nomenclature and ependymomas be classified as supratentorial, posterior fossa, and spinal ependymoma. Supratentorial ependymomas include supratentorial ependymoma RELA fusion-positive and YAP1 fusion-positive. Posterior fossa (PF) ependymomas include posterior fossa ependymoma pediatric type/PFA, and posterior fossa ependymoma adult-type/PFB. Since subependymomas occur in all three compartments and have a similar clinical course, they are included in all three compartments.
It was recommended that the names of tumors be kept as simple as possible and site, age, or genetic signatures be included in the name only if there was clear clinical utility.
It was stressed that methylome profiling could provide powerful information for the classification and diagnosis of CNS tumors and can reliably identify the same. Relatively user-friendly applications are now available which only require the user to upload data with automated analysis and results that are simple to interpret.
Recommendations proposed by Update 5 that Roman numerals be replaced by Arabic numerals (1, 2, 3, 4) for all WHO CNS tumor grades were endorsed.
The main features of Update 6 are summarized below:
- The recommendations of a working group meeting held in 2019 in Utrecht, the Netherlands in preparation for the new WHO classification of CNS tumors were summarized.
- Recommendations on a number of emerging new entities, their nomenclature, and diagnostic principles for the new WHO classification were made.
- The utility of DNA methylation profiling in the classification and diagnosis of CNS tumors was acknowledged while recognizing that adoption of such advanced technologies will require adaptation of clinical practice and the pace of such adaptation will vary in different areas of the world according to the resources available. The generous use of NOS designation for such new entities in the transition period in countries and centers with fewer resources was recommended.
- Definitions were proposed and described characteristic features were described for a number of new entities, many of which can be accurately diagnosed by their specific molecular features.
- The recommendation of update 5 to use the presence of homozygous deletion of CDKN2A/B as a marker for grade 4 even in the absence of histologic criteria for grade 4 in IDH-mutant diffuse astrocytomas was approved. It was proposed that IDH-mutant astrocytomas which are grade IV by histology and/or molecular criteria be called “astrocytoma, IDH mutant, grade 4” rather than “Glioblastoma, IDH mutant” in order to distinguish them from the much more frequent and even more aggressive IDH-wild-type glioblastomas.
cIMPACT-NOW update 7: Advances in the understanding of the biological basis and molecular characteristics of ependymal tumors led cIMPACT-NOW to recommend a new classification. Ependymal tumors constitute a heterogeneous category. A poor association has been found between the grading of these tumors and the clinical outcome. Recent studies using either DNA methylation profiling to demonstrate the molecular groups of ependymomas or genome-wide sequencing to determine the genomic landscape of these tumors supported the principle that ependymomas with similar histological features from across the neuraxis have distinct origins and oncogenic events of clinicopathological significance and potential therapeutic utility. Separation of ependymal tumors by anatomic site was an important principle of the new classification proposed by Update 7 which was prompted by methylome profiling data which indicated that molecular groups of ependymal tumors in the posterior fossa and supratentorial and spinal compartments were distinct.64
Methylation and gene expression profiling studies provided evidence for at least nine molecular groups of ependymomas across the three principle anatomic compartments of the CNS. Each of the nine molecular groups was found to have distinct molecular alterations and clinicopathological characteristics and identifying them as distinct entities was found to have clinical utility.
One molecular group in each of the three compartments is made up of tumors with the histologic features of subependymoma. The supratentorial (ST) compartment is dominated by ependymomas with C11 or f-95-RELA fusion genes. The third group in the supratentorial compartment is that of ependymomas with a high frequency of YAP1-MAMLD1 fusions. Methylation profiling divides posterior fossa (PF) ependymomas into 2 main groups PFA and PFB. PFA tumors have low levels of the epigenetic marker histone H3K27 trimethylation, while high levels are found in PFB ependymomas. PFA ependymomas occur mainly in infants, and most have anaplastic features and have a significantly worse prognosis than PFB ependymomas which occur mainly in older children and adults who do not usually show features of anaplasia and have a better prognosis.58–61,65–68 Among spinal cord (SC) ependymomas, myxopapillary ependymomas predominate in adults forming one methylation group. The other major group in this compartment is the classic ependymoma, while subependymoma constitutes a rare third group. Recently, an aggressive spinal cord ependymoma was recognized. It typically shows an anaplastic morphology, is characterized by MYCN amplifications, and disseminates early throughout the neuraxis.69
It was recommended that subependymomas continue to be diagnosed based on histologic criteria. No clinically significant molecular groups have been identified in any of the three CNS compartments.
It was recommended that spinal cord myxopapillary ependymomas be diagnosed based on histologic criteria but noted that they have a clinical outcome similar to classic ependymoma and should be assigned WHO grade 2 rather than grade 1.
It was proposed that histologic subtypes of classic ependymoma (papillary, clear cell, tanycytic) be recognized as distinctive patterns in the histopathological description but should no longer be included in the classification of ependymomas as they have no clinical utility.
It was proposed that when molecular testing is unavailable, ependymomas be diagnosed as NOS. If testing was performed but was unable to classify these tumors into any of the distinct molecular types, the term NEC should be used.
The distinction between classic and anaplastic ependymoma was dropped. Anaplastic ependymoma will not be listed in the classification. Grade 2 or 3 was allowed to be assigned to ependymomas not defined by molecular status. The new Ependymoma Classification recommended by Update 7 is shown in Table 3.
It was proposed the consideration of DNA methylation profiling as a frontline diagnostic test when ependymoma is included in the differential diagnosis of a CNS tumor. The inclusion of gain of chromosome 1q (seen in 15 to 20% of PF ependymomas) as a molecular marker in the classification of PF ependymomas was recommended as it has been reliably shown to be associated with a poor prognosis. It recommended an integrated and tiered approach to report the diagnosis of ependymomas for incorporating molecular characteristics and histopathological features in the final diagnosis was recommended.13,64
The main features of Update 7 are summarized below:
● Update 7 used DNA methylation profiling to advance the classification of ependymomas.
● A classification of ependymal tumors based on the principle of defining CNS tumors by characteristic molecular alterations was proposed.
● An integrated, layered approach to the diagnosis of ependymal tumors combining the histopathological features and the molecular characteristics was recommended.
● It was recommended that ependymal tumors be classified by both anatomic site and molecular features thus allowing improved assessment of prognostic and predictive information.
● Based on these principles, ependymomas were classified into supratentorial, posterior fossa, and spinal ependymomas along with the characteristic molecular signatures.
● A new entity “ependymoma of the spinal cord with MYCN amplifications” was recognized which is associated with poor prognosis.
● It was recommended that ependymomas be graded 2 or 3 across main groups.
● It was recommended that myxopapillary ependymoma be graded as grade 2 rather than grade 1.
● It was recommended that papillary, clear cell and tanycytic variants be only described in histologic features and not be listed as specific entities in the classification owing to the absence of any clinical utility.
 |
Table 3 Ependymoma Classification Recommended by Update 7 |
Data Sets and Templates for integrated reporting of CNS tumors: Following the publication of 2016 WHO CNS, an expert ICCR (International Collaboration on Cancer Registry) designated DAC (Data Set Authorizing Committee) designed and developed standard templates for diagnostic pathology reporting of CNS tumors. “Core” elements defined as those that were unanimously agreed to be mandatory for diagnosis, clinical management and prognostication, and “non-core” elements defined as clinically important and recommended for good practice which should ideally be included in the report, but which may not yet be validated or regularly used in patient management were identified. The data sets were kept deliberately flexible so that these can be used for histologic-molecular or histology only reporting of CNS tumors (when molecular testing is not available or not required). The “Notes (Commentaries)” appended with the data sets provide clarity and extensive practical guidance to pathologists. It was hoped that the consistent use of these easy-to-use, highly reproducible templates will allow pathologists to report CNS tumors in consort with the 2016 WHO CNS and will prove useful for patient care, clinical trials, epidemiologic studies, and monitoring of neuro-oncologic care around the world.70
Molecular Workup of CNS Tumors
Molecular testing has become an important tool in the workup of CNS tumors in order to provide accurate diagnosis and appropriate prognostic and predictive information. A good neuropathology department now needs to be equipped with a modern molecular diagnostic laboratory that can identify a wide array of important single nucleotide variants (SNVs), insertions and deletions, fusions, copy number variants (CNVs), DNA methylation changes, etc. Tumor location, age of the patient and histological diagnosis are important features in selecting appropriate tests such as FISH, PCR, DNA and RNA sequencing, etc. Next-generation sequencing (NGS) is becoming widely accepted as a cost-effective and reliable method for evaluating multiple genes simultaneously. A combination of next-generation sequencing (NGS) panel and copy number array appears to be ideal for diffuse gliomas and embryonal tumors. For tumors in which histologic diagnosis is ambiguous, genomic array-based DNA methylome profiling is getting popular and is revolutionizing how primary CNS tumors as well as tumors metastasizing to the CNS are being diagnosed.71–73
Surrogate IHC markers (low cost, only one tissue section needed per marker, quick turnaround time), fluorescent in-situ hybridization (FISH) – still the most widely used tool for assessing single copy number variants and fusions – are especially well suited when the biopsy material is very small or when only a small portion of the tissue contains tumor or when searching for specific fusions or copy number gains such as EGFR amplification or when there is intratumoral heterogeneity. Next-generation sequencing (NGS) is cost-effective and can perform rapid sequencing of DNA and/or RNA efficiently on formalin-fixed paraffin-embedded tissue and allows evaluation of the majority of the most common molecular alterations including fusions, SNVs, CNVs, and small indels (insertions and deletions) simultaneously.
In some settings, IHC and FISH can first be used for screening molecular alterations and definitive methods can be used subsequently for confirmation. In the near future, genome-wide profiling will probably become more widely used. It is critical that pathologists and clinicians recognize the diversity of nuclear testing available, know which tests to order and how to use molecular data for optimal care of patients.1,24,35,58,67,74–83 Examples of application of molecular diagnostics in some common adult and pediatric CNS tumors are shown in Table 4.
 |
Table 4 Some Examples of the Application of Molecular Diagnostics in More Common Adult and Pediatric CNS Tumors |
DNA Methylation Profiling
DNA methylation profiling assesses methylation at hundreds or thousands of CPG loci across the entire genome. CNS tumors with different molecular signatures or characteristics possess different methylation “fingerprints” allowing accurate diagnosis even between tumors with similar histologic appearances. Metastatic tumors retain the methylation “fingerprint” of their tissue of origin.
Molecular profiling can reliably subtype medulloblastomas (for example, group 3 versus group 4) and ependymomas (PFA versus PFB) and is superior to histologic WHO grade for prognostic stratification of meningiomas. It has proven essential for the discovery of new CNS tumor entities such as CNS embryonal tumors with alterations of BCOR, FOXR2, CIC or MN1, etc. Its essential role in CNS tumor diagnosis and prognosis has been recognized by the various cIMPACT updates which recommend its increased and widespread use in the near future.
The cancer methylome combines characteristics reflecting the cells from which the tumor arises as well as somatically acquired changes in the cells. Thus, unknown primary sites of highly undifferentiated metastatic cancers can be traced. As discussed above, DNA methylation profiling has been shown to be highly robust and reproducible even on poor quality sections and small-sized samples.
The most important work in DNA methylation-based classification of CNS tumors was done by Capper et al.74 They published a comprehensive CNS tumor reference cohort (minimum eight cases per group) representing all WHO defined CNS tumor entities. In addition, the authors profiled diffuse large B cell lymphoma, melanoma, mesenchymal tumors, pituitary adenomas and plasma cell neoplasms which occur in the CNS. All these were analyzed by unsupervised clustering within each separate entity as well as across histologically similar tumor entities. Based on this process, 82 CNS tumor classes characterized by distinct DNA methylation profiles were designated.
For evaluation of clinical utility, a series of 1155 (adult 71% and pediatric 29%) CNS tumors comprising 64 specific entities were prospectively analyzed along with the histopathological workup. Histopathological evaluation was performed blinded to DNA methylation profiling results and included standard molecular testing. In 88% of cases, the histopathology matched to an established DNA methylation class with a calibrated classifier score >0.9 and results obtained by pathology and DNA methylation profiling were concordant. In most of the concordant cases, it was possible to assign an unambiguous molecular subgroup (eg, in medulloblastoma and ependymoma) which would not have been possible on histological evaluation alone. Subsequently, Capper et al published another paper reporting finding from multiple institutions.84
A study by Jaunmuktane et al85 shared their experience of combining a conventional molecular diagnostic approach with DNA methylation-based classification in adult brain tumors at their own institutions as well as cases referred from other centers. The diagnosis was changed in 25% cases, refined in approximately 50% and confirmed in 25%. In a proportion of cases, where there was a change in diagnosis, there was also a significant impact on treatment and clinical management, while in others, the DNA methylation profiling and integrated diagnosis prevented unnecessary and potentially harmful treatment. The authors proposed a diagnostic algorithm for routine use that integrated histology, conventional molecular tests and methylation arrays.
A significant proportion of pediatric CNS tumors undergo methylation-based classification for risk stratification (for example, medulloblastoma and ependymoma), but this has been done only in a minority of adult CNS tumors, for which a combination of histopathology and conventional molecular testing (IHC, targeted sequencing, copy number assays) remains the first-line diagnostic approach and is adequate for the majority of adult gliomas. In adults, DNA methylation-based profiling is most commonly used to obtain a more accurate and clinically relevant diagnosis for tumors with non-specific or non-representative or unusual histology or when conventional molecular testing does not yield diagnostically informative results.74,86
Gliomas with DNA MMR Defects (MSI Instability) Leading to Hypermutated Genomes
DNA mismatched repair (MMR) is crucial for the integrity of the genome. The primary function of the MMR pathway is to identify and correct base-base mismatches and insertions/deletions which occur during DNA replication and recombination. MMR deficiency results in instability (gains or losses) in microsatellite regions of the genome. Immunohistochemistry is used to determine the expression of the four MMR proteins ie MSH2, MSH6, MLH1, and PMS2. Loss of MLH1/PMS2 and/or MSH2/MSH6 expression are the most common patterns seen in tumors with MMR deficiency. Next-generation sequencing (NGS), however, is the gold standard for the detection of DNA MMR defects.
A subset of gliomas also show DNA mismatch repair defects leading to hypermutated genomes. When these gliomas are challenged with temozolomide (TMZ), recurrent subclones with inactivating mutations often emerge in genes encoding one or more DNA MMR enzymes. This inactivation causes failure of MMR mechanisms which are essential for inducing programmed cell death in glioma cells damaged by TMZ and leads to TMZ resistance in these tumors. While such tumors are resistant to alkylating chemotherapies like TMZ, they may be more responsive to immunotherapies as they express more mutant neoantigens on their cell surfaces. MMR IHC is very useful as a viable frontline screening test for gliomas in which immunotherapy is being considered.87,88
It appears that when MSH6 is inactivated in high-grade gliomas, alkylating agents convert from induction of tumor cell death to promotion of neoplastic progression. In a study by Johnson et al, 6% of pediatric high-grade gliomas were hypermutated harboring deleterious mutations. Tumor Mutational Burden (TMB) can be a biomarker to identify pediatric glioblastoma patients who may benefit from immunotherapy.89 A study by Hodges et al concluded that only small subsets of glioma patients are likely to benefit from monotherapy immune checkpoint inhibition.90 A recent randomized, multi-institutional clinical trial to evaluate the immune response and survival following neoadjuvant therapy with pembrolizumab in 35 patients with recurrent, surgically resectable glioblastomas showed that neoadjuvant administration of PD-1 blockade enhanced the local and systemic anti-tumor response and may represent an effective approach to the treatment of glioblastomas.91
Increased Tumor Mutational Load (TML) has been shown to correlate with higher levels of neoantigens and is indicative of the potential to induce a durable response to immunotherapy. Following treatment with TMZ, a subset of gliomas recurs with increased TML. These hypermutated recurrent gliomas represent a subtype of recurrence with unique molecular vulnerabilities.92–94 A study by Thuijl et al demonstrated that gliomas with MSI instability and MGMT methylation can lead to resistance to TMZ.95
WHO CNS Fifth Edition (2021 WHO CNS 5)
As the 2021 WHO CNS5 came out recently, we believe that our review on advancements in CNS tumors will hopefully be of benefit to pathologists and clinicians in understanding many of the crucial changes and new additions in the new WHO CNS classification.96–98
2021 WHO CNS 5 has incorporated most of the recommendations included in the seven cIMPACT-NOW Updates and groups tumors into more biologically and molecularly defined entities, thus building on the 2016 WHO CNS. For CNS tumor nomenclature, it follows the recommendations of the 2019 cIMPACT-NOW Utrecht meeting (Update 6). Several new entities have been added based on cIMPACT-NOW recommendation. Key diagnostic genes, molecular pathways, and/or combinations in common primary CNS tumors have been specified. Arabic numerals are now used for grading, and grades of some tumors have been modified (eg, myxopapillary ependymoma). Names of some tumors have been simplified (HPC/SFT has been renamed simply as a solitary fibrous tumor with changes to grading within tumor types, modifier terms like “anaplastic” have been excluded). Common names like “anaplastic astrocytoma” and “anaplastic oligodendroglioma” have been excluded. Diffuse midline glioma is now designated as “H3 K27-altered” rather than “H3 K27M mutant” as the pathogenic pathway in these tumors can be altered various ways. Astroblastoma has been specified as “MN1-altered” to improve diagnostic focus for this tumor entity. There is an emphasis on layered reporting, and the use of the terms NOS and NEC has been clarified (in line with cIMPACT recommendations). The aim is to achieve better prognostication and optimal therapy for specific CNS tumors. These changes will also allow more homogeneous patient populations to be enrolled in clinical trials for the development of novel therapies.
The most significant changes involve the differentiation of adult gliomas from pediatric gliomas. The separation of gliomas into an adult and pediatric types based on their well-established molecular genetic differences will help clinicians in prognostication and therapeutics. The greatest practical implications stem from the changes in the classification of glioblastoma in adults. The separation of astrocytomas into IDH-wild type and IDH-mutated is important. The identification of 10% astrocytomas with non-canonical IDH mutations which are not detected by IDHR132 IHC as well as those astrocytomas with the molecular features of glioblastoma will require access to adequate molecular testing. 2021 WHO CNS5 further separates pediatric low-grade gliomas from pediatric high-grade gliomas which have different molecular signatures affecting prognosis and resulting in different molecularly targeted approaches for individual tumor entities. Ultra-high risk posterior fossa ependymoma (PFA) which is characterized by loss of chromosome 6q is recognized.
The layered reporting incorporating histologic type and molecular details of medulloblastomas is emphasized as crucial for a better conceptualization of these tumors. Tremendous challenges remain in developing optimal molecular-based therapies for non-medulloblastomatous embryonal tumors.
The 2021 WHO CNS5 remains a work in progress. The correct balance needs to be found between developing molecularly targeted therapies based on the new classification and avoiding the potentially detrimental effect of adding ineffective molecular targeted therapies based on an incomplete biological understanding of specific tumors. It remains a great challenge, especially in pediatric tumors.97–101
Conclusions
The cIMPACT-NOW Updates effectively laid the foundation for the 2021 WHO CNS5. Most of the proposed recommendations, including the use of Arabic numerals, grading within tumor types, newly recognized tumor entities, and revised nomenclature, have been incorporated in 2021 WHO CNS5. Similarly, key diagnostic genes, molecules, pathways, and/or combinations in major primary CNS tumors have been incorporated. We have discussed the seven Updates, the key molecular diagnostics, and the importance of DNA methylation profiling in CNS tumors in detail. We also discussed the integrated reporting of CNS tumors. We analyzed in detail specific recommendations made in the Updates especially the division of gliomas into adult diffuse gliomas; pediatric type diffuse high-grade gliomas (which are expected to behave aggressively); pediatric type diffuse low-grade gliomas (which are expected to have a good prognosis); circumscribed astrocytic gliomas; glioneuronal and neuronal tumors and ependymomas (now classified by site as well as histological and molecular features). We also discussed hypermutated gliomas and the importance of identifying such tumors, especially for therapeutic purposes. Finally, we presented an overview of salient features of 2021 WHO CNS5. CNS tumor classification is still imperfect. However, 2021 WHO CNS5 has attempted to include newly recognized entities, phase out obsolete tumor types and adjust the taxonomic structure. It incorporates numerous molecular changes with a clinicopathologic utility which have been shown to be important for more accurate classification of CNS tumors with the ultimate aim of benefiting patients. We hope that our comprehensive and detailed review of recent relevant advances in CNS tumors will help readers in understanding the new WHO Classification of CNS tumors.
Abbreviations
WHO, World Health Organization; CNS, central nervous system; ISN, International Society of Neuropathology; cIMPACT-NOW, Consortium to inform molecular and practical approaches to CNS tumor taxonomy not official WHO; ICCR, International Collaboration on Cancer Reporting; MSI, microsatellite instability; NOS, not otherwise specified; NEC, not elsewhere classified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; AT/RT, atypical teratoid/rhabdoid tumor; IHC, immunohistochemistry; EGFR, epidermal growth factor receptor; CNV, copy number variation; IARC, International Agency for Research on Cancer; DNT, dysembryoplastic neuroepithelial tumor; PLNTY, polymorphous low grade neuroepithelial tumor of the young; MVNT, multinodular and vacuolating neuronal tumor; WC2, Working Committee 2; SC, spinal cord; FISH, fluorescent in-situ hybridization; NGS, next-generation sequencing; AI, artificial intelligence; DAC, data set authorizing committee; MMR, mismatched repair; TMZ, temozolomide; TMB, tumor mutation burden; SNV, single nucleotide variants; CNV, copy number variants.
Data Sharing Statement
Data and materials of this work are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This is a review article, and no procedures were performed.
Funding
No financial support was provided for this study.
Disclosure
It is declared that all authors have no conflict of interest.
References
1. Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System. Revised 4th Edition. Lyon: International Agency for Research on Cancer (IARC); 2016.
2. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
3. Sonoda Y, Yokoo H, Tanaka S, Kinoshita M, Nakada M, Nishihara H. Practical procedures for the integrated diagnosis of astrocytic and oligodendroglial tumors. Brain Tumor Pathol. 2019;36(2):56–62. doi:10.1007/s10014-019-00337-y
4. Onizuka H, Masui K, Komori T. Diffuse gliomas to date and beyond 2016 WHO classification of tumours of the central nervous system. Int J Clin Oncol. 2020;25(6):997–1003. doi:10.1007/s10147-020-01695-w
5. Komori T. The anticipated revision of the grading criteria for adult isocitrate dehydrogenase-mutant diffuse glioma within the neuro-oncology community. Neuro Oncol. 2019;21(12):1485–1486. doi:10.1093/neuonc/noz186
6. Kondo T. Molecular mechanisms involved in gliomagenesis. Brain Tumor Pathol. 2017;34(1):1–7. doi:10.1007/s10014-017-0278-8
7. Louis DN, Aldape K, Brat DJ, et al. cIMPACT-NOW (the consortium to inform molecular and practical approaches to CNS tumor taxonomy): a new initiative in advancing nervous system tumor classification. Brain Pathol. 2017;27(6):851–852. doi:10.1111/bpa.12457
8. Louis DN, Aldape K, Brat DJ, et al. Announcing cIMPACT-NOW: the consortium to inform molecular and practical approaches to CNS tumor taxonomy. Acta Neuropathol. 2017;133(1):1–3. doi:10.1007/s00401-016-1646-x
9. Yeaney GA, Brat DJ. What every neuropathologist needs to know: update on cIMPACT-NOW. J Neuropathol Exp Neurol. 2019;78(4):294–296. doi:10.1093/jnen/nlz012
10. Kristensen BW, Priesterbach-Ackley LP, Petersen JK, Wesseling P. Molecular pathology of tumors of the central nervous system. Ann Oncol. 2019;30(8):1265–1278. doi:10.1093/annonc/mdz164
11. Louis DN, Wesseling P, Paulus W, et al. cIMPACT-NOW update 1: Not Otherwise Specified (NOS) and Not Elsewhere Classified (NEC). Acta Neuropathol. 2018;135(3):481–484. doi:10.1007/s00401-018-1808-0
12. Louis DN, Ellison DW, Brat DJ, et al. cIMPACT-NOW: a practical summary of diagnostic points from round 1 updates. Brain Pathol. 2019;29(4):469–472. doi:10.1111/bpa.12732
13. Castro LNG, Wesseling P. The cIMPACT-NOW updates and their significance to current neuro-oncology practice. Neurooncol Pract. 2020;8(1):4–10. doi:10.1093/nop/npaa055
14. Louis DN, Perry A, Burger P, et al. International society of neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014;24(5):429–435. doi:10.1111/bpa.12171
15. Louis DN, Giannini C, Capper D, et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 2018;135(4):639–642. doi:10.1007/s00401-018-1826-y
16. Gessi M, Capper D, Sahm F, et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol. 2016;132(4):635–637. doi:10.1007/s00401-016-1608-3
17. Hochart A, Escande F, Rocourt N, et al. Long survival in a child with a mutated K27M-H3.3 pilocytic astrocytoma. Ann Clin Transl Neurol. 2015;2(4):439–443. doi:10.1002/acn3.184
18. Kleinschmidt-DeMasters BK, Donson A, Foreman NK, Dorris K. H3 K27M mutation in gangliogliomas can be associated with poor prognosis. Brain Pathol. 2017;27(6):846–850. doi:10.1111/bpa.12455
19. Morita S, Nitta M, Muragaki Y, et al. Brainstem pilocytic astrocytoma with H3 K27M mutation: case report. J Neurosurg. 2018;129(3):593–597. doi:10.3171/2017.4.JNS162443
20. Orillac C, Thomas C, Dastagirzada Y, et al. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun. 2016;4(1):84. doi:10.1186/s40478-016-0361-0
21. Pagès M, Beccaria K, Boddaert N, et al. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018;28(1):103–111. doi:10.1111/bpa.12473
22. Pratt D, Natarajan SK, Banda A, et al. Circumscribed/non-diffuse histology confers a better prognosis in H3K27M-mutant gliomas. Acta Neuropathol. 2018;135(2):299–301. doi:10.1007/s00401-018-1805-3
23. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–612.
24. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018;136(5):805–810. doi:10.1007/s00401-018-1913-0
25. Korshunov A, Capper D, Reuss D, et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016;131(1):137–146. doi:10.1007/s00401-015-1493-1
26. Ceccarelli M, Barthel FP, Malta TM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. doi:10.1016/j.cell.2015.12.028
27. Stichel D, Ebrahimi A, Reuss D, et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018;136(5):793–803. doi:10.1007/s00401-018-1905-0
28. Wijnenga MMJ, Dubbink HJ, French PJ, et al. Molecular and clinical heterogeneity of adult diffuse low-grade IDH wild-type gliomas: assessment of TERT promoter mutation and chromosome 7 and 10 copy number status allows superior prognostic stratification. Acta Neuropathol. 2017;134(6):957–959. doi:10.1007/s00401-017-1781-z
29. Tesileanu CMS, Dirven L, Wijnenga MMJ, et al. Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: a confirmation of the cIMPACT-NOW criteria. Neuro Oncol. 2020;22(4):515–523. doi:10.1093/neuonc/noz200
30. Ellison DW, Hawkins C, Jones DTW, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 2019;137(4):683–687. doi:10.1007/s00401-019-01987-0
31. Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33(9):1015–1022. doi:10.1200/JCO.2014.58.3922
32. Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131(6):833–845. doi:10.1007/s00401-016-1539-z
33. Smits A, Jakola AS. Clinical presentation, natural history, and prognosis of diffuse low-grade gliomas. Neurosurg Clin N Am. 2019;30(1):35–42. doi:10.1016/j.nec.2018.08.002
34. Brown NF, Carter T, Kitchen N, Mulholland P. Dabrafenib and trametinib in BRAFV600E mutated glioma. CNS Oncol. 2017;6(4):291–296. doi:10.2217/cns-2017-0006
35. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139(3):603–608. doi:10.1007/s00401-020-02127-9
36. Cimino PJ, Holland EC. Targeted copy number analysis outperforms histologic grading in predicting patient survival for WHO grades II/III IDH-mutant astrocytomas. Neuro Oncol. 2019;21(6):819–821. doi:10.1093/neuonc/noz052
37. Yoda RA, Marxen T, Longo L, et al. Mitotic index thresholds do not predict clinical outcome for IDH-mutant astrocytoma. J Neuropathol Exp Neurol. 2019;78(11):1002–1010. doi:10.1093/jnen/nlz082
38. Olar A, Wani KM, Alfaro-Munoz KD, et al. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol. 2015;129(4):585–596. doi:10.1007/s00401-015-1398-z
39. Shirahata M, Ono T, Stichel D, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136(1):153–166. doi:10.1007/s00401-018-1849-4
40. Duregon E, Bertero L, Pittaro A, et al. Ki-67 proliferation index but not mitotic thresholds integrates the molecular prognostic stratification of lower grade gliomas. Oncotarget. 2016;7(16):21190–21198. doi:10.18632/oncotarget.8498
41. Reuss DE, Mamatjan Y, Schrimpf D, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 2015;129(6):867–873. doi:10.1007/s00401-015-1438-8
42. Aoki K, Nakamura H, Suzuki H, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018;20(1):66–77. doi:10.1093/neuonc/nox132
43. Korshunov A, Casalini B, Chavez L, et al. Integrated molecular characterization of IDH-mutant glioblastomas. Neuropathol Appl Neurobiol. 2019;45(2):108–118. doi:10.1111/nan.12523
44. Reis GF, Pekmezci M, Hansen HM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization grades II-III) astrocytomas. J Neuropathol Exp Neurol. 2015;74(5):442–452. doi:10.1097/NEN.0000000000000188
45. Weller M, Weber RG, Willscher E, et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015;129(5):679–693. doi:10.1007/s00401-015-1409-0
46. Yang RR, Shi ZF, Zhang ZY, et al. IDH mutant lower grade (WHO grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. 2020;30(3):541–553. doi:10.1111/bpa.12801
47. Appay R, Dehais C, Maurage CA, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019;21(12):1519–1528. doi:10.1093/neuonc/noz126.000
48. Phillips JJ, Aranda D, Ellison DW, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 2013;23(5):565–573. doi:10.1111/bpa.12043
49. Richardson TE, Sathe AA, Kanchwala M, et al. Genetic and epigenetic features of rapidly progressing IDH-mutant astrocytomas. J Neuropathol Exp Neurol. 2018;77(7):542–548. doi:10.1093/jnen/nly026
50. Li KK, Shi ZF, Malta TM, et al. Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neuro Oncol Adv. 2019;1(1):vdz015. doi:10.1093/noajnl/vdz015
51. Louis DN, Wesseling P, Aldape K, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30(4):844–856. doi:10.1111/bpa.12832
52. Haque F, Varlet P, Puntonet J, et al. Evaluation of a novel antibody to define histone 3.3 G34R mutant brain tumours. Acta Neuropathol Commun. 2017;5:45. doi:10.1186/s40478-017-0449-1
53. Yoshimoto K, Hatae R, Sangatsuda Y, et al. Prevalence and clinicopathological features of H3.3 G34-mutant high-grade gliomas: a retrospective study of 411 consecutive glioma cases in a single institution. Brain Tumor Pathol. 2017;34(3):103–112. doi:10.1007/s10014-017-0287-7
54. Reinhardt A, Stichel D, Schrimpf D, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 2018;136(2):273–291. doi:10.1007/s00401-018-1837-8
55. Reinhardt A, Stichel D, Schrimpf D, et al. Tumors diagnosed as cerebellar glioblastoma comprise distinct molecular entities. Acta Neuropathol Commun. 2019;7(1):163. doi:10.1186/s40478-019-0801-8
56. Lehman NL, Usubalieva A, Lin T, et al. Genomic analysis demonstrates that histologically defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun. 2019;7(1):42. doi:10.1186/s40478-019-0689-3
57. Godfraind C, Kaczmarska JM, Kocak M, et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;124(2):247–257.
58. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. doi:10.1016/j.ccell.2015.04.002
59. Pajtler KW, Mack SC, Ramaswamy V, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 2017;133(1):5–12. doi:10.1007/s00401-016-1643-0
60. Pajtler KW, Wen J, Sill M, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136(2):211–226. doi:10.1007/s00401-018-1877-0
61. Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. doi:10.1016/j.ccr.2011.07.007
62. Witt H, Gramatzki D, Hentschel B, et al. DNA methylation-based classification of ependymomas in adulthood: implications for diagnosis and treatment. Neuro Oncol. 2018;20(12):1616–1624. doi:10.1093/neuonc/noy118
63. Upadhyaya SA, Robinson GW, Onar-Thomas A, et al. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol. 2019;21(10):1319–1330. doi:10.1093/neuonc/noz069
64. Ellison DW, Aldape KD, Capper D, et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020;30(5):863–866. doi:10.1111/bpa.12866
65. Ellison DW, Kocak M, Figarella-Branger D, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed. 2011;10:7. doi:10.1186/1477-5751-10-7
66. Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. doi:10.1038/nature13108
67. Panwalkar P, Clark J, Ramaswamy V, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134(5):705–714. doi:10.1007/s00401-017-1752-4
68. Cavalli FMG, Hübner JM, Sharma T, et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018;136(2):227–237. doi:10.1007/s00401-018-1888-x
69. Ghasemi DR, Sill M, Okonechnikov K, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138(6):1075–1089. doi:10.1007/s00401-019-02056-2
70. Louis DN, Wesseling P, Brandner S, et al. Data sets for the reporting of tumors of the central nervous system: recommendations from the international collaboration on cancer reporting. Arch Pathol Lab Med. 2020;144(2):196–206. doi:10.5858/arpa.2018-0565-OA
71. Rushing EJ. WHO classification of tumors of the nervous system: preview of the upcoming 5th edition. Memo. 2021;14(2):188–191. doi:10.1007/s12254-021-00680-x
72. Cooney TM, Lubanszky E, Prasad R, Hawkins C, Mueller S. Diffuse midline glioma: review of epigenetics. J Neurooncol. 2020;150(1):27–34. doi:10.1007/s11060-020-03553-1
73. Deng MY, Sill M, Sturm D, et al. Diffuse glioneuronal tumour with oligodendroglioma-like features and nuclear clusters (DGONC) - a molecularly defined glioneuronal CNS tumour class displaying recurrent monosomy 14. Neuropathol Appl Neurobiol. 2020;46(5):422–430. doi:10.1111/nan.12590
74. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. doi:10.1038/nature26000
75. Feldman AZ, Jennings LJ, Wadhwani NR, Brat DJ, Horbinski CM. The essentials of molecular testing in CNS tumors: what to order and how to integrate results. Curr Neurol Neurosci Rep. 2020;20(7):23. doi:10.1007/s11910-020-01041-7
76. Horbinski C, Ligon KL, Brastianos P, et al. The medical necessity of advanced molecular testing in the diagnosis and treatment of brain tumor patients. Neuro Oncol. 2019;21(12):1498–1508. doi:10.1093/neuonc/noz119
77. Orozco JIJ, Knijnenburg TA, Manughian-Peter AO, et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat Commun. 2018;9(1):4627. doi:10.1038/s41467-018-06715-y
78. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–848. doi:10.1007/s00401-015-1432-1
79. Brat DJ, Verhaak RG, Aldape KD, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498.
80. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):775–788. doi:10.1007/s00401-015-1410-7
81. Vaubel RA, Caron AA, Yamada S, et al. Recurrent copy number alterations in low-grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol. 2018;28(2):172–182. doi:10.1111/bpa.12495
82. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. doi:10.1007/s00401-012-0958-8
83. Kumar R, Liu APY, Northcott PA. Medulloblastoma genomics in the modern molecular era. Brain Pathol. 2020;30(3):679–690. doi:10.1111/bpa.12804
84. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136(2):181–210. doi:10.1007/s00401-018-1879-y
85. Jaunmuktane Z, Capper D, Jones DTW, et al. Methylation array profiling of adult brain tumours: diagnostic outcomes in a large, single centre. Acta Neuropathol Commun. 2019;7(1):24. doi:10.1186/s40478-019-0668-8
86. Tanboon J, Williams EA, Louis DN. The diagnostic use of immunohistochemical surrogates for signature molecular genetic alterations in gliomas. J Neuropathol Exp Neurol. 2016;75(1):4–18. doi:10.1093/jnen/nlv009
87. Salem ME, Bodor JN, Puccini A, et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int J Cancer. 2020;147(10):2948–2956. doi:10.1002/ijc.33115
88. McCord M, Steffens A, Javier R, Kam KL, McCortney K, Horbinski C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun. 2020;8(1):15. doi:10.1186/s40478-020-0892-2
89. Johnson A, Severson E, Gay L, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22(12):1478–1490. doi:10.1634/theoncologist.2017-0242
90. Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19(8):1047–1057. doi:10.1093/neuonc/nox026
91. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Randomized Controlled Trial Nat Med. 2019;25(3):477–486. doi:10.1038/s41591-018-0337-7
92. Gorsi HS, Malicki DM, Barsan V, et al. Nivolumab in the treatment of recurrent or refractory pediatric brain tumors: a single institutional experience. J Pediatr Hematol Oncol. 2019;41(4):e235–e241. doi:10.1097/MPH.0000000000001339
93. Daniel P, Sabri S, Chaddad A, et al. Temozolomide Induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol. 2019;9:41. doi:10.3389/fonc.2019.00041
94. Hunter C, Smith R, Cahill DP, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66(8):3987–3991. doi:10.1158/0008-5472.CAN-06-0127
95. van Thuijl HF, Mazor T, Johnson BE, et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015;129(4):597–607. doi:10.1007/s00401-015-1403-6
96. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–1251. doi:10.1093/neuonc/noab106
97. Wen PY, Packer RJ. The 2021 WHO classification of tumors of the central nervous system: clinical implication. Neuro Oncol. 2021;23(8):1215–1217. doi:10.1093/neuonc/noab120
98. WHO Classification of Tumours. Central Nervous System Tumours. Edited by the WHO Classification of Tumours Editorial Board.
99. Baroni LV, Sundaresan L, Heled A, et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol. 2021;23(8):1360–1370. doi:10.1093/neuonc/noab034
100. Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastoma revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer. 2020;20(1):42–56. doi:10.1038/s41568-019-0223-8
101. Fukuoka K, Mamatjan Y, Tatevossian R, et al. Clinical impact of combined epigenetic and molecular analysis of pediatric low-grade gliomas. Neuro Oncol. 2020;22(10):1474–1483. doi:10.1093/neuonc/noaa077
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.