")
Back to Journals » Infection and Drug Resistance » Volume 13
Evaluation of the Anti-Leishmanial Effect of Recombinant Clostridium α-Toxin
Authors Namdar F , Khanahmad H, Ghayour Z, Mirzaei F, Namdar A, Aghaei M, Izadi S, Khamesipour F, Hejazi SH
Received 9 April 2020
Accepted for publication 24 June 2020
Published 15 July 2020 Volume 2020:13 Pages 2355—2364
DOI https://doi.org/10.2147/IDR.S257561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Fatemeh Namdar,1 Hossein Khanahmad,2 Zahra Ghayour,1 Farzaneh Mirzaei,3 Azam Namdar,4 Maryam Aghaei,5 Shahrokh Izadi,6 Faham Khamesipour,7,8 Seyed Hossein Hejazi1,9
1Department of Parasitology and Mycology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran; 2Department of Genetics and Molecular Biology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran; 3Department of Parasitology and Mycology, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 4Department of Community Medicine, School of Medicine, Fasa University of Medical Sciences, Fasa, Iran; 5Skin Diseases and Leishmaniasis Research Center, Isfahan University of Medical Sciences, Isfahan, Iran; 6Department of Medical Parasitology and Mycology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran; 7Cellular and Molecular Research Center, Sabzevar University of Medical Sciences, Sabzevar, Iran; 8Shahid Beheshti University of Medical Sciences, Tehran, Iran; 9Skin Diseases and Leishmaniasis Research Center, Department of Parasitology and Mycology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
Correspondence: Seyed Hossein Hejazi
Skin Diseases and Leishmaniasis Research Center, Department of Parasitology and Mycology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
Tel +98 9133118711
Email [email protected]
Background: Leishmaniasis is an infectious disease common in tropical and subtropical regions caused by the genus Leishmania, which is transmitted by the bite of female sandflies. In this study, we evaluate the anti-leishmanial effect of recombinant Clostridium α-toxin protein alone and the combination with glucantime through in vitro and in vivo.
Materials and Methods: Production, expression, and purification of recombinant α-toxin were evaluated by SDS-PAGE and Western blotting techniques. The antileishmanial activities of the purified α-toxin plus and without glucantime were examined in vitro and in vivo.
Results: The results indicated successful expression of α-toxin as a 48 kDa band on SDS-PAGE and Western blot methods. Also, evaluation of α-toxin IC50 showed the strong fatal effect of it, and glucantime on medium proliferated Leishmania promastigotes at lower concentrations compared with glucantime or α-toxin alone. Moreover, in vivo surveys showed that at the end of treatment courses, the mean of lesion size diminished in glucantime plus α-toxin treated mice versus negative control groups (p < 0.001). Also, there was a significant difference in the parasite burden of the spleen and liver of the control versus the test groups (p < 0.001).
Conclusion: The results showed recombinant α-toxin has synergistic effects with glucantime in destroying Leishmania parasites.
Keywords: α-toxin, Clostridium septicum, parasitic diseases, Leishmania, in vitro, in vivo
Introduction
Leishmaniasis is an infectious disease common in tropical and subtropical regions that are caused by parasites of the genus Leishmania, which transmitted by the bite of female sandflies.1
The motile promastigotes multiply in the gut of the female sand-fly and spread to the mammalian host during the blood meal. Inside the body of mammalian hosts, the entered parasites infect macrophages of the reticuloendothelial system, and into them that contain high temperature and low pH, get transformed into non-motile amastigotes in the period of 12–24 h. Amastigotes multiply by dual splitting in the phagolysosomal vacuoles, and after macrophage collapse, rereleased parasites infect adjacent healthy macrophages and cause leishmaniasis.2
Conventional drugs for the treatment of leishmaniasis are pentavalent antimonials that have side effects such as toxicity to other cells.3 Meglumine antimoniate (glucantime) is pentavalent antimony approved by the World Health Organization as the first-choice drug for the treatment of all types of leishmaniasis; while various side effects are reported.3
In recent years, in addition to synthetic and herbal medicines, some bacterial metabolites such as toxins alone or in combination with routine medications against various infections have also been examined.
One of the significant bacterial toxins against the Leishmania parasites is α-toxin originated from Clostridium septicum (C. septicum).4 The organism is a bar-shaped, anaerobic, and gram-positive bacterium of the natural flora of the human and animal’s intestine.5 This motile and fermentative bacterium produces molecular hydrogen gas and carbon dioxide as a sub-product of cellular respiration.6 Serology or enzymatic studies indicate that it contains four toxins, such as lethal and necrotizing toxin (α-toxin), DNase (beta-toxin), hyaluronidase (gamma-toxin) and the thiol-activated or septicolysin (δ-toxin) toxin.7 Other enzymes, such as proteases and neuraminidase, are also produced by C. septicum.8 This organism is the most important cause of inflammation in gangrene of the intestinal wall (visceral necrotizing colitis) and visceral blood poisoning in animals. It also causes pericarditis, non-injurious muscle necrosis and gangrene in humans that later lead to colon malignancy.5 C. septicum cause muscle necrosis through the secretion of its exotoxins such as α-toxin.9 This toxin is a pore-forming secretory exotoxin belonging to the aerolysin-like family that is expressed by the Csa gene as passive protoxin (Atpro, 443 amino acids, 46.5 kDa) and possibly through the secretory pathway of type III.8,10 The protoxin is activated by proteolytic cleavage near the C terminus and an active monomer of 41.3-kDa cytolytically active form (ATact) which eventually causes the release of a 45-amino-acid fragment.11 Various proteases such as trypsin and proteinase K can activate C. septicum. Still, it seems that furin present on the surface of the eukaryotic cells is the primary activator of α-protoxin in vivo.12 Then, monomeric subunits of α-toxin oligomer on the detergent-resistant membranes (DRMs) lead to the incorporation of this complex into the plasma membrane of mammalian cells and generate a pore (1.3–1.6 nm in diameter) that stimulates the selective permeability of small ions such as potassium and induces hemolysis by breaking the ionic balance.13,14 Also, this toxin can induce cellular necrosis with rapid K+ efflux, deregulation of mitochondrial activity leading to increased ROS levels and dramatically reduced levels of ATP.15
Because the use of a variety of drugs in the treatment of leishmaniasis has not been effective so far, it seems that the accompanying safety dose of a compound such as Clostridium α-toxin as a synergistic factor with regards to biological properties and considering that it leads to non-specific stimulation of the immune system and immunity against various infectious agents is beneficial. Due to low scientific researches on the effect of α-toxin of C. septicum on parasitic diseases, in this study, α-toxin was produced recombinantly, and its killing effects alone or in combination with glucantime on Leishmania parasites were investigated in vitro. Also, its properties to accelerate lesion improvement by itself or plus glucantime on cutaneous leishmaniasis (CL) of mouse model was studied. A compelling hypothesis is that α-toxin has a synergistic effect with glucantime.
Materials and Methods
Ethics Statement
Experimental protocols of this study were approved by the Institutional Research and Ethics Committee of Medical Sciences from Isfahan University of Medical Science (IR.Mui.REC.1395.3.360). Also, all experimental procedures were in accordance with the guidelines for the Care and Use of Laboratory Animals published by National Institutes of Health (NIH Publication) and approved by the Research Council of Isfahan University of Medical Sciences.
Construction of the Recombinant Vector
Host and vector were used, E. coli BL21 (DE3) F− ompT gal dcm hsdSB (rB − mB −) and pET28a (+), respectively. Coding sequence (CDS) of the α-toxin gene (Accession number, Q53482) was obtained via the NCBI data bank.
Two web servers, optimizer (http://www.genoms.uvr.es), and E. coli rare codon analyzer 2 (http://www.faculty.ucr.edu), were performed on α-toxin CDS. The optimized sequences (1250 bp) were ordered from the GeneCust (Luxembourg), and synthesized DNA fragment had been inserted into the pET28a (+) vector into sites. The recombinant vector was transformed in chemically competent E. coli BL21 by heat shock protocol and spread on Luria-Bertani (LB) agar medium containing 50 µg/mL kanamycin.
Plasmid Extraction
A colony of bacteria with pET28a (+) α-toxin was cultured and shakes in LB broth medium with 50µg/mL kanamycin overnight at 37°C and 200 rpm. The plasmid DNA was extracted according to the kit instructions (Vivantis, Malaysia, #GF- PL-050). Subsequently, the plasmid was digested with Nco I/Xho I enzymes, and the digested vector was electrophoretically separated on 1% agarose gel.
Protein Expression
The transformed recombinant bacteria were cultured overnight in the LB broth medium contained Kanamycin (50 µg/mL) at 37°C and 200 rpm. The overnight culture was transferred to 50 mL of LB broth medium. After reaching the culture optical density (OD600) to 0.4–0.6, the expression of α-toxin was induced by the addition of 1 mM of isopropyl-b-D-thiogalactopyranoside (IPTG; CinnaGen, Iran). Again the optical density was measured by the spectrophotometer (Shimadzu, Japan), and the temperature was set to 30°C. After the 16hrs incubation period, the cells were harvested by centrifugation at 12,000g (4°C), and the total protein lysate was obtained via the five times of sonication for the 30s at 50/60Hz.
Purification of Recombinant Protein
To purify the recombinant α-toxin, which had the C-terminal His-tag, Ni-NTA resin (Qiagen) was used.16 The column was washed with five volumes of Wash Buffer (10 mM Tris base, 100 mM NaH2Po4, and 8 M urea, pH: 6.3) and double-distilled H2O, respectively. 10 mL of total protein lysate sample (obtained from 300 mL of recombinant bacterial cultures) was passed through the column. To remove non-specific bound proteins, the column was washed with the washing buffer and the recombinant protein eluted by adding elution buffer (10 mM Tris base, NaH2Po4, and 8 M urea, pH: 5.9 and pH: 4.5).17 Finally, SDS-PAGE was applied to detect the purified α-toxin band. To remove urea, the recombinant protein solution was maintained in the dialysis bag containing PBS for 24hrs at 4°C, and the protein concentration was measured using a BCA kit (Parstous, Iran).18
SDS-PAGE and Western Blot Analysis
The sample protein fraction was subjected in an SDS-PAGE system with 10% running gel and 4% stacking gel (Laemmli method)19 and then transferred at 80 V for 90 min to a nitrocellulose membrane. After blocking the membrane with TBS skim milk 5% (w/v) for 1 hr at 25°C, the membrane was incubated with a dilution of anti-his-horseradish peroxidase antibody (HRP) (1:1000, Sigma, USA) for 1 hr at room temperature. The HRP bound to His-tagged protein was visualized by adding the tetramethylbenzidine (TMB) substrate.20
Parasite Culture
L. major parasites (MRHO/IR/75/ER) were maintained at the Department of Parasitology and Mycology, Isfahan University of Medical Sciences, Isfahan, Iran on susceptible Balb/c mice and cryopreserved vials in liquid nitrogen. A cryopreserved bottle was passaged in complete RPMI 1640 (Gibco, USA) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, USA) at 25°C.
Cytotoxicity Assessment
The J744-A1 cell line was purchased from the Pasteur Institute of Tehran, Iran, in the form of cryopreserved vials. The cells were washed after de-freezing and poured into 75mL culture flasks containing RPMI 1640 medium supplemented with FCS 20%, 100U/mL penicillin and 100μg/mL streptomycin and incubated at 37°C in the presence of 5% CO2 for 5–6 days. At about 100% confluency, Trypsin-EDTA was used to detach the cells from the floor of the flask, washed with RPMI 1640 medium, and counted with the hemocytometer chamber. The harvested cells were divided into a 96 well plate 2×104 cells per well along with complete RPMI 1640 and incubated at 37°C and 5% CO2 for 24 hrs. Afterward, the supernatant was removed, and the wells were treated with different dilutions of α-toxin (0.0003–200 μg/mL) in complete medium and incubated at 37°C for 1hr under a humidified air/5% CO2. Each treatment was repeated three times, and the wells without α-toxin were considered as a negative control. The CC50 of α-toxin on macrophages was determined using the MTS kit (Promega, Madison, USA) and the GraphPad Prism 7 software.21
Infection of Macrophages with Leishmania major Promastigotes
The sterile coverslips were placed at the bottom of the six-well chamber glasses, and 2×105 macrophages were seeded on into wells. The plates were incubated under 37°C and 5% CO2 until the cells adhere entirely to the coverslips. After 24 hrs of incubation, the metacyclics were added to the wells with approximately 10:1 parasite per cell. Then, plates were incubated under 34±1°C and 5% CO2 as long as the parasites enter into the macrophages.
Anti-Amastigote Assessment
The infected macrophages were treated with eighteen dilutions of α-toxin (0.001 to 200 μg/mL), glucantime with the similar eighteen dilutions (0.001–200 μg/mL), α-toxin plus glucantime (with the same concentrations) and finally α-toxin 0.003 μg/mL (non-toxic dose) plus glucantime (0.001–200 μg/mL). For evaluating the IC50, three wells were not exposed to the glucantime or α-toxin as negative controls. The coverslips were exited after 48 hrs of treatment, washed, and placed on slides. They were dried, fixed with methanol and stained with Giemsa. The amastigote number of 100 parasite-infected macrophages were counted and compared with the negative controls. The concentration of the drug or α-toxin, which able to eliminate 50% of the parasites, was considered as IC50. The test was performed in triplicate for each concentration, and the mean number of amastigotes was reported. To investigate the synergistic effect of glucantime and α-toxin, pre-infected macrophages in 6-wells chamber were treated with 0.001 to 200 μg/mL α-toxin plus 0.001 to 200 μg/mL glucantime. Also, the infected macrophages were treated with a non-cytotoxic concentration of α-toxin (0.003μg/mL) plus different concentrations of glucantime (0.001–200μg/mL).
In vivo Experiments
All procedures verified according to the guidelines of the Institutional Animal Care and Ethics Committee of Isfahan University of Medical Sciences, Isfahan, Iran (IR.MUI.REC.1395.3.360), and measures are taken to protect animals from pain or discomfort.
Forty female Balb/c mice, 4–6 weeks old, were purchased from the Pasteur Institute of Tehran, Iran. To produce CL infection in mice, each of them was inoculated 1x106 metacyclics intradermally at the base of the tail. After 3 weeks of parasite inoculation and simultaneously with the onset of CL lesions, they were divided into four groups of ten mice as follows: 1). 100 mg/kg glucantime, 2). 10 μg/kg α-toxin, 3). glucantime plus α-toxin (20 mg/kg and 10μg/kg, respectively), 4). without treatment (negative control). The method of treatment was a daily intra-lesional injection and the duration of treatment was 3 weeks. Before starting the treatment and then during the treatment period, the ulcers were measured weekly, using a Vernier Caliper.
Evaluation of the Parasite Burden
After the end of the treatment course, five mice from each group were sacrificed. The mice were autopsied, spleen, and liver of them were removed and weighed in a sterile condition, homogenized by tissue grinder in 2 and 4 mL of Schneider’s medium, including FBS 20% and gentamicin 0.1%, respectively, and were diluted from 1 to 10−10 in a 96 well plate. After 7 days of incubation at 25°C, the presence or absence of motile promastigotes in each well was recorded. The end titer was the dilution with at least one living parasite, and the parasite burden/mg of tissue was calculated as follows: Parasite burden =-log 10 (parasite dilution/tissue weight).
Statistical Analysis
Values were presented as the mean ± SEM. The significance of differences was determined by Analysis of Variances (ANOVA) and Student’s t-test using SPSS software version 22 with (p<0.05).
Result
The Result of Digestion
The authenticity of pET28a -α-toxin plasmid confirmed using Nco I/Xho I restriction enzyme digestion. The result of digestion showed two fragments of 5234 bp and 1250.
Expression and Purification of Recombinant Protein
Protein production was carried out through vector transformation into the E. coli. This was proved by SDS-PAGE on both bacterial lysate and purified protein derived from it. After completion of the analysis, the investigation of the polyacrylamide gel confirmed the existence of a 48-kDa band related to α-toxin, which was fused with 6×His tags (Figure 1). To ensure the detected bands were α-toxin-6×His tags, Western blotting was carried out by implementing an antibody against 6×His tags that were shown as a single band with an apparent MW of 48 kDa. The protein concentration was measured to be about 1 mg/mL (Figure 2).
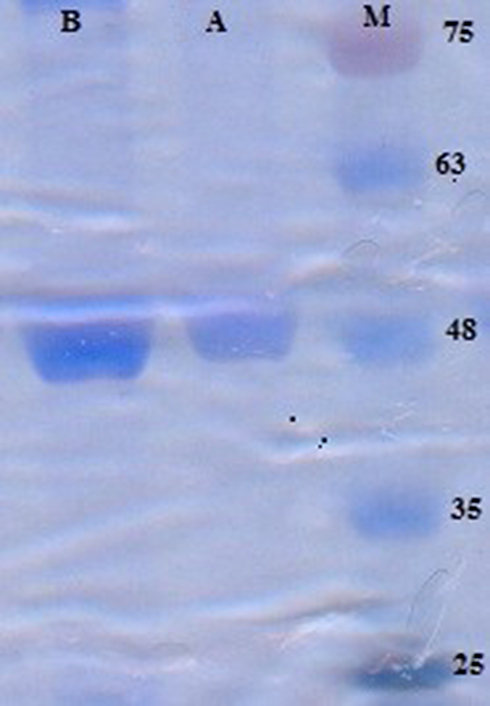 |
Figure 1 SDS-PAGE analysis of purified recombinant protein of α-toxin by Ni-NTA resin M: prestained Protein Ladder (Sinaclon, Iran) (10–170 kDa), Cat No. SL7011 (PR911654); Lanes A and B indicate second and fourth eluted fraction of recombinant protein of E. coli BL21 (DE3) transformed with pET28a/-α- toxin-6×His tags. |
 |
Figure 2 Western blot analysis of purified recombinant protein of α-toxin-6×His tags. M: prestained Protein Ladder (Sinaclon, Iran). Lane A: Negative control E. coli strain BL21 (DE3). Lane B purified recombinant protein of E. coli BL21 (DE3) transformed with pET28a/-α-toxin-6×His tags shows one band at 48 kDa. |
The α-Toxin Cytotoxicity in J774-A1 Macrophages and Anti-Amastigote Assessment
The level of CC50 of α-toxin on the J774-A1 cell line was evaluated by GraphPad prism 7.01 software, which was 0.03 ± 0.002 µg/mL. This protein was not found to be fatal for cells at concentrations of ≤ 0.003 μg/mL. After examining the effect of each dilution (0.001–200 μg/mL) of glucantime and also α-toxin, on 100 macrophages in comparison with negative control, the level of glucantime and α-toxin IC50 has obtained 4.27± 0.55 µg/mL and 0.008 ± 0.001 µg/mL, respectively. The mean of glucantime IC50 plus α-toxin was found 0.004± 0.002 µg/mL. Also, the level of glucantime IC50 along with 0.003 µg/mL non-toxic α-toxin was obtained 0.3± 0.001 µg/mL (Table 1 and Figure 3A).
 |
Table 1 The Mean of the IC50 Level of Glucantime and α-Toxin (µg/Ml) in vitro |
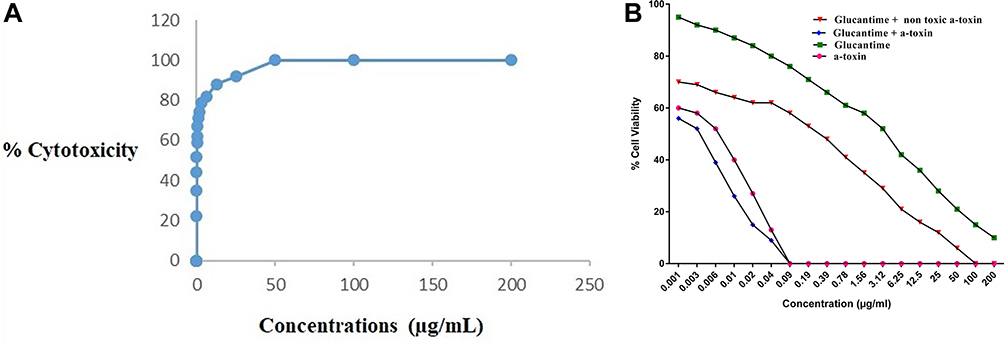 |
Figure 3 The CC50 of α-toxin on macrophages (A) The calculation of CC50 of α-toxin, using the results of different concentrations of α-toxin on macrophage (J774-A1) at 20 concentrations in serial dilution starting from 0.0003 ug mL−1. The percentage of cellular cytotoxicity, determined by MTS assay after treatment with the indicated doses of α-toxin after 1hr at 37°C. (B) IC50 of α-toxin, glucantime, α-toxin + glucantime and α-toxin (non-toxic dose)+ glucantime. The calculation of IC50 of glucantime and α-toxin using the results of different concentrations of glucantime or α-toxin on amastigote forms of L. major at 18 concentrations in serial dilution starting from 0.001 ug/mL. Moreover, for calculation of IC50 of glucantime + α-toxin, using the results of different concentrations of glucantime (0.001–200 ug mL−1) along with non-cytotoxic concentration of α-toxin (0.003 ug mL−1) on amastigote forms of L. major. The percentage of cellular viability, measured by counting the number of amastigotes in each macrophage by examining 100 macrophages on each coverslip and comparing them with those obtained in positive control with light microscope after 48 h incubation at 37°C, 5% CO2. |
The results of the mean number of amastigotes in each macrophage showed that glucantime plus the non-cytotoxic concentration of α-toxin (0.003 µg/mL) efficiently reduced the number of amastigotes forms of L. major in each macrophage in all concentration compared with glucantime or α-toxin alone (p < 0.05). The concentration of glucantime plus α-toxin, necessary to reduce by 50% of survival index was 0.004 µg/mL ± 0.002 that is indicated more powerful anti-leishmanial effects in the amastigotes form of L. major when compared with glucantime (IC50 = 4.27 µg/mL ± 0.55) alone. In addition, results demonstrated that α-toxin (IC50 = 0.008 µg/mL ± 0.001) alone induced more anti-amastigote effect than glucantime alone (Figure 3B).
In vivo Assessment
The ulcers size was measured on day zero and during 3 weeks of the treatment course. The repeated-observations ANOVA test showed that both the time (p < 0.001) and the different treatment regimens (p = 0.001) had significant differences in the size of the ulcers. As seen in Table 2, over time, the ulcer sizes have changed, but these changes have not been the same in four groups. A significant reduction in lesion size was observed in groups of α-toxin, glucantime, and glucantime plus α-toxin, which was the smallest in the last group (p < 0.001). There were no significant differences in treatment responses between the two first groups. In the negative control group, with time, the ulcer size has increased significantly in comparison with the three test groups.
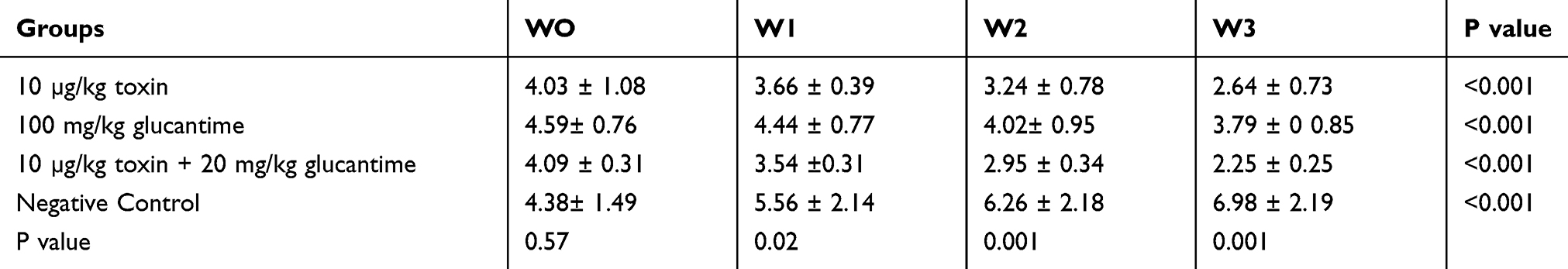 |
Table 2 Frequency Distribution of the Mean Size ± SD of the Ulcers in Different Groups (mm) |
The One Way ANOVA test showed that before the treatment course, the mean size of the ulcers was not significantly different among the four groups (p = 0.57). Still, it became significantly different at the end of the first, second and third weeks of treatment course (p=0.02, p<0.001 and p<0.001, respectively) (Table 2, Figure 4).
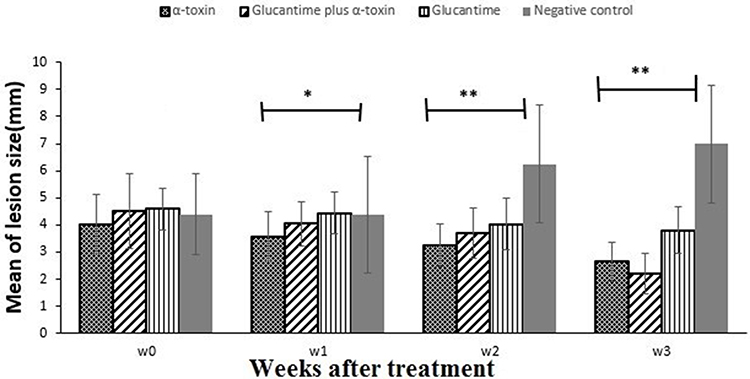 |
Figure 4 The mean size of lesions (mm) in control and test groups weekly. Diagrams show the therapeutic effect of treatment with α-toxin and glucantime on lesions due to infection of BALB/c mice L. major. Mice were infected with 1× 106 stationary phase of L. major promastigotes in the tail base. After 21 days post-infection, the treatment course was started. Values show the mean lesion size ±SD of 10 mice per group (“*”shows significant difference (p < 0.05) and “**” shows significant difference (p < 0.001) among control and all other test groups). |
Parasite Burden Assessment
At the end of the treatment course, the parasite burden was determined via titration method. The results showed significant differences in parasite burden of the spleen (p < 0.001) and liver (p < 0.001) among the groups exposed to different therapy protocols. There were significant differences in the parasite burden of all treated groups with negative control (Table 3 and Figure 5).
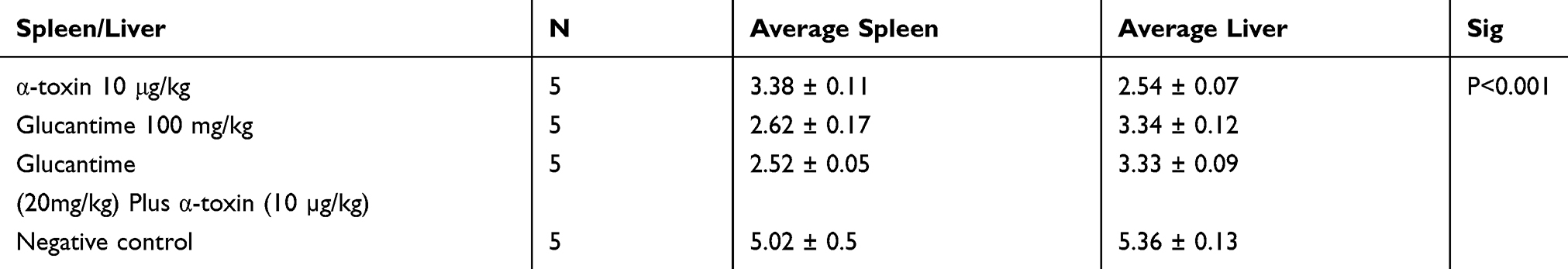 |
Table 3 The Mean Burden of Parasite in the Spleen/Liver of BALB/c Mice in Different Therapy Groups |
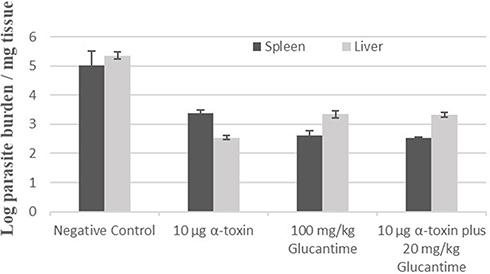 |
Figure 5 The mean burden of the parasite in the spleen and liver of BALB/c mice of different groups. After the end of the treatment course, parasite numbers recovered from spleen and liver were evaluated by limiting-dilution assay. The error bars represent standard deviations (SD) of the mean. |
Discussion
The α-toxin is a secretory pore-forming exotoxin of C. septicum responsible for muscle necrosis.9 Glycosylphosphatidylinositols (GPIs) in the protozoan L.major can be detected by α-toxin. The α-toxin by inducing small ion-selective permeability and rapid K+ efflux binding to L. major gp63 and GPIs and inject into the plasma causing hemolysis and cellular necrosis.4
Recombinant DNA technology luckily provided resources that can play a significant role in the use of products manufactured by this technique alone or in conjunction with drugs of choice when treating serious illnesses. One of the essential products produced by this method is some of the desired bacterial toxins which can be removed the cell barriers to the definitive impact of the drugs of choice and facilitate an effective drug delivery system. It has enabled researchers to treat infections like leishmaniasis, which could not be cured using traditional methods.22 Since codon optimization could be adopted as an excellent method to increase production yield,23 in this study pET28a plasmid includes the construction of the codon-optimized synthetic sequence of α-toxin was provided and transformed into BL21 (DE3) cells. Whereas the recombinant pET28a-α-toxin plasmid can be lethal to E. coli at high levels, inducing α-toxin expression with IPTG did not continue more than 2 h. The molecular weight of the designed construct was estimated at around 48 kDa, as SDS-PAGE and Western blot confirmed the specific 48 kDa protein band.
Despite codon optimization in our study, the expression level of α-toxin protein was measured by about 1 mg/mL. This amount was relatively low, and this defect was resolved by further experiments so that further optimization of expression and culture conditions significantly improved protein production.
So far, several studies have been carried out on recombinant α-toxin production. For example, in 2006, Xu et al colonized the α-toxin gene of C. perfringens type A in the Pgem-T vector and expressed optimally in E. coli. Also, immunization in a mouse model was obtained using this recombinant protein.24 In 2007, Inoue et al showed the α-toxin gene of C. perfringens in E. coli.25 In the same year, Zhang et al cloned the α-toxin gene of C. septicum in an expression vector of pQE30 and expressed in E. coli. They also produced a toxoid vaccine by adding 0.3% formaldehyde into expressed α-toxin and showed that the recombinant strain M15 (pQE30-alpha) could be as a candidate to provide a protective immune response against C. septicum infection.26
Some studies have been conducted about α-toxin’s lethal effects on cells and various infectious agents. For example, Gordon et al (1999) described the fatal effect of α-toxin on Chinese hamster ovary cells (CHO) transfected with GPI-anchored folate receptors.11 Wichroski et al investigated the effect of intracellular morphology from α-toxin on unicellular protozoan cells of the Toxoplasma gondii parasite.13 They observed that toxin targets members of the SAG family of GPI-anchored surface proteins and toxin-treated cells showed the plasma membrane wrinkle along with swelling and fragmentation of the endoplasmic reticulum.27 In another study by Podešvová et al using snake venom a “suicidal” program developed in L. mexicana. This mechanism is based on the expression and (de)stabilization of a simple phospholipase A2 toxin from the Bothrops pauloensis snake venom, resulting in inducible cell death in vitro of the parasites. The suicidal strain during macrophage infection was strongly attenuated. The authors stated that such a deliberately attenuated parasite may be used to vaccinate the host, because its viability is controlled by toxin stabilization, resulting in a significantly reduced pathogenesis.28
Evaluation of α-toxin IC50 in our study showed that there is a significant difference between the mean number of amastigotes from 100 macrophages in groups exposed to α-toxin plus glucantime in comparison with negative control. As the strong fatal effect of the two mentioned components on cultivated Leishmania parasites was found at lower optimal concentrations (0.03 ± 0.002 µg/mL) compared with glucantime plus non–toxic α-toxin or each of them alone. The research of Zheng et al in 2005, on live parasites, showed that α-toxin binds to gp63 and GPI from L. major and has a fatal effect at a concentration of 0.77 ng (50%).4 These results are reminiscent of the effect of α-toxin on Toxoplasma gondii, which also has a high concentration of GPI-anchored proteins in its plasma membrane. As Wichroski study results showed that tachyzoites of Toxoplasma were sensitive to low concentrations of α-toxin (50% effective concentration [EC50] of 9 ng/mL [0.2 nM]; EC99 of 135 ng/mL [3 nM]).13
The results of in vivo study of the healing effect of α-toxin on mouse CL was caused by L. major showed that using α-toxin as a synergistic agent plus glucantime could significant therapeutic implications. As in the control group, the mean of the wound area was higher than the group receiving α-toxin and glucantime. It seems that α-toxin has an impressive effect on the cytoplasm of macrophages and even the parasites. On the other hand, because of the biological properties of the α-toxin, including the ability to create pores in the cell wall, this toxin may play an extraordinary role in the transfer of drugs, especially in the infections that have intracellular agents and the drug delivery system has a vital role in the treatment of such diseases. So the decisive results of healing of the CL ulcers in the mice receiving glucantime plus α-toxin can emphasize the synergistic role of α-toxin with the selected drug in the recovery of CL. Furthermore, the mice without treatment developed larger lesions throughout the treatment course, as with time, the diameter of the ulcers continued to increase, but the lesion size of the other three groups, especially the group of glucantime plus α-toxin diminished.
In this way, another study was aimed to design a different method of drug delivery for increased transfer of the choice drug (meglumine antimoniate) within the host cells. Therefore, listeriolysin O (LLO), a bacterial product which is a member of pore-forming peptides was produced recombinant and used as an enhancer factor with meglumine antimoniate in order to facilitate the transition of the drug in an in vitro model of macrophage-L. major amastigote infection. The results showed a combination of pore-forming proteins with anti-leishmanial agents could increase the killing effect of the medicinal agents.29 Also, there was a significant difference in the parasitic burden of the spleen and liver of the control group versus the group receiving α-toxin plus glucantime. Thus, mice treated with the α-toxin plus glucantime showed significantly higher resistance to infection compared with the other groups.
Concerning, the lowest effective dose of the toxin (without any side effects) is an essential medical aid, in this study, the optimal dose of the toxin was determined 10 µg/kg after doing the basic plots on different mouse groups. Also, Tweten et al (2001) showed that intravenous injection of α-toxin (10µg/Kg) in rats and mice resulted in shock and death.27 They suggested that such studies on α-toxin could develop a new generation of drugs that could have synergistic effects with glucantime in the treatment of CL.
Conclusion
In this study, expression and purification of C. septicum α-toxin were successfully done. Also, the anti-Leishmania effect of α-toxin plus glucantime was evaluated in vitro and in vivo. The results showed the most effective way to control the proliferation and eliminate of parasites is to use a combination of glucantime with α-toxin. Despite the popular activity for recombinant α-toxin, more efforts should be carried out on the characterization of α-toxin to obtain a realistic view of valuable data on the expression and antimicrobial properties of it and this activity should be quantified compared to the conventional treatment protocols. It is hoped that this achievement could be used as an experimental model for drug development studies and generating better alternatives to common cures in human cases.
Abbreviations
C. septicum, Clostridium septicum; DRMs, detergent-resistant membranes; GPIs, glycosylphosphatidylinositols; CHO, Chinese hamster ovary cells; CDS, coding sequence; TMB, tetramethyl benzidine.
Acknowledgments
This study was conducted at the Department of Parasitology and Mycology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran, with the grant awarded by the Research Vice-Presidency from Isfahan University of Medical Sciences, Isfahan, Iran. The results presented in this article are part of a Ph.D. thesis under the last author’s supervision.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests in this work.
References
1. Sharma U, Singh S Immunobiology of leishmaniasis. 2009.
2. Sacks D, Kamhawi S. Molecular aspects of parasite-vector and vector-host interactions in leishmaniasis. Ann Rev Microbiol. 2001;55(1):453–483. doi:10.1146/annurev.micro.55.1.453
3. Singh N, Kumar M, Singh RK. Leishmaniasis: current status of available drugs and new potential drug targets. Asian Pac J Trop Med. 2012;5(6):485–497. doi:10.1016/S1995-7645(12)60084-4
4. Zheng Z, Tweten RK, Mensa-Wilmot K. Intracellular glycosylphosphatidylinositols accumulate on endosomes: toxicity of alpha-toxin to Leishmania major. Eukaryot Cell. 2005;4(3):556–566. doi:10.1128/EC.4.3.556-566.2005
5. Tellez G, Pumford NR, Morgan MJ, Wolfenden AD, Hargis BM. Evidence for Clostridium septicum as a primary cause of cellulitis in commercial turkeys. J Veterinary Diagn Invest. 2009;21(3):374–377. doi:10.1177/104063870902100313
6. Liechti M, Schöb O, Kacl G, Caduff B. Clostridium septicum aortitis in a patient with colon carcinoma. Eur J Clin Microbiol Infect Dis. 2003;22(10):632–634. doi:10.1007/s10096-003-1009-0
7. Koransky JR, Stargel MD, Dowell V
8. Ballard J, Bryant A, Stevens D, Tweten R. Purification and characterization of the lethal toxin (alpha-toxin) of Clostridium septicum. Infect Immun. 1992;60(3):784–790. doi:10.1128/IAI.60.3.784-790.1992
9. Hickey MJ, Kwan RY, Awad MM, et al. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008;4(4):e1000045. doi:10.1371/journal.ppat.1000045
10. Melton-Witt JA, Bentsen LM, Tweten RK. Identification of functional domains of Clostridium septicum alpha toxin. Biochemistry. 2006;45(48):14347–14354. doi:10.1021/bi061334p
11. Gordon VM, Benz R, Fujii K, Leppla SH, Tweten RK. Clostridium septicum alpha-toxin is proteolytically activated by furin. Infect Immun. 1997;65(10):4130–4134. doi:10.1128/IAI.65.10.4130-4134.1997
12. Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK. The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J Biol Chem. 2004;279(14):14315–14322. doi:10.1074/jbc.M313758200
13. Wichroski MJ, Melton JA, Donahue CG, Tweten RK, Ward GE. Clostridium septicum alpha-toxin is active against the parasitic protozoan Toxoplasma gondii and targets members of the SAG family of glycosylphosphatidylinositol-anchored surface proteins. Infect Immun. 2002;70(8):4353–4361. doi:10.1128/IAI.70.8.4353-4361.2002
14. Sellman BR, Kagan BL, Tweten RK. Generation of a membrane‐bound, oligomerized pre‐pore complex is necessary for pore formation by Clostridium septicum alpha toxin. Mol Microbiol. 1997;23(3):551–558. doi:10.1046/j.1365-2958.1997.d01-1876.x
15. Knapp O, Maier E, Mkaddem SB, et al. Clostridium septicum alpha-toxin forms pores and induces rapid cell necrosis. Toxicon. 2010;55(1):61–72. doi:10.1016/j.toxicon.2009.06.037
16. Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol. 2001;12(2):195–201. doi:10.1016/S0958-1669(00)00199-3
17. Li Y, Li X, Wang G. Cloning, expression, isotope labeling, and purification of human antimicrobial peptide LL-37 in Escherichia coli for NMR studies. Protein Expr Purif. 2006;47(2):498–505. doi:10.1016/j.pep.2005.10.022
18. Das S, Chatterjee N, Bose D, et al. Lipid isolated from a Leishmania donovani strain reduces Escherichia coli induced sepsis in mice through inhibition of inflammatory responses. Mediators Inflamm. 2014;2014.
19. Uk L. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(259):680–685. doi:10.1038/227680a0
20. Sanbrook J, Fritsch E, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989.
21. Morais-Teixeira E, Damasceno QS, Galuppo MK, Romanha AJ, Rabello A. The in vitro leishmanicidal activity of hexadecylphosphocholine (miltefosine) against four medically relevant Leishmania species of Brazil. Mem Inst Oswaldo Cruz. 2011;106(4):475–478. doi:10.1590/S0074-02762011000400015
22. Taheri T, Rafati S. Leishmaniasis: recombinant DNA vaccination and different approaches for vaccine development. Clin Investig (Lond). 2013;3(11):1023–1044. doi:10.4155/cli.13.99
23. Yadava A, Ockenhouse CF. Effect of codon optimization on expression levels of a functionally folded malaria vaccine candidate in prokaryotic and eukaryotic expression systems. Infect Immun. 2003;71(9):4961–4969. doi:10.1128/IAI.71.9.4961-4969.2003
24. Xu C, Xu C, Zhao Z. Expression of alpha-toxin gene of Clostridium perfringens type A and its primary immunological protective function. Wei Sheng Wu Xue Bao= Acta Microbiologica Sinica. 2006;46(4):624–628.
25. Inoue M, Kikuchi M, Komoriya T, Kohno H. Cloning of Clostridium perfringens alpha-toxin gene and extracellular expression in Escherichia coli. Rinsho Biseibutsu Jinsoku Shindan Kenkyukai Shi= JARMAM: Journal of the Association for Rapid Method and Automation in Microbiology. 2007;18(2):127–135.
26. Zhang Y, Bian Y, Zhao B. The study on the cloning and expression of alpha toxin gene of Clostridium septicum and the immunity of the toxoid. Sheng Wu Gong Cheng Xue Bao= Chinese Journal of Biotechnology. 2007;23(1):67–72.
27. Tweten RK. Clostridium perfringens beta toxin and Clostridium septicum alpha toxin: their mechanisms and possible role in pathogenesis. Vet Microbiol. 2001;82(1):1–9. doi:10.1016/S0378-1135(01)00372-8
28. Podešvová L, Leštinová T, Horáková E, Lukeš J, Volf P, Yurchenko V. Suicidal Leishmania. Pathogens. 2020;9(2):79. doi:10.3390/pathogens9020079
29. Mirzaei F, Khanahmad H, Namdar F, Izadi S, Hejazi SH. A novel strategy for enhance potentiation of meglumine anti-moniate against leishmania major in vitro. Iran J Parasitol. 2019;14(4):542–551.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.