Back to Journals » Drug Design, Development and Therapy » Volume 8
Evaluation of the biological activity of novel monocationic fluoroaryl-2,2’-bichalcophenes and their analogues
Authors Hussin WA, Ismail M, Alzahrani A, El-Sayed W ![]()
Received 18 April 2014
Accepted for publication 16 May 2014
Published 17 July 2014 Volume 2014:8 Pages 963—972
DOI https://doi.org/10.2147/DDDT.S66469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Warda A Hussin,1,2,*,† Mohamed A Ismail,1,3,* Abdullah M Alzahrani,1 Wael M El-Sayed,1,4
1King Faisal University, College of Science, Departments of Chemistry and Biological Sciences, Hofuf, Saudi Arabia; 2Al-Azhr University, Faculty of Science, Department of Botany and Microbiology, Cairo, Egypt; 3Department of Chemistry, Faculty of Science, Mansoura University, Mansoura, Egypt; 4University of Ain Shams, Faculty of Science, Department of Zoology, Abbassia, Cairo, Egypt
*These authors contributed equally to this work
†Warda A Hussin passed away on May 21, 2014
Abstract: A series of bichalcophene fluorobenzamidines 5a–e was synthesized from the corresponding mononitriles 4a–e via a direct reaction with lithium bis(trimethylsilyl)amide LiN(TMS)2 followed by de-protection with ethanolic HCl (gas). Bichalcophene fluorobenzonitriles 4a–e were prepared adopting a Stille coupling reaction between the bromo compounds 3a–c and 2-(tri-n-butylstannyl)furan or analogues. As an approach to drug discovery, the structure–antimutagenicity relationship of novel fluoroarylbichalcophenes was examined using the Ames Salmonella/microsomal assay. At nontoxic concentrations (10 and 20 µM), all derivatives alone or in combination with sodium azide (NaN3; 2 µg/plate) or benzo[a]pyrene (B[a]P; 20 µM) in the presence of S9 mix were not mutagenic. The fluoroaryl derivatives significantly reduced the NaN3-induced and B[a]P-induced mutagenicity under pre-exposure and co-exposure conditions. The recorded antimutagenic activity of fluoroaryl derivatives varied depending on the kind of mutagen and the exposure regimen. Monocationic fluoroarylbichalcophenes were superior to the corresponding mononitriles in reducing B[a]P-induced mutagenicity. Nevertheless, mononitriles were more active against NaN3, especially at low concentrations and under pre-exposure treatments. The antimutagenic activity was congruent with a high antioxidant activity that could promote the DNA repair system. The fluorine substitution changed the antimutagenic signature of bichalcophenes. Some of these compounds could be selected for further anticancer studies.
Keywords: fluoroaryl-2,2’-bichalcophenes, stille coupling, Salmonella typhimurium, sodium azide, benzo[a]pyrene, antimutagenicity
Introduction
New anticancer compounds without undesirable side effects are urgently needed. Aromatic diamidines, such as pentamidine, are used extensively against several human ailments.1,2 However, some pentamidine derivatives have been withdrawn from further human trials due to associated renal and hepatic toxicities.3 A series of furamidine derivatives was synthesized by replacing the phenyl group(s) with pyridyl group(s). Several of these aza-analogues were more active than the furamidine itself.4,5 Recently, bifuran diamidine I (Figure 1) has been shown to recognize G-quadruplex DNA.6 More recently, monocationic bifuran II was proven to be more effective against methicillin-resistant Staphylococcus aureus infection in mice than was vancomycin.7 Promising antimutagenic activity against sodium azide (NaN3) and benzo[a]pyrene (B[a]P)-induced mutagenicity was shown for many of these bichalcophenes.8 Although thiophene and furan rings are known for their broad biological activities,9 we have shown that replacing the furan ring with a thiophene ring increased the activity.10 To improve the pharmacological properties, and as an approach to drug discovery of the previously investigated compounds, we synthesized a series of fluoroarylbichalcophenes. Fluorine substitution can alter the biological and chemical properties of compounds, and this led to the development of a vast number of novel fluorinated drugs. The high electronegativity of fluorine substituent can modify the electron distribution in a molecule, which affects its absorption, distribution, and metabolism.11 The presence of a fluorine atom usually increases lipophilicity and hence biological availability.12 Through microsomal inhibition, fluorine substitution was shown to abolish quinolone mutagenicity.13
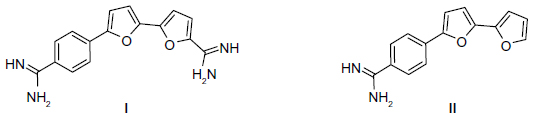 | Figure 1 Structure of some biologically important cationic bichalcophene compounds. |
Novel potential drugs are usually screened for their possible toxicity and mutagenic and antimutagenic activities in many systems, including the Salmonella/microsomal assay. There is a strong relation between mutagenicity in Salmonella and carcinogenicity in animal models.14 Given that introduction of a fluorine atom onto a molecule modifies its mutagenic potency,15 we investigated the structure–mutagenicity relationship profile of fluoroarylbichalcophenes and the effect of fluorine on their antimutagenic potency using the Ames Salmonella/microsomal assay. We also investigated the antioxidant activity of these novel compounds.
Materials and methods
Chemistry
Melting points were recorded using a Gallenkamp melting-point apparatus and were uncorrected. Thin-layer chromatography analysis was carried out on silica gel 60 F254 pre-coated aluminum sheets and detected under ultraviolet light. Infrared (IR) spectra were recorded using the potassium bromide (KBr) wafer technique on a Shimadzu 5800 Fourier transform (FT)-IR spectrometer. 1H nuclear magnetic resonance (NMR) spectra were recorded employing a Varian Mercury VX-300 spectrometer, and chemical shifts (δ) were in parts per million (ppm) relative to tetramethylsilane (TMS) as the internal standard. Mass spectra were recorded on a gas chromatography–mass spectrometry (GC-MS) (Schimadzu QP-1000 EX) spectrometer. Elemental analyses were performed at the micro-analytical laboratories of the Faculty of Science, Cairo University (Cairo, Egypt) and are within ±0.4 of the theoretical values. All chemicals and solvents were purchased from Aldrich Chemical Co (St Louis, MO, USA). N-Bromosuccinimide (NBS) was recrystallized from nitromethane prior to use. All solvents were reagent grade.
Procedure for preparation of fluoroarylchalcophene carbonitriles 2a–c
3-Fluoro-4-(furan-2-yl)benzonitrile (2a)
A mixture of 4-bromo-3-fluorobenzonitrile 1a (3.98 g, 20 mmol), 2-(tri-n-butylstannyl)furan (7.15 g, 20 mmol), and tetrakis(triphenylphosphine) palladium (300 mg) in dry dioxane (20 mL) was heated under nitrogen at reflux (90°C–100°C) for 24 hours. The solvent was evaporated under reduced pressure, the solid was dissolved in ethyl acetate, and the solution was passed through celite to remove Pd. The solution was evaporated, and the solid was collected to give 2a as an off-white solid in 83% yield, mp 73°C–73.5°C (hexanes). Rate of flow (Rf)=0.71, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3117, 3083, 3058 (CH), 2229 (CN), 1643, 1617, 1585 (C=C) cm−1. 1H NMR (CDCl3); δ 6.57–6.59 (m, 1H), 7.01–7.04 (m, 1H), 7.39–7.58 (m, 3H), 7.93 (t, J=7.8 Hz, 1H). Anal Calcd for C11H6FNO: C 70.59; H 3.23; N 7.48. Found: C 70.65; H 3.14; N 7.31.
2-Fluoro-4-(furan-2-yl)benzonitrile (2b)
A white solid in 74% yield, mp 77°C–78°C (hexanes). Rf=0.52, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3113, 3076, 3045 (CH), 2232 (CN), 1620, 1559 (C=C) cm−1. 1H NMR (CDCl3); δ 6.54–6.56 (m, 1H), 6.86 (d, J=3.3 Hz, 1H), 7.46–7.64 (m, 4H). Anal Calcd for C11H6FNO: C 70.59; H 3.23; N 7.48. Found: C 70.51; H 3.18; N 7.65.
2-Fluoro-4-(thiophen-2-yl)benzonitrile (2c)
A yellow solid in 81% yield, mp 99°C–99.5°C (hexanes). Rf=0.55, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3120, 3081 (CH), 2227 (CN), 1614, 1557 (C=C) cm−1. 1H NMR (CDCl3); δ 7.12–7.14 (m, 1H), 7.40–7.48 (m, 4H), 7.58–7.61 (m, 1H). MS (EI) m/e (rel int); 203 (M+, 100), 171 (6), 158 (15). Anal Calcd for C11H6FNS: C 65.01; H 2.98; N 6.89. Found: C 64.78; H 3.07; N 6.93.
Procedure for preparation of bromochalcophenes 3a–c
4-(5-Bromofuran-2-yl)-3-fluorobenzonitrile (3a)
To a solution of 2a (2.81 g, 15 mmol) in dimethylformamide (DMF) (15 mL), we added portion-wise NBS (2.67 g, 15 mmol) with stirring. The reaction mixture was stirred overnight at room temperature, and then poured onto cold water. The precipitate formed was collected, washed with water, and dried to give 3a as a red-pink powder in 91% yield, mp 138°C–138.5°C (EtOH). Rf=0.68, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3095, 3074, 3052 (CH), 2228 (CN), 1615, 1558 (C=C) cm−1. 1H NMR (DMSO-d6); δ 6.86 (d, J=3.0 Hz, 1H), 7.11 (d, J=3.0 Hz, 1H), 7.76 (d, J=8.1 Hz, 1H), 7.89–8.00 (m, 2H). Anal Calcd for C11H5BrFNO: C 49.65; H 1.89; N 5.26. Found: C 49.31; H 2.12; N 5.03.
4-(5-Bromofuran-2-yl)-2-fluorobenzonitrile (3b)
A pink powder in 86% yield, mp 123°C–124°C (EtOH). Rf=0.43, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3109 (CH), 2233 (CN), 1618, 1553 (C=C) cm−1. 1H NMR (CDCl3); δ 6.47 (d, J=3.0 Hz, 1H), 6.80 (d, J=3.0 Hz, 1H), 7.42–7.48 (m, 2H), 7.59–7.62 (m, 1H). MS (EI) m/e (rel int); 266 (M+, 6), 267 (M+ +1, 43), 158 (100).
4-(5-Bromothiophen-2-yl)-2-fluorobenzonitrile (3c)
A pale-yellow powder in 89% yield, mp 108°C–109°C (EtOH). Rf=0.47, petroleum ether (40°C–60°C)-EtOAc (95:5). IR (KBr) ν’ 3120, 3083 (CH), 2232 (CN), 1615, 1560 (C=C) cm−1. 1H NMR (CDCl3); δ 7.09–7.16 (m, 2H), 7.31–7.47 (m, 2H), 7.58–7.61 (m, 1H). MS (EI) m/e (rel int); 282 (M+, 15), 283 (M+ +1, 100), 158 (81).
General procedure for bichalcophene fluorobenzonitrile synthesis 4a–e
4-(2,2′-Bifuran-5-yl)-3-fluorobenzonitrile (4a)
Adopting the same procedure used for the preparation of 2a, a Stille coupling reaction was performed using bromo compound 3a to furnish bifuran fluorobenzonitrile 4a as a golden-yellow solid in 73% yield, mp 127°C–127.5°C (EtOH/petroleum ether). Rf=0.72, petroleum ether (40°C–60°C)-EtOAc (9:1). IR (KBr) ν’ 3131, 3080 (CH), 2234 (CN), 1616, 1575 (C=C) cm−1. 1H NMR (DMSO-d6); δ 6.67–6.69 (m, 1H), 6.93 (d, J=3.3 Hz, 1H), 6.98 (d, J=3.3 Hz, 1H), 7.17–7.20 (m, 1H), 7.78–7.84 (m, 2H), 7.97–8.08 (m, 2H). MS (EI) m/e (rel int); 253 (M+, 100), 254 (M+ +1, 17), 224 (15), 196 (29). Anal Calcd for C15H8FNO2: C 71.14; H 3.18; N 5.53. Found: C 70.81; H 3.35; N 5.37.
4-(2,2′-Bifuran-5-yl)-2-fluorobenzonitrile (4b)
A golden-yellow solid in 77% yield, mp 130°C–131°C (EtOH/petroleum ether). Rf=0.51, petroleum ether (40°C–60°C)-EtOAc (9:1). IR (KBr) ν’ 3128, 3090 (CH), 2229 (CN), 1616, 1579 (C=C) cm−1. 1H NMR (DMSO-d6); δ 6.66–6.68 (m, 1H), 7.45 (d, J=3.9 Hz, 1H), 6.89 (d, J=3.9 Hz, 1H), 6.97 (d, J=3.0 Hz, 1H), 7.74–7.98 (m, 4H). MS (EI) m/e (rel int); 253 (M+, 100), 254 (M+ +1, 17), 224 (19), 196 (30). Anal Calcd for C15H8FNO2: C 71.14; H 3.18; N 5.53. Found: C 71.02; H 3.25; N 5.57.
3-Fluoro-4-(5-(thiophen-2-yl)furan-2-yl)benzonitrile (4c)
A yellow solid in 68% yield, mp 124°C–125°C (EtOH/petroleum ether). Rf=0.70, petroleum ether (40°C–60°C)-EtOAc (9:1). IR (KBr) ν’ 3105, 3078 (CH), 2228 (CN), 1613, 1598 (C=C) cm−1. 1H NMR (DMSO-d6); δ 7.01 (d, J=3.9 Hz, 1H), 7.16–7.20 (m, 2H), 7.60 (d, J=3.9 Hz, 1H), 7.65 (d, J=3.9 Hz, 1H), 7.78–7.81 (m, 1H), 7.96–8.05 (m, 2H). MS (EI) m/e (rel int); 269 (M+, 100), 270 (M+ +1, 94), 253 (22), 240 (91), 222 (11). Anal Calcd for C15H8FNOS: C 66.90; H 2.99; N 5.20. Found: C 67.04; H 3.08; N 5.15.
2-Fluoro-4-(5-(thiophen-2-yl)furan-2-yl)benzonitrile (4d)
A yellow solid in 71% yield, mp 149°C–149.5°C (EtOH/petroleum ether). Rf=0.63, petroleum ether (40°C–60°C)-EtOAc (9:1). IR (KBr) ν’ 3105, 3085 (CH), 2227 (CN), 1619, 1597 (C=C) cm−1. 1H NMR (DMSO-d6); δ 6.97 (d, J=3.3 Hz, 1H), 7.15–7.18 (m, 1H), 7.44 (d, J=3.9 Hz, 1H), 7.57–7.64 (m, 3H), 7.72–7.98 (m, 2H). MS (EI) m/e (rel int); 269 (M+, 100), 270 (M+ +1, 18), 253 (2), 240 (20). Anal Calcd for C15H8FNOS: C 66.90; H 2.99; N 5.20. Found: C 66.93; H 3.11; N 5.13.
4-(2,2′-Bithiophen-5-yl)-2-fluorobenzonitrile (4e)
A yellow solid in 76% yield, mp 155°C–156°C (EtOH/petroleum ether). Rf=0.70, petroleum ether (40°C–60°C)-EtOAc (9:1). IR (KBr) ν’ 3102, 3080 (CH), 2227 (CN), 1612, 1554 (C=C) cm−1. 1H NMR (DMSO-d6); δ 7.10–7.12 (m, 1H), 7.39–7.40 (m, 1H), 7.57 (d, J=4.2 Hz, 1H), 7.63–7.65 (m, 1H), 7.79 (d, J=3.9 Hz, 1H), 7.86–7.92 (m, 3H). MS (EI) m/e (rel int); 285 (M+, 100), 240 (10). Anal Calcd for C15H8FNS2: C 63.13; H 2.83; N 4.91. Found: C 62.91; H 2.80; N 4.67.
General procedure for bichalcophene fluorobenzamidine synthesis 5a–e
4-(2,2′-Bifuran-5-yl)-3-fluorobenzamidine hydrochloride salt (5a)
The bifuran mononitrile 4a (381 mg, 1.5 mmol), suspended in freshly distilled tetrahydrofuran (THF) (8 mL), was treated with LiN(TMS)2 (1M solution in THF, 4 mL, 4 mmol), and the reaction was allowed to stir overnight. The reaction mixture was then cooled to 0°C and a hydrogen chloride ethanolic solution (12 mL, 1.25 M) was added, whereupon a precipitate started forming. The mixture was left to run overnight, whereafter it was diluted with ether and the resultant solid was collected by filtration. The monoamidine was purified by neutralization with 1N NaOH, followed by filtration of the resultant solid and washing with water (3× 8 mL). Finally, the free base was stirred with hydrogen chloride ethanolic solution overnight and diluted with ether; the solid formed was filtered and dried to furnish the monoamidine 5a hydrochloride salt as a yellow solid in 69% yield, mp 302°C–304°C (EtOH/H2O). IR (KBr) ν’ 3343, 3266 (NH, NH2), 3060, 3038 (CH), 1667, 1617 (C=N, NH bending, C=C) cm−1. 1H NMR (DMSO-d6); δ 6.67–6.69 (m, 1H), 6.94 (d, J=3.6 Hz, 1H), 7.00 (d, J=3.6 Hz, 1H), 7.18–7.21 (m, 1H), 7.80–7.94 (m, 3H), 8.10 (t, J=7.8 Hz, 1H), 9.44 (br s, 4H, exchangeable with D2O). MS (EI) m/e (rel int); 270 (M+, 100), 271 (M+ +1, 18), 254 (26), 253 (10). Anal Calcd for C15H11FN2O2-1.0HCl: C 58.73; H 3.94; N 9.13. Found: C 58.67; H 4.16; N 8.92.
4-(2,2′-Bifuran-5-yl)-2-fluorobenzamidine hydrochloride salt (5b)
A yellow solid in 65% yield, mp 277°C–278.5°C (EtOH/H2O). IR (KBr) ν’ 3360, 3298, 3193 (NH, NH2), 3117, 3092 (CH), 1679, 1619 (C=N, NH bending, C=C) cm−1. 1H NMR (DMSO-d6); δ 6.64–6.65 (m, 1H), 6.87 (d, J=3.1 Hz, 1H), 6.95 (d, J=3.8 Hz, 1H), 7.41 (d, J=3.8 Hz, 1H), 7.71–7.85 (m, 4H), 9.43 (br s, 4H, exchangeable with D2O). MS (EI) m/e (rel int); 270 (M+, 100), 271 (M+ +1, 19), 254 (42). Anal Calcd for C15H11FN2O2-1.0HCl: C 58.73; H 3.94; N 9.13. Found: C 58.45; H 4.21; N 8.93.
3-Fluoro-4-(5-(thiophen-2-yl)furan-2-yl)benzamidine hydrochloride salt (5c)
A yellow solid in 72% yield, mp 282°C–284°C (EtOH/H2O). IR (KBr) ν’ 3335, 3246 (NH, NH2), 3131, 3059 (CH), 1679, 1619, 1598 (C=N, NH bending, C=C) cm−1. 1H NMR (DMSO-d6); δ 7.03 (d, J=3.3 Hz, 1H), 7.18–7.21 (m, 2H), 7.61–7.67 (m, 2H), 7.82–7.93 (m, 2H), 8.08 (t, J=7.8 Hz, 1H), 9.42 (br s, 4H, exchangeable with D2O). MS (EI) m/e (rel int); 286 (M+, 100), 287 (M+ +1, 19), 270 (31), 269 (15). Anal Calcd for C15H11FN2OS-1.0HCl-0.75H2O: C 53.57; H 4.04; N 8.32. Found: C 53.63; H 4.18; N 7.99.
2-Fluoro-4-(5-(thiophen-2-yl)furan-2-yl)benzamidine hydrochloride salt (5d)
A yellow solid in 52% yield, mp 268°C–269.5°C (EtOH/H2O). IR (KBr) ν’ 3296, 3185 (NH, NH2), 3111, 3033 (CH), 1662, 1622 (C=N, NH bending, C=C) cm−1. 1H NMR (DMSO-d6); δ 6.98 (d, J=3.3 Hz, 1H), 7.17–7.20 (m, 1H), 7.42 (d, J=3.6 Hz, 1H), 7.59–7.65 (m, 2H), 7.76–7.82 (m, 3H), 9.36 (br s, 2H, exchangeable with D2O), 9.49 (br s, 2H, exchangeable with D2O). MS (EI) m/e (rel int); 286 (M+, 100), 287 (M+ +1, 20), 270 (53), 269 (16). Anal Calcd for C15H11FN2OS-1.0HCl-0.5H2O: C 54.30; H 3.95; N 8.44. Found: C 54.34; H 3.97; N 8.49.
4-(2,2′-Bithiophen-5-yl)-2-fluorobenamidine hydrochloride salt (5e)
A yellowish-brown solid in 73% yield, mp 289°C–291°C (EtOH/H2O). IR (KBr) ν’ 3298, 3197 (NH, NH2), 3098, 3018 (CH), 1656, 1620 (C=N, NH bending, C=C) cm−1. 1H NMR (DMSO-d6); δ 7.10–7.11 (m, 1H), 7.39 (s, 2H), 7.57 (d, J=4.2 Hz, 1H), 7.65–7.84 (m, 4H), 9.50 (br s, 4H, exchangeable with D2O). MS (EI) m/e (rel int); 302 (M+, 100), 286 (42). Anal Calcd for C15H11FN2S2-1.0HCl: C 53.17; H 3.57; N 8.27. Found: C 52.86; H 3.61; N 8.44.
Biology
The Salmonella typhimurium TA1535 bacterial strain was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). NaN3, B[a]P, and nicotinamide adenine dinucleotide phosphate reduced (NADPH) were obtained from Sigma-Aldrich (St Louis, MO, USA). The S9 mix was prepared as described elsewhere.8
Cytotoxicity assays
The effect of fluoroarylbichalcophenes on the viability of S. typhimurium TA1535 was investigated by growing the bacteria under two exposure conditions: broth microdilution and colony plate counting methods. The details have been described elsewhere.8
Combined cytotoxic effect of fluoroaryl-2,2′-bichalcophenes and selected mutagens
Prior to mutagenicity/antimutagenicity assay evaluation, exclusion of the possible toxic effect that could arise from the combination of mutagens used with fluoroarylbichalcophenes was performed. Cytotoxicity assays were performed as previously mentioned, with some modifications as previously described elsewhere.8
Mutagenicity/antimutagenicity assays
The potential mutagenic activity of the fluoroarylbichalcophenes was assayed according to Maron and Ames14 and fully described elsewhere.8 To assess the antimutagenic potential of fluoroarylbichalcophenes toward NaN3 or B[a]P, pre-exposure and co-exposure assays were carried out simultaneously via a method modified from Maron and Ames.14 Under pre-exposure conditions, each fluoroarylbichalcophene was incubated with S. typhimurium TA1535 at 37°C for 30 minutes on shaking incubator before the addition of the mutagen. Co-exposure assays were performed by incubating the bacteria, the mutagen, and each fluoroarylbichalcophene at 37°C for 30 minutes prior to plating on minimal glucose agar (MGA). Positive and negative controls were performed in all assays. All antimutagenesis determinations were performed in triplicate. The number of revertant colonies was counted after 48 hours of incubation, and the antimutagenic potential of the tested compounds was expressed as a percentage of reduction in mutagenicity,16 and calculated according to the following equation:

where Rm is the number of revertants/plate in the presence of mutagen, Rs is the number of spontaneous revertants/plate, and Ra is the number of revertants/plate in the presence of fluoroarylbichalcophenes.
A 20% or less reduction means no antimutagenic activity, 20%–40% reduction means moderate activity, and 40% or more reduction means strong antimutagenic activity.
The mutant frequency or mutation rate was then calculated from the mutant colonies/viable colonies for both exposure conditions for the mutagens investigated. All procedures were approved by the University of King Faisal Committee of Scientific Research Ethics.
Determination of total antioxidant activity
The antioxidant activity of fluoroarylbichalcophenes was measured spectrophotometrically using a phosphomolybdenum method,17 based on the reduction of Mo(VI) to Mo(V) and the subsequent formation of specific green phosphate/Mo(V) compounds. A 0.3 mL aliquot of sample solution (20 μM final concentration) was combined with 2.7 mL of the reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate). The sample was capped and incubated in a boiling water bath at 95°C for 90 minutes. After cooling to room temperature, the absorbance was measured at 695 nm. Stock solution of ascorbate was freshly prepared and used as a standard antioxidant.
Statistical analysis of data
Statistical analyses of data were performed using analysis of variance (ANOVA), followed by Fisher’s protected least significant difference multiple range test. Differences were considered significant at P-values of <0.05.
Results and discussion
Chemistry
A series of bichalcophene fluorobenzamidines 5a–e was prepared from the corresponding mononitriles 4a–e by direct reaction with LiN(TMS)2 (Figure 2). Thus, compound 4-(2,2′-bifuran-5-yl)-3-fluorobenzamidine (5a) was obtained from the corresponding mononitrile 4a by treatment with LiN(TMS)2 followed by deprotection with ethanolic HCl (gas). The structures of fluorobenzamidines 5a–e were identified by their spectroscopic and elemental analyses. Thus, 1H NMR spectrum of compound 5a displayed singlet signal at δ 9.44 (4H) characteristic for the cationic amidine group in addition to the signals corresponding to the trisubstituted benzene ring and bifuran moiety. Fluoroarylbichalcophene mononitriles 4a–e were prepared adopting a Stille coupling reaction between the corresponding bromo compounds 3a–c and 2-(tri-n-butylstannyl)furan or analogues using our previously described methodology for the preparation of non-fluorinated bichalcophene analogues.18 The structures of compounds 4a–e were assigned based on their spectral and elemental analyses.
 | Figure 2 Synthesis of novel monocationic fluoroaryl-2,2′-bichalcophene derivatives. |
Biology
Cytotoxicity
Investigating novel compounds for therapeutic and pharmacological activities should be parallel with omitting their toxicological effects. In addition, the determination of the potential mutagenic effect of any drug under development is mandatory. In our previous studies, ten novel bichalcophenes showed no genotoxic effects and were effective in reducing the mutagenicity induced by NaN3 and B[a]P in the Ames microsomal assay.8 A series of fluoroarylbichalcophene derivatives was synthesized and examined for their antimutagenic potency. To rule out the possible toxic effect exerted by the investigated fluoroarylbichalcophenes on the viability of S. typhimurium TA1535, the non-toxic/non-growth inhibitory concentrations were determined by growing the bacteria under two exposure conditions: broth microdilution and colony plate counting methods. In the broth microdilution method, all fluoroarylbichalcophenes caused a significant reduction in the viability of S. typhimurium TA1535 at 50 and 100 μM as compared with control untreated cells (Table 1). No significant toxicity was reported with all compounds at 25 μM. The effect of fluoroarylbichalcophenes on the total viable count of TA1535 was performed at 10 and 20 μM to avoid any toxicity concerns that may arise with higher concentrations. All fluoroarylbichalcophenes were non-toxic and had no significant reducing effect on the total viable count of S. typhimurium TA1535 at both concentrations investigated. In a previous study on non-fluorinated bichalcophenes,8 compounds that correspond to compounds 5a and 5e in the current study were toxic at 25 μM to Salmonella in liquid medium. The addition of a fluorine atom reduced the toxicity of these compounds (Table 1). This allowed the examination of the antimutagenic activity of these compounds at the highest concentration possible without compromising the bacterial growth due to toxicity effect.
 | Table 1 Cytotoxicity of fluoroarylbichalcophenes to Salmonella typhimurium TA1535 in liquid medium |
Effect of combination of fluoroarylbichalcophenes and mutagens on Salmonella typhimurium TA1535 viability
The combined toxic effect of the investigated compounds along with two selected mutagens was investigated to validate the antimutagenic assays and to ensure that the reduction in the number of revertant colonies was not related to any overt toxicity. A direct-acting mutagen (NaN3; 2 μg/plate) and a polycyclic aromatic indirect hydrocarbon mutagen (B[a]P; 20 μM) were selected for this purpose. Both mutagens mutate S. typhimurium TA1535 with base-pair substitution mutation through different pathways. NaN3 does not require metabolic activation; however, B[a]P mutates TA1535 in the presence of rat liver microsomal enzymes (S9 fraction). The majority of mutations induced by B[a]P in various systems is at guanine, with most of the mutations being GC to TA transversions.19 None of the investigated fluoroarylbichalcophenes had a significant toxic effect on the viability of TA1535 when used along with NaN3 or B[a]P (data not shown).
The mutagenic/antimutagenic potential of fluoroarylbichalcophenes against NaN3 and B[a]P
A gene mutation is a permanent DNA sequence change, and the accumulation of genetic errors results in cancer development. The Ames test was developed for the screening of chemical mutagenicity/carcinogenicity. The test is useful in detecting frame shift mutation or base substitution of DNA.20 This assay is very competent in screening for anticancer activity of novel compounds. This assay also enables the screening of a large number of compounds within a reasonable time frame and cost and helps identify the potential anticancer hits. On investigating the mutagenic effect of fluoroaryl compounds at non-growth inhibitory and non-toxic concentrations, all compounds were non-mutagenic to TA1535 in the presence or absence of S9 as compared with negative spontaneous revertant or positive control bacteria treated with B[a]P (Table 2). The assay was performed in the absence of S9 mix, and similar results were obtained (data not shown). The potential antimutagenic activity of fluoroaryl derivatives was evaluated using a modified Ames assay under pre-exposure and co-exposure treatments. Almost all fluoroarylbichalcophenes exerted a strong antimutagenic activity (>40%) against NaN3-induced mutagenicity in pre-exposure and co-exposure assays (Table 3). Under the pre-exposure condition, the investigated compounds were antimutagenic at 10 and 20 μM except for compound 5b and 5c, which exerted a strong antimutagenic potential only at 20 μM. The recorded antimutagenic activity was concentration-dependent with monocationic fluoroaryl derivatives (5a–5e) under pre-exposure conditions. However, the difference in concentration had no significant effect on the recorded activity in co-exposure experiments, except with derivatives 5a and 5d (Table 3). In terms of the difference between mononitrile and the corresponding monocationic, mononitrile derivatives (4b–4e) were more active than the corresponding monocationic derivatives under pre-exposure treatment at the low concentration investigated. For much similar bichalcophenes, the monocationic derivatives were more effective than the corresponding mononitrile derivatives.8 This rule still stands in the present study but was contradicted once at the low concentration and under the pre-exposure conditions against azide-mutagenicity (Table 3). A similar trend of inhibition of mutagenicity against B[a]P was also recorded. When evaluating the antimutagenic potency of fluoroarylbichalcophenes against B[a]P-induced mutagenicity, all compounds caused a non-concentration-dependent reduction (21%–91%) in the induced mutagenicity under the pre-exposure condition (Table 4). In the co-exposure experiment, all compounds showed a low to strong antimutagenic potential at 10 and 20 μM, except derivatives 4c, 4d, and 4e, which had no significant activity at the low concentration investigated. The investigated monocationic derivatives were more effective than the corresponding mononitrile in both pre- and co-treatment conditions. It seems that the bacteria were able to metabolize these fluorinated bichalcophenes much faster than the corresponding non-fluorinated bichalcophenes. This resulted in less efficiency in the antimutagenic activity of fluorinated compounds against NaN3-mutagenicity, especially at the low concentration (Table 3), ie, the metabolites were less active antimutagenic agents than the parent compounds. This hypothesis was also supported by the data from the co-exposure regimen where there was almost no difference between the fluorinated and non-fluorinated compounds. In this exposure regimen, the mutagen, bacteria, and the fluoroarylbichalcophenes were mixed at the same time. The fluorine substitution elevated the antimutagenic behavior of bichalcophenes against B[a]P mutagenicity. Fluorinated monocationic bichalcophenes showed a remarkable antimutagenic profile, reflecting the ability of these compounds to interfere with the cytochrome P450 (CYP450)-dependent activation of B[a]P (Table 4). The P450 system was blocked by many fluorinated compounds.12,21 B[a]P is known to be metabolized by CYP1A1/2 followed by epoxide hydrolysis; both steps were suppressed by fluorine-substituted compounds.22 In a previous study,10 there was an evident structural-activity relationship, where the activity was elevated by the presence of a thiophene ring. This phenomenon could not be discerned in the current study where the presence of thiophene ring(s) in compounds 4 and 5 (c–e) had sporadic effects on the antimutagenic activities of these compounds.
 | Table 2 Determination of mutagenic activity of fluoroaryl-2,2′-bichalcophenes in Salmonella typhimurium TA1535 in presence of S9 mix |
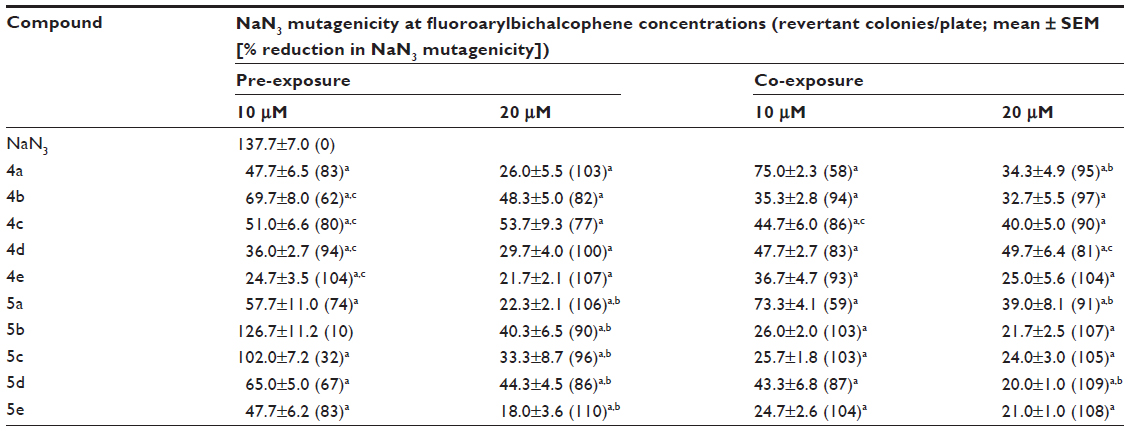 | Table 3 Determination of antimutagenic activity of fluoroaryl-2,2′-bichalcophenes in Salmonella typhimurium TA1535 against sodium azide (2 μg/plate) |
 | Table 4 Determination of antimutagenic activity of fluoroaryl-2,2′-bichalcophenes in Salmonella typhimurium TA1535 against benzo[a]pyrene 20 μM in the presence of S9 mix |
Mutation frequency is highly correlated to mutation rate. All fluoroarylbichalcophenes reduced the mutant frequency caused by NaN3 by 8%–87% (Table 5). Most of the monocationic compounds were more effective than the mononitrile compounds in reducing the mutant frequency under co-exposure conditions. With regard to the reduction of mutant frequency with B[a]P, the investigated compounds were not as effective as they were against NaN3 and caused a reduction between 6% and 69% in the recorded mutant frequency (Table 6). The recorded antimutagenic activity of the investigated compounds could be attributed to various mechanisms. These compounds could interfere with the azide absorption into the bacteria by modifying the cell membrane or they could prevent the azide binding to DNA. Another mechanism is the direct binding and protection of DNA from the electrophilic mutagen or its metabolites,23 given that the fluoroaryl derivatives are nucleophilic. One of the possible mechanisms is the inhibition of CYP4501A activity that metabolically activates B[a]P, based on the inhibition of enzymatic oxidation at the site of F-substitution due to its electron-withdrawing nature.13,24 Another possible mechanism could be the elevation in the antioxidant milieu of the cells, thus promoting the DNA repair system.25 Testing the total antioxidant activity of fluoroaryl compounds showed that compounds 5a–e were in the lead, with antioxidant activity of ~46%–73% of that of ascorbic acid at the same concentration (Figure 3). The promising antioxidant activity could provide a protective effect of fluoroaryl derivatives against oxidative DNA damage.26 In summary, novel fluoroarylbichalcophenes in the current study provided a significant antigenotoxic activity against the DNA-intercalation caused by NaN3 and prevented the adduct formation for B[a]P metabolites probably through inhibiting the microsomal-dependent activation of the mutagen.
 | Table 5 Effects of fluoroaryl-2,2′-bichalcophenes on sodium azide mutant frequency |
 | Table 6 Effects of fluoroaryl-2,2′-bichalcophenes on benzo[a]pyrene mutant frequency |
 | Figure 3 Total antioxidant activity of fluoroaryl-2,2′-bichalcophene derivatives at 25 μM expressed as the percentage of equivalent ascorbate activity at the same concentration. |
Acknowledgment
This project was supported by King Faisal University, Deanship of Scientific Research, Grant 140078.
Disclosure
The authors report no conflicts of interest in this work.
References
Connor TH, Trizna Z. Pentamidine isethionate is negative in tests for microbial mutagenicity and chromosomal breakage in vitro. Toxicol Lett. 1992;63(1):69–74. | |
Stauffert I, Paulini H, Steinmann U, Sippel H, Estler CJ. Investigations on mutagenicity and genotoxicity of pentamidine and some related trypanocidal diamidines. Mutat Res. 1990;245(2):93–98. | |
Thuita JK, Karanja SM, Wenzler T, et al. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop. 2008;108(1):6–10. | |
Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. Synthesis and antiprotozoal activity of aza-analogues of furamidine. J Med Chem. 2003;46(22):4761–4769. | |
Wenzler T, Boykin DW, Ismail MA, Hall JE, Tidwell RR, Brun R. New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289. Antimicrob Agents Chemother. 2009;53(10):4185–4192. | |
White EW, Tanious F, Ismail MA, et al. Structure-specific recognition of quadruplex DNA by organic cations: influence of shape, substituents and charge. Biophys Chem. 2007;126(1–3):140–153. | |
El-Sayed WM, Hussin WA, Ismail MA. Efficacy of two novel 2,2′-bifurans to inhibit methicillin-resistant Staphylococcus aureus infection in male mice in comparison to vancomycin. Drug Des Devel Ther. 2012;6:279–287. | |
El-Sayed WM, Hussin WA. Antimutagenic and antioxidant activity of novel 4-substituted phenyl-2,2′-bichalcophenes and aza-analogs. Drug Des Devel Ther. 2013;7:73–81. | |
Boschi D, Guglielmo S, Aiello S, Morace G, Borghi E, Fruttero R. Synthesis and in vitro antimicrobial activities of new (cyano-NNO-azoxy)pyrazole derivatives. Bioorg Med Chem Lett. 2011;21(11):3431–3434. | |
Hussin WA, Ismail MA, El-Sayed WM. Novel 4-substituted phenyl-2,2′-bichalcophenes and aza-analogs as antibacterial agents: a structural activity relationship. Drug Des Devel Ther. 2013;7:185–193. | |
Wakselman C. Fluorinated organic compounds: synthesis and biological applications. Ann Pharm Fr. 1999;57(2):108–115. | |
Park BK, Kitteringham NR, O’Neill PM. Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol. 2001;41:443–470. | |
Kato T, Hakura A, Mizutani T, Saeki K. Anti-mutagenic structural modification by fluorine-substitution in highly mutagenic 4-methylquinoline derivatives. Mutat Res. 2000;465(1–2):173–182. | |
Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutat Res. 1983;113:173–215. | |
Saeki K. Anti-carcinogenic structural modification by fluorine-substitution in aza-polycyclic aromatic hydrocarbons. Yakugaku Zasshi. 2000;120(12):1373–1385. | |
Hong CE, Cho MC, Jang HA, Lyu SY. Mutagenicity and anti-mutagenicity of Acanthopanax divaricatus var. albeofructus. J Toxicol Sci. 2011;36:661–668. | |
Prieto P, Pineda M, Aguilar M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex: specific application to the determination of vitamin E. Anal Biochem. 1999;269(2):337–341. | |
Youssef MM, Al-Omair MA, Ismail MA. Synthesis, DNA affinity, and antimicrobial activity of 4-substituted phenyl-2,2′-bichalcophenes and aza-analogues. Med Chem Res. 2012;21(12):4074–4082. | |
DeMarini DM, Hanley NM, Warren SH, Adams LD, King LC. Association between mutation spectra and stable and unstable DNA adduct profiles in Salmonella for benzo[a]pyrene and dibenzo[a,l]pyrene. Mutat Res. 2011;714(1–2):17–25. | |
Zeiger E. Identification of rodent carcinogens and noncarcinogens using genetic toxicity tests: premises, promises, and performance. Regul Toxicol Pharmacol. 1998;28(2):85–95. | |
Hartmann RW, Palusczak A, Lacan F, Ricci G, Ruzziconi R. CYP 17 and CYP 19 inhibitors. Evaluation of fluorine effects on the inhibiting activity of regioselectively fluorinated 1-(Naphthalen-2-ylmethyl)imidazoles. J Enzyme Inhib Med Chem. 2004;19(2):145–155. | |
Buhler DR, Unlu F, Thakker DR, et al. Metabolism and tumorigenicity of 7-, 8-, 9-, and 10-fluorobenzo(a)pyrenes. Cancer Res. 1982;42(11):4779–4783. | |
Ismail MA, Bialy SA, Brun R, et al. Dicationic phenyl-2,2′-bichalcophenes and analogues as antiprotozoal agents. Bioorg Med Chem. 2011;19(2):978–984. | |
Katoh Y, Nemoto N, Tanaka M, Takayama S. Inhibition of benzo[a]pyrene-induced mutagenesis in Chinese hamster V79 cells by hemin and related compounds. Mutat Res. 1983;121(2):153–157. | |
Collins AR, Azqueta A, Langie SA. Effects of micronutrients on DNA repair. Eur J Nutr. 2012;51(3):261–279. | |
Muzandu K, El Bohi K, Shaban Z, Ishizuka M, Kazusaka A, Fujita S. Lycopene and beta-carotene ameliorate catechol estrogen-mediated DNA damage. Jpn J Vet Res. 2005;52(4):173–184. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.