")
Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 13
Evaluation of Platelet Activation by HIV Protease Inhibitors – The HIV-PLA II Study
Authors Kann G, Owasil J, Kuczka K, Haberl A, Wolf T, Khaykin P, Harder S, Stephan C , von Hentig N
Received 26 May 2020
Accepted for publication 13 November 2020
Published 6 August 2021 Volume 2021:13 Pages 789—800
DOI https://doi.org/10.2147/HIV.S262282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bassel Sawaya
Gerrit Kann,1,* Junaid Owasil,1,* Karina Kuczka,2 Annette Haberl,1 Timo Wolf,1 Pavel Khaykin,1 Sebastian Harder,2 Christoph Stephan,1 Nils von Hentig1
1HIVCENTER, Medical HIV Treatment and Research Unit, Johann Wolfgang Goethe University Frankfurt, Frankfurt am Main, Germany; 2Pharmazentrum Frankfurt, Institute of Clinical Pharmacology, Johann Wolfgang Goethe University Frankfurt, Frankfurt am Main, Germany
*These authors contributed equally to this work
Correspondence: Nils von Hentig
HIVCENTER, Medical HIV Treatment and Research Unit, Johann Wolfgang Goethe University, Theodor-Stern-Kai 7, Frankfurt am Main, 60590, Germany
Tel +49-69-63017680
Fax +49-69-630183442
Email [email protected]
Background: In the past, protease inhibitors (PIs) and the reverse transcriptase inhibitor abacavir were identified increasing the risk for thromboembolic complications and cardiovascular events (CVE) of HIV infected patients taking a combination antiretroviral therapy (cART). Results of the previous HIV-PLA I-study lead to the assumption that platelet activation could play a substantial role in increasing CVE risks.
Methods: The open label, monocentric HIV-PLA II-study investigated HIV-1-infected, therapy-naïve adults (n=45) starting with cART, consisting either of boosted PI (atazanavir, n= 6, darunavir, n=11), NNRTI (efavirenz, n=14) or integrase inhibitor (raltegravir, n=14), each plus tenofovir/emtricitabine co-medication. Main exclusion criteria were tobacco smoking, the intake of NSAIDs or abacavir or past CVE. Platelet adhesive molecule p-selectin (CD62P) and FITC anti-human Integrin α-IIb/Integrin β-3 (CD41/CD61) antibody (PAC-1) binding, monocyte CD11b/monocyte-associated CD41 expression and the endogenous thrombin potential (ETP) were assessed ex vivo-in vitro at baseline, weeks 4, 12 and 24. Therapy regimens were blinded to the investigators for laboratory and statistical analyses.
Results: CD11b and ETP showed no significant changes or differences between all study groups. In contrast, the mean + SD mean fluorescence units (MFI) of CD62P and PAC-1 increased significantly in patients taking PI, indicating an enhanced potential for thrombocyte activation and aggregation.
Conclusion: CD62P expression, detecting the ɑ-platelet degranulation of pro-inflammatory and pro-thrombotic factors and adhesive proteins, and PAC-1 expression, representing a marker for conformation changes of the GIIb/IIIa receptor, increased significantly in patients taking HIV protease inhibitors. The findings of this study revealed a yet unknown pathway of platelet activation, possibly contributing to the increased risk for CVE under HIV protease inhibitor containing cART.
Clinical Trial Registration No.: DRKS00000288.
Keywords: HIV protease inhibitors, platelets, leucocytes, PAC-1, GIIb/IIIa-receptor
Introduction
In the past, protease inhibitors (PIs) and the reverse transcriptase inhibitor abacavir were identified increasing the risk for thromboembolic complications and cardiovascular events (CVE) of HIV infected patients taking a combination antiretroviral therapy (cART). Even after adjusting statistical analyses for CVE risk factors of the endothelial side, ie smoking, hyperlipidemia, age etc., the reasons remained unknown. Results of the previous HIV-PLA I-study lead to the assumption that platelet activation could play a yet unknown role in increasing CVE risks in patients taking cART.
HIV Infection and Platelets
The possible relevance of platelets for propagating the inflammatory process is increasingly recognized. A constituent of the α-granule membrane CD62P or sP-selectin is expressed on the platelet surface upon activation. CD62P though is capable to interact with different partners such as neutrophils and monocytes and its ligand P-selectin glycoprotein ligand-1 (PSGL-1). This results in the formation of platelet-leukocytes aggregates (PLA)1 and an upregulation of the MAC-1 endothelial cell ligand (CD11b/CD18) leading to interaction of the partners. The formation of PLA increases on the surface of diseased vessels throughout lesion development, thus promoting the progression of atherosclerosis.3,4
The HIV infection itself is known to trigger thrombocytopenia.5 On the other hand, HIV-patients show an enhanced platelet activation with increased CD62P-expression and release of chemokines (eg, RANTES) from the thrombocyte granules.16 Also, a platelet-independently enhanced activation of pro-coagulation factors such as D-dimer and thromboembolic complications in HIV patients have been described.7,8 Furthermore, circulating immune complexes in HIV-infected patients include antibodies against Glycoprotein IIb/IIIa determining the extent of the thrombocytopenia.
In the course of thromboembolic events circulating thrombocytes are being activated eg by collagen as a component of the sub-endothelial matrix (pro-aggregation stimulus). Furthermore, CD62, so-called P-Selectin, as a member of platelet alpha-granule is being expressed on the platelets surface representing a neo-antigen. CD62 mediates thrombocyte–leucocyte interaction. The correspondent PSGL-1 (P-selectin glycoprotein ligand-1) is expressed on leucocytes’ surfaces triggering an inflammatory answer.2–4
The growth of smooth muscle tissue in terms of vascular remodelling is initiated by growth factors of thrombocytes. The inflammatory reaction is sustained by both the forming of platelet-leucocyte aggregates and consecutively the release of leukocyte mediators as well as up-regulation of adhesive receptors on leucocytes’ surfaces, namely MAC-1 (Macrophage-1-antigen), and those of endothelium cells, ICAM-1 (Intercellular Cell Adhesion Molecule).5–12
Usually, the reduction of the viral load under antiretroviral therapy (ART) goes along with a decrease of thrombocytopenia9 and is able to at least decrease the release of RANTES, and of further endothelium activation markers (eg, von-Willebrand Factor) and D-dimer.
cART and Platelets
But also cART itself may trigger cardiovascular risk in patients on therapy. Data from the D:A:D-study have shown very early an independent risk for CVE under cART even after adjustment for most of the CVE cofactors, a signal that exists constantly through to 2016. The same counts for CVE data from the INSIGHT Smart-study,13,14 especially when PI and Abacavir15–17 were taken.
While the endothelial part of this complication has been discussed sufficiently,10,11 and recent publications again have shown the coincidence of enhanced highly soluble c-reactive protein (hsCRP), interleukin-6 (IL-6), D-dimer and CVE risk,18–24 the cellular side of atherosclerotic inflammation has been neglected, and data regarding PLA-formation, CD62-expression and platelet-induced thrombin generation in HIV-patients under cART are scarce.
Likewise, the coagulatory reaction is maintained. On the one hand leucocyte adhesion on the damaged endothelial surface induces the release of proteolytic enzymes and toxic substances leading to further uncovering of pro-aggregatory molecules. On the other hand, tissue factor present in subendothelial tissue and leukocytes contributes to coagulation by inducing thrombin generation by which thrombocyte activation and the thrombotic process is amplified.10,25 Thrombin cleaves the N-terminus of the fibrinogen alpha and beta chains to fibrin peptide A and B respectively. The hereby resulting fibrin monomers polymerize to form protofibrils, which associate laterally to form fibrin fibres; finally a thrombus is formed.26 According to Hemker et al coagulation already starts when only 5% of the fibrinogen has been converted. At this point common coagulation diagnostic techniques apply – such as prothrombin Time (PT) and partial thromboplastin time (PTT). While these routine diagnostic tests measure the time until a clot is formed they seem not to be neither suitable to point out an increased coagulability nor sensitive enough to detect minor bleeding diathesis. A testing system that records the potential of the thrombin is Hemker’s thrombin generation assay which is part of this work.27,28
The HIV-PLA2 Study
Thrombocytes are part of a reaction chain that leads to cardiovascular events and progress of arteriosclerotic diseases. The thrombocyte–leucocyte interaction contributes to vascular inflammatory reactions. These issues are objectives of this study on 45 HIV-1 infected adult outpatients, who started antiretroviral therapy (ART); study visits were performed at the beginning of therapy (visit 1) and throughout 24 weeks (visits 2–4) of treatment. The participants were divided into three groups, treated with either protease inhibitors (PIs, n=17) or non-nucleoside reverse-transcriptase inhibitors (NNRTIs, n=14) or integrase inhibitors (INI, n=14).
Following the protocol and the methods of the HIV-PLA1 pilot study,29 we evaluated markers for thrombocyte activation (CD 62P and PAC-1-binding), markers for leucocyte and platelet–monocyte interaction (CD11b, and CD41), and the influence of platelets on coagulation (endogenous thrombin potential, ETP) in 45 adult HIV-1 infected patients, naïve to therapy, before and 4, 12 and 24 weeks after the initiation of cART.
Methods
Patients and Study Protocol
The study had been approved by the Medical Faculty Ethics Review Board of the Johann Wolfgang Goethe University Frankfurt am Main, Germany (No. 290/09) and was conducted following the Declaration of Helsinki (version 2013) for medical research involving human subjects. Patients had provided written informed consent prior to enrolment. The study is registered in the German Freiburg Study Base.
Blood samples were drawn from therapy-naïve patients immediately before starting a first line therapy with either HIV protease inhibitors (PI, darunavir 600mg BID, n=11; atazanavir 300mg QD, n=6), non-nucleoside reverse transcriptase inhibitors (NNRTI, efavirenz 600mg QD, n=14) or integrase inhibitors (INI, Raltegravir 400mg BID, n=14) together with a standardized backbone of emtricitabine/tenofovir-disoproxil fumarate (FTC/TDF) 200/245mg.
Baseline lab parameters obtained were RBC, WBC, platelet count, haemoglobin, AST, ALT, γGT and creatinine. Furthermore, the expression of CD62P, PAC-1-binding, PLA formation (via CD41-positive monocytes), CD11b expression and thrombin generation (endogenous thrombin potential, ETP) were determined. After 4, 12 and 24 weeks on therapy, all analyses described above were repeated.
Therapy regimens were blinded to the pharmacological investigators for laboratory and statistical analyses.
Patients who took medication known to influence platelet aggregation, eg NSAIDs, or who reported cigarette or tobacco smoking or had a CVE in their history were not included in this study. Also, abacavir was not allowed as part of the cART.30,31
Laboratory
Citrated whole blood (3.13%, Sarstedt, Nürmbrecht, Germany) was assessed from patients and transferred to the laboratory within 20 minutes avoiding agitation and subsequent platelet activation. The Appendix provides a summary of publications regarding the applied laboratory methods.
Flow Cytometric Methods
Preparation of blood samples was adapted from a consensus protocol1 and modified for assessment of PLA-formation2 and leukocyte activation. Samples were analyzed on a FACS (Beckman Coulter, Krefeld, Germany). Staining and acquisition protocols were adopted for measurement of platelet (CD62P, PAC-1, CD41) and monocyte (CD11b, CD14) activation markers and formation of PLAs (CD41 bearing leukocytes) using double- and triple-color immunofluorescence staining. All antibodies were from Beckman Coulter (Miami, FL, USA) except PAC-1 and its isotype control which were from BD Pharmingen (San Diego, CA, USA). All incubations were performed at room temperature in the dark for 10 min.
Equal amounts of whole blood and HEPES buffer (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; AppliChem, Darmstadt, Germany) were mixed. For platelet activation, thrombin receptor activating peptide (TRAP-6, H-Ser-Phe-Leu-Leu-Arg-Asn-OH, Bachem, Heidelberg, Germany) at a final concentration of 5 (for platelet testing) or 50 µM (leucocytes, PLA forming) was added and incubated for 10 min; in addition aqua bi-dest was added for testing in non-activated samples. The concentrations of these inducers had been chosen since they are intermediate and sensitive enough to indicate drug-induced changes of platelet activation in both directions eg activation by the immunosuppressant as well as inhibition by antiplatelet therapy.3
Expression of CD 62P and PAC-1
In the flow cytometric determination of platelet activation status we focused on parameters that represented different platelet functions: CD62-expression is a marker of platelet degranulation, whereas PAC-1 reflects the activation induced conformational change of the GPIIb/IIIa receptor, leading to fibrinogen binding and subsequent aggregation.
For analysis of platelet CD62P and PAC-1 expression, 5µL of activated or non-activated pre-diluted samples was incubated with saturating concentrations of anti-CD41-PC5, anti-CD62P-PE and PAC-1-FITC. The reaction was stopped by fixing the cells for 30 min with 1% formaldehyde. Samples were further diluted, washed and analysed on a flow cytometer. Platelets were defined as CD41 positive and within the platelet cluster in a log side-scatter (SSC) versus log CD41-PC5 dot plot. Data on 3000 platelets respectively events of measurement were acquired from each sample. Both, the percentage of platelets expressing CD62P and the mean of CD62 positive events were analysed and corrected for nonspecific binding of equimolar concentrations of mouse IgG isotype conjugates. For estimation of PAC-1 expression, CD41-PC5 positive platelet population was analysed with respect to PAC-1 positive events, given as mean values. All mean values refer to logarithmic data.
PLA Formation and CD11b Expression
Adhesion of leukocytes to platelets leads to the formation of platelet-leukocyte aggregates (PLA). These are deposited at sites of vascular injury and represent an important mechanism by which leukocytes contribute to thrombosis and platelets contribute to inflammation.4,5,32 CD 41 represents the platelet mass attached to monocytes and neutrophils; CD 11b is an activation marker for the monocytes, as it refers to phagocytosis, but is expressed on natural killer cells and neutrophils as well. Functionally, CD11b regulates leukocyte adhesion and migration to mediate the inflammatory response; CD11b is directly involved in cellular adhesion. CD14 is expressed by monocytes and macrophages and at a considerably lesser extent by neutrophils, which helped in differentiating leucocyte subpopulations rather than using side scatter (SSC) alone.
For the measurement of leucocyte-platelet formation, 50µL of activated (TRAP) or non-activated samples were stained with saturating concentrations of anti-CD14-FITC, anti-CD11b-PE and anti-CD 41-PC5 for 10 minutes. Erythrocytes were lysed using FACSlysing solution (Becton Dickinson, San Jose, CA, USA). Analysis was carried out by acquisition and processing of data from 10,000 leucocytes (monocytes and neutrophil granulocytes).33
PLA-formation was estimated by gating on leukocyte subpopulations (identification of monocytes by CD14 positivity in a FSC versus log CD14-FITC dot plot). Percentages of monocytes and neutrophils with platelets attached were defined as those events in the respective gate which had an anti-CD41-PC5 fluorescence intensity above background level determined by IgG isotype control. In addition, CD41 positive events in each leucocyte sub-population were measured as the mean of its logarithmic scale.Furthermore, monocyte activation was analysed in the total monocyte population with respect to CD11b (mean of logarithmic data).
Measurement of Thrombin Generation
Platelet Rich Plasma was obtained according the subsequently described process: the whole blood was centrifuged (Megafuge 1.0 R; Heraeus/Hanau) 7 minutes at 1400 U/min (equates 609g), at 21°Celsius. No brake was used stopping this process in order to avoid further uncontrolled activation of platelets. The PRP was pipetted into a separate container (Polystyrol; Greiner/Pleidelsheim) and stored at room temperature (22–25°Celsius). The sample was further analysed within 120 minutes.
Eighty microliters of PRP and 20 µL of diluted thrombin generation trigger (Innovin®, Dade Behring, Marburg, Germany; collagen Horm® NYCOMED, Linz, Austria) were added to each well of a 96-well microtiter plate. The reaction was started by adding 20 µL of substrate solution containing 0.1 M CaCl2. The final concentrations therefore were: PRP, diluted 2:3, substrate 833µM, DMSO 1.67%, CaCl2 16.7 mM. The microtiter plate fluorometer used was the Fluoroskan Ascent Type 374 (Labsystems, Finland), with excitation wavelength 390 nm, emission wavelength 460 nm, and a measurement integration time per well of 20 ms. Fluorescence was measured in each well at 20 s interval a total of 200 times, which means that each run lasted 66 min. Four identical experiments were run in parallel, allowing 24 different experiments to be carried out simultaneously.
Data Management of ETP
The course of the fluorescent signal from each well is retrieved in an Excel spreadsheet. The mean of 4 parallel experiments is obtained as follows: The lag-time of thrombin formation is determined defined as the point where the fluorescence signal deviates from a horizontal (base-) line by more than two SDs [Hemker at al. 2000]. The lag-time is measured for each of the curves and the mean (± SD) calculated. The lag-times are superimposed and the fluorescence signal (mean ± SD) at each time point calculated. The time course of thrombin activity is obtained as the first derivative of the mean curve. Correction for the residual alpha 2-macroglobulin-thrombin activity was carried out using an algorithm published earlier,6 available at www.thrombin.com. The first derivative of the fluorescence–time curve reflects the course of thrombin activity in sample, analogous to the former chromogenic method. Parameters of interest are the integral of this curve (ETP-AUC) and the maximal generation rate which is the peak of the first derivative (ETP-peak).
Statistics
Patients´ baseline parameters were subject to descriptive statistics. The thrombocyte-leucocyte aggregation and activation markers were analyzed by means of repeated measures ANOVA, comparing baseline with week 4, 12 and 24 values, and comparing groups at each time-point, using SPSS®20.0 for Windows. Starting from the measured values of the HIV-PL I Study (Graff J, von Hentig et al 2008 JAC) we estimated a sample size of 13 patients in each group being satisfactory for proving a statistical significant difference between the groups at a level of significance of ɑ=0.05.
Results
Patients
Forty-five HIV-1 infected adults (female/male) aged between 28 and 62 years were enrolled to the study consecutively according to their attendance in the primary HIV care if meeting the inclusion criteria. All patients were therapy-naïve. Baseline demographic and laboratory parameters are shown in Table 1.
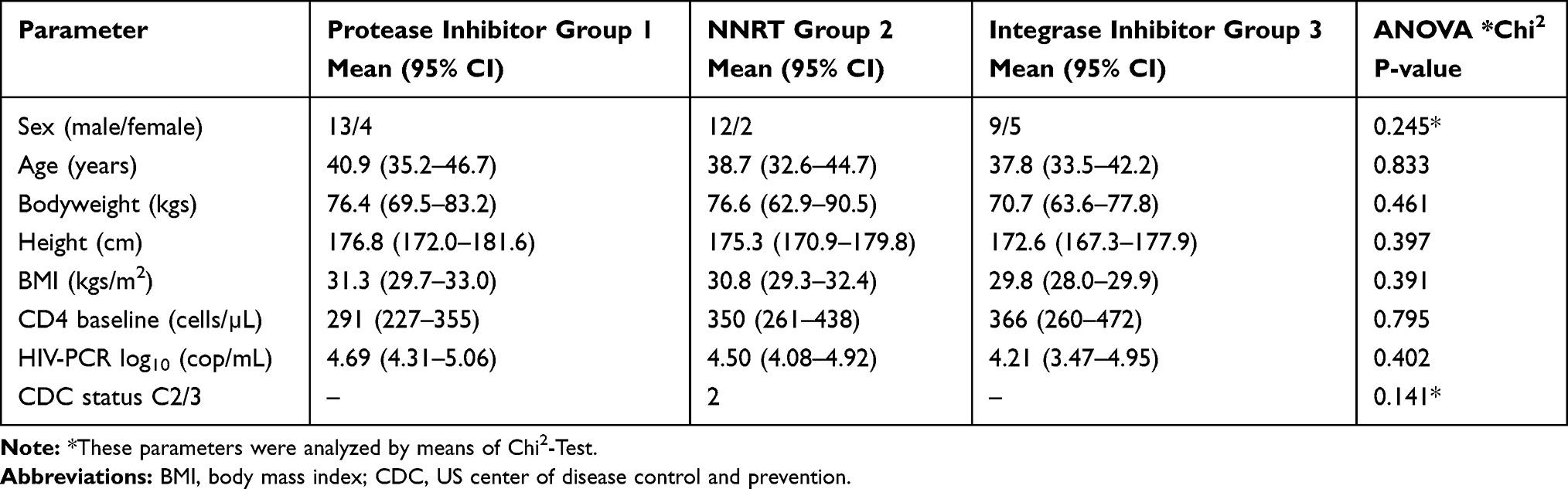 |
Table 1 Baseline Parameters of 45 Patients Starting with cART, Previously HIV Protease Inhibitor, NNRTI or Integrase Inhibitor Therapy-Naïve |
Seventeen patients initiated antiretroviral therapy with ritonavir 100mg-boosted HIV protease inhibitors at standard doses (darunavir 600mg BID, n=11; atazanavir 300mg QD, n=6), fifteen patients started with the NNRTI Efavirenz 600mg QD and thirteen patients received the INI raltegravir 400mg BID. The standardized backbone for all patients was emtricitabine/Tenofovir-DF 200/245mg.
Platelet and Monocyte Activation and Formation of Platelet-Monocyte Aggregates
After the initiation of ART, we could only detect significantly enhanced CD11b activity on platelets in patients taking HIV protease inhibitors at single time points after week 12 (CD11b_xmed_monos_bsl, P=0.005) and week 24 (CD11b_xmed_granulos_bsl, p=0.038), whereas CD41 activity on platelets was not enhanced. Also, the maximal thrombin generation rate (ETP-peak) did not show significant alterations, and the ETP time to peak decreased without showing statistical significance (Tables 2–4).
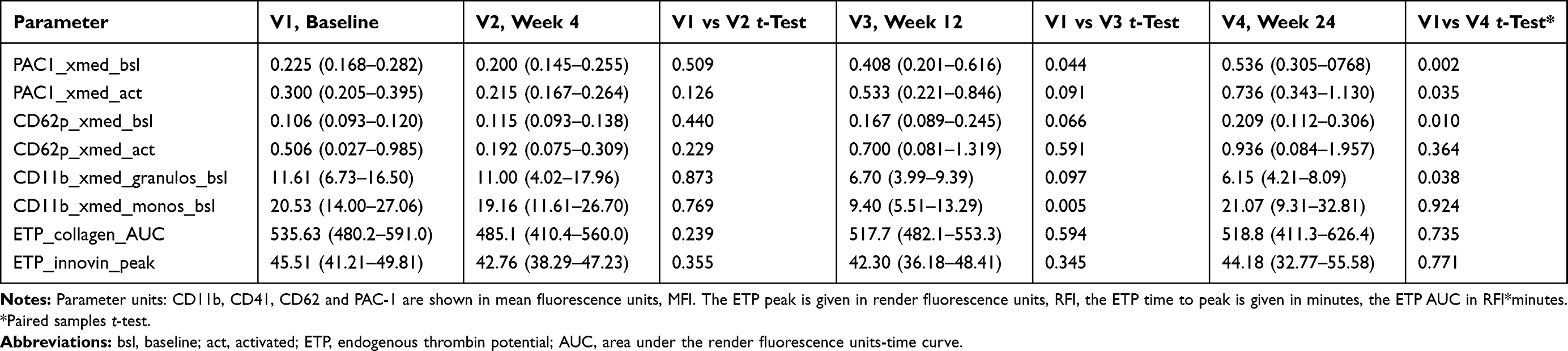 |
Table 2 Platelet-Leucocyte Adhesion, and Platelet and Leucocyte Activation Markers in Seventeen Adult HIV-1 Infected Patients Taking Low-Dose Ritonavir-Boosted HIV Protease Inhibitors (Darunavir 600mg BID, n=11; Atazanavir 300mg QD, n=6) Plus Emtricitabine/Tenofovir-DF 200/245, Before and 4, 12 and 24 Weeks After Therapy Onset. (Mean, 95% CI) |
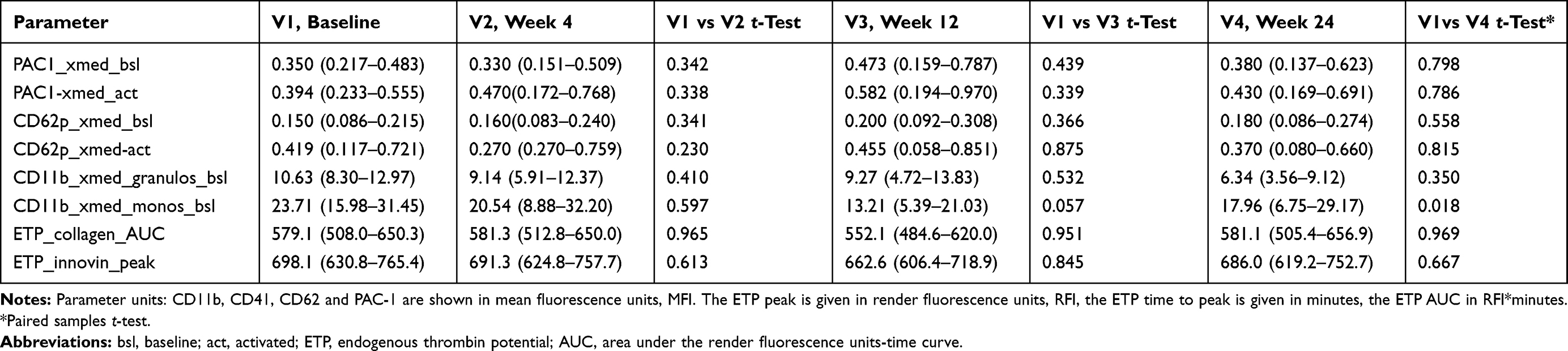 |
Table 3 Platelet-Leucocyte Adhesion, and Platelet and Leucocyte Activation Markers in Fourteen Adult HIV-1 Infected Patients Taking the Non-Nucleoside Reverse Transcriptase Inhibitor Efavirenz 600mg Plus Emtricitabine/Tenofovir-DF 200/245, Before and 4, 12 and 24 Weeks After Therapy Onset. (Mean, 95% CI) |
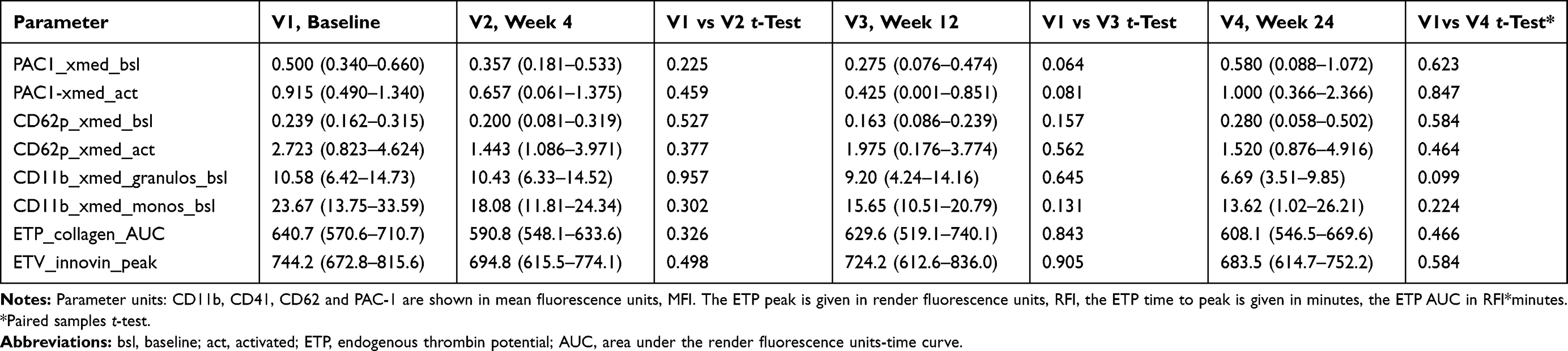 |
Table 4 Platelet-Leucocyte Adhesion, and Platelet and Leucocyte Activation Markers in Fourteen Adult HIV-1 Infected Patients Taking the HIV Integrase Inhibitor Raltegravir 400mg Plus Emtricitabine/Tenofovir-DF 200/245, Before and 4, 12 and 24 Weeks After Therapy Onset. (Mean, 95% CI) |
PAC1 and CD62p
Figure 1 shows the expected pre-activation of platelets in therapy-naïve HIV+ patients, which decreases in the first weeks of cART as previously described and then increases subsequently to week 24 in patients taking HIV protease inhibitors.
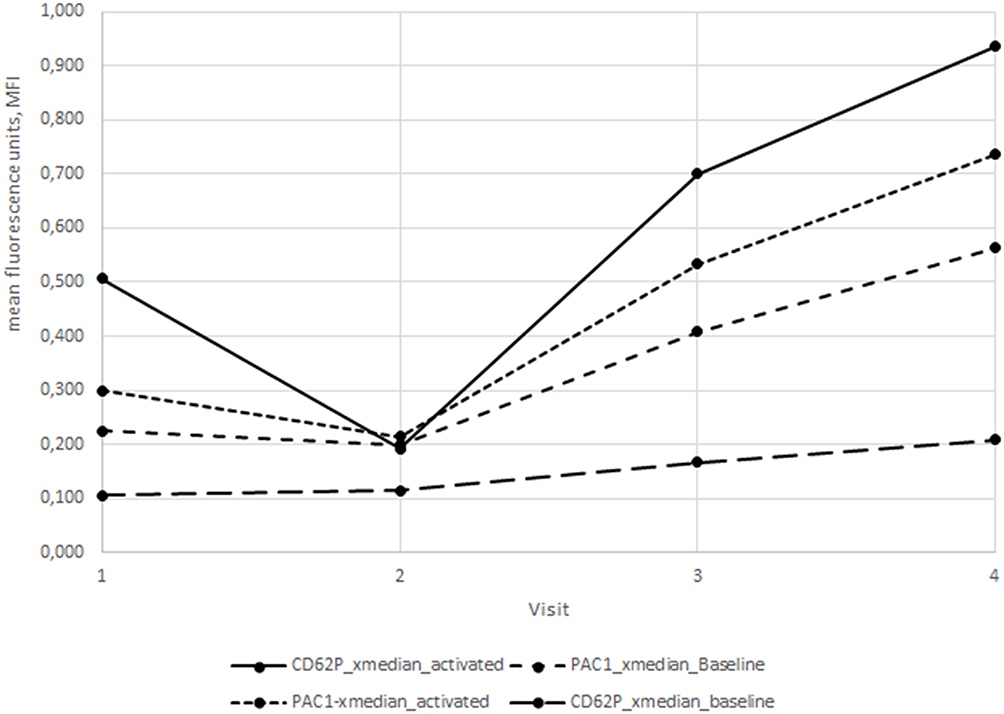 |
Figure 1 X-median PAC-1 and CD62p data from patients taking HIV protease inhibitors (n=17) at baseline (V1), week 4 (V2), week 12 (V3) and week 24 (V4) after initiation of cART. |
Subsequently, we could find significant changes in CD62p and PAC1 in the HIV protease inhibitor-group: Both parameters showed a significant increase at 12 and 24 weeks on therapy after an initial decrease a week 4 (Table 2, Figure 2A and B). CD62p changed from 0.106 (0.093–0.120) MFI at baseline to 0.209 (0.112–0.306) MFI at week 24 (p=0.010) and PAC1 increased from 0.225 (0.168–0.282) MFI at baseline to 0.536 (0.305–0768) MFI at week 24 (p=0.002), respectively. However, if visualized in whisker-box plots (Figure 3A and B), it becomes clear, that only 50% of patients show an increase in both parameters, while the 3rd and 4th quartile remain on their baseline values. A further analysis, trying to correlate any of the documented patient´s characteristics age, sex, weight, BMI, baseline CD4 cell count, baseline viral load could only detect a non-significant difference between these two groups regarding the baseline CD4 cell count, with a tendency towards higher baseline CD4 cell counts in the group with activated platelets (CD62P: 215 vs 319 cells/mm3, p=0.089; PAC1: 240 vs 315 cells/mm3, p=0.09).
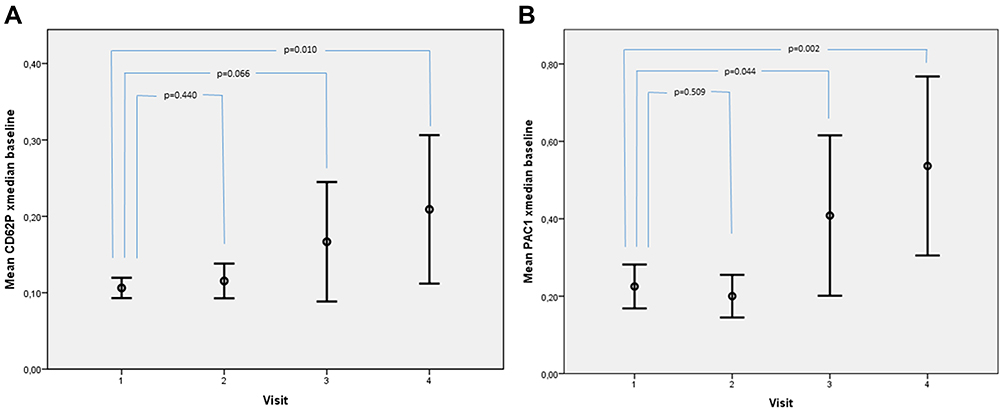 |
Figure 2 Mean (95% CI) PAC1 (A) and CD62p (B) MFI, when activated with collagen. The horizontal lines show the 95% confidence interval, circles represent the mean values. |
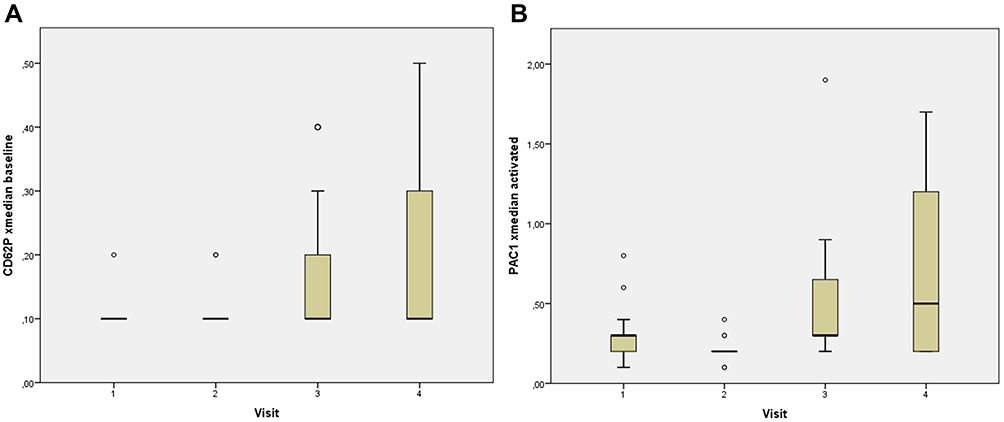 |
Figure 3 Whisker-box plots of PAC1 (A) and CD62p (B) MFI, when activated with collagen. The horizontal line shows the median, boxes represent the 2nd and 3rd quartile, whiskers represent the 1st and 4th quartile. Outliers are shown as circles. Abbreviation: MFI, mean fluorescence unit. |
Regression Analysis
A Pearson regression analysis could not detect apparent correlations between baseline patient´s characteristics and any of the measured variables. An initially significant correlation between CD62p and PAC1and baseline viral load (log10 copies/mL, p = 0.035, r2=0.28) or baseline CD4 cell count (cells/mm3, p=0.002, r2=0.35) could not be confirmed in a visual scatter plot.
Discussion
It was shown before that the platelet aggregation which was activated before therapy onset decreased after four weeks on therapy with the HIV protease inhibitor indinavir.6 ART could also decrease previously enhanced endothelial activation markers such as VCAM-1, ICAM-1 and vWF. However, a reduction of soluble CD62 (sP-selectin) as a marker for platelet activation could not be shown in the same study.14
Also, a number of publications gave a strong hint towards a decreased risk for CVE after therapy onset since HIV itself activates platelets and increases inflammation markers, eg hsCRP, IL-6 and D-dimer.18–22 This could be confirmed in our study for the evaluated platelet aggregation markers PAC1 and also for the conformal changes of the GIIb/IIIa-receptor. However, after 12 and 24 weeks on HIV protease inhibitors the ex vivo-in vitro samples showed again a significant increase in platelet activation. Also, the increase of CD11b at single time points without showing a general increase over time, might be a hint towards enhanced platelet activation, as CD11b is one marker for an increased formation of PLA aggregates in TRAP-activated samples.
Since platelet aggregation itself is considered a relatively crude marker for platelet activation, we used a more sensitive approach for platelet testing. In this study we found evidence for enhanced platelet activation during a 24 week-course antiretroviral therapy with HIV protease inhibitors by determining surface expression of activation markers and markers for the conformal changes of the GIIb/IIIa-receptor. These are (i) capable of initiating various inflammatory responses, eg the production of chemokines, cytokines or matrix metalloproteinases which promote the development and progression of atherosclerosis and (ii) direct thrombocyte aggregation.4
The study groups of patients receiving either NNRTI or INI-based cART did not show marked differences in all platelet activation markers compared to those who were on a HIV-protease inhibitor containing therapy.
It has been shown, that HIV infected subjects under PI-based cART have a cumulative risk of developing atherosclerosis, myocardial infarction and other thromboembolic events. As the reasons for this are supposed to be multifactorial, eg, a combination of the metabolic syndrome, hyperlipidemia and lipodystrophy,13,14,34–42 the increased platelet-leucocyte aggregation may further contribute to the genesis of cardiovascular diseases or the increasing incidence of cardiovascular events through years on therapy.13
Two studies of the INSIGHT-START study group presented at CROI 2017 evaluated the correlation between the initiation of cART and various parameters of inflammation coagulation and vascular injury and their clinical impact during the START trial. In this trial, altogether 468 adult HIV-1 positive patients with a baseline CD4-cell count of over 500/µL were randomized into two groups, one starting cART immediately after inclusion (n=2325), the other starting in delay with antiretrovirals, if the CD4-cell count dropped below 350/µL (n=2359). All patients were evaluated regarding the effect on the incidence of AIDS, AIDS-related death or severe AIDS-related illness on non-AIDS related death. The results were clearly remarkable: The authors detected a significant correlation between the increase of certain inflammation and coagulation markers such as D-dimer, IL6 or the D-dimer/IL6 score and the incidence of AIDS, AIDS-related mortality, non-AIDS-related mortality, tuberculosis and cardiovascular events. The authors conclude that there is a consistent relation between the increase of IL6 and D-dimer across all CD4-cell count strata and43,44 A Spanish group also presented new data on the correlation between abacavir plasma exposure and the incidence of thrombosis in a mouse-model: By inducing the formation of thrombus by application of 25micM FeCl3, the time to formation decreased significantly at abacavir plasma concentrations >5micg/mL. This animal study again confirms mechanistically the data of the earlier D:A:D-publication.30
Furthermore, Wouter et al investigated the impact of a therapy switch from a NNRT/PI-based regarding alterations in platelet activation, but could not find any correlation: Of 41 patients, 20 were switched to an INI-based regimen, while the remaining 21 patients continued their cART. Biomarkers measured representing an enhanced platelet activation according the authors were PF4, Beta-TG and hsCRP. However, the reason for a missing of correlations between cART and platelet activation in this study could be the fact that (1) a switch from NNRTI to Raltegravir does not show any alterations in platelet activation as NNRTI do not active platelet formation. And (2) it could be also the fact, that the authors did not measure more sensitive and direct markers for platelet activation such as CD40, CD41 or CD62P as done in our study.29
Finally, Ryom et al from the D:A:D-study group showed most recent data of this huge HIV cohort, confirming a correlation between the incidence of new cardiovascular events and the intake of HIV-second generation protease inhibitors, especially dolutegravir. Data from 2009 to 2016 revealed an increase in the 5-year risk of developing a CVE according to the D:A:D risk score, when taking dolutegravir of 59% (CI95% 1.33–1.91) other than with atazanavir (3%-increase of 5-year risk of CVE, CI95% 0.90–1.18). The CVE endpoint was adjusted for co-factors, such as age, sex, ethnic heritage, smoking, enhanced cholesterol and others and events were pooled from myocardial infarction, stroke, cardiac arrest or invasive cardiac interventions. However, the authors weren´t able to adjust for cofactors such as diabetes or the intake of comedication such as NSAIDs due to the data structure of this observational cohort. Nevertheless, these data confirm a relation between the intake of PI and CVE, although future investigations are warranted.45
Conclusion
Complementing the results of the previous pilot study, the currently presented results reveal strong evidence for a yet unknown enhancement of platelet activation by HIV protease inhibitors: the accelerated degranulation of inflammation markers from thrombocytes into blood plasma and conformal changes of the GIIb/IIIa-receptor most probably lead to enhanced thrombocyte aggregation under HIV protease inhibitor containing cART.
CD62P expression, detecting the ɑ-platelet degranulation of pro-inflammatory and pro-thrombotic factors and adhesive proteins, and PAC1 expression, representing a marker for conformation changes of the GbIIb/IIIa receptor, increased significantly in patients taking HIV protease inhibitors. This has not been evaluated by any of the previous studies discussed above revealing the pathway of platelet activation contributing to the increased risk for cardiovascular events under HIV protease inhibitor containing cART. Those detected changes in platelet activation deserve further attention.
The increase of PAC1 and CD62p in the visits 3 and 4 of this study in the upper 1st and 2nd quartile of patients did not show any statistically significant correlation to any of the documented patient´s characteristics and in this case deserves further evaluation for reasons which weren´t recorded in this study.
However, the limitations of this study are also obvious and therefore the results must be appraised with caution:
(1) As the number of participants in this study was small and the observation period comparably short, long-term evaluations of the detected effects in larger patient populations using standardized platelet activation testing are warranted.
(2) These studies should use means of thrombocyte testing which are available in clinical routine, eg the Multiplate®-test, as the methods used in this study are time- and work-intensive, expensive and vulnerable to lab test bias.
(3) Identifying the origin and pathway of thrombocyte activation does not mean that its cause has been identified: Still, we have no mechanism-based proof for the role of HIV-protease inhibitors and their chemical structures being the reason for thrombocyte aggregation. Only, the singular effect of the low-dose ritonavir booster, which was coadministered together with all PIs in the PLA-I- and PLA-II-studies could be excluded after the recent presentation of the D:A:D study data, were ritonavir-boosted atazanavir did not show an impact on the incidence of CVE.46
Another possibility could be the activation of yet unknown mediators of thrombocyte activation by HIV protease inhibitors without a direct acting role of the drugs themselves.
(4) It furthermore remains to be elucidated whether patients with enhanced platelet activation but no presence of cardiovascular disease might benefit from an anti-platelet therapy with eg P2Y12-antagonists like Clopidogrel18 or whether patients after CVE on a dual anti-platelet therapy with ASS100mg/clopidogrel 75mg should remain on clopidogrel-based secondary prophylaxis.
Clinical endpoint studies ought to be performed to evaluate this question.
Data Sharing Statement
Data, ie anonymized patient´s characteristics and laboratory raw data, can be accessed by an official request (c. Person, Institution, Direction, Contact Data) via email to the corresponding author until 30.06.2021. Deidentified participant data or any other study documents will not be shared due to the European and German data safety regulations (General Data Protection Regulation, GDPR, Art. No.3).
Funding
The authors are grateful for the support of this study provided by the Fritz- und Heinrich Riese Foundation.
Disclosure
Dr Karina Kuczka reports grants from Heinrich- und Fritz Riese-Stiftung (Foundation) during the conduct of the study. Dr Timo Wolf reports personal fees from Gilead, during the conduct of the study; personal fees from Gilead, Merck Sharp Dome, outside the submitted work. Dr Pavel Khaykin reports personal fees from Gilead, AbbVie, Sanofi, ViiV, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Gresele P, Falcinelli E, Sebastiano M, Baldelli F. Endothelial and platelet function alterations in HIV-infected patients. Thromb Res. 2012;129(3):301–308. doi:10.1016/j.thromres.2011.11.022
2. Zahler S, Massoudy P, Hartl H, Hahnel C, Meisner H, Becker BF. Acute cardiac inflammatory responses to postischemic reperfusion during cardiopulmonary bypass. Cardiovasc Res. 1999;41(3):722–730. doi:10.1016/S0008-6363(98)00229-6
3. Furie B, Furie BC. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytes: role of P-selectin and the P-selectin ligand, PSGL-1. Thromb Haemost. 1995;74(1):224–227. doi:10.1055/s-0038-1642681
4. Evangelista V, Manarini S, Rotondo S, et al. Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: evidence of adhesion cascade and cross talk between P-selectin and the beta 2 integrin CD11b/CD18. Blood. 1996;88(11):4183–4194. doi:10.1182/blood.V88.11.4183.4183
5. Graff J, Klinkhardt U, Schini-Kerth VB, et al. Close relationship between the platelet activation marker CD62 and the granular release of platelet-derived growth factor. J Pharmacol Exp Ther. 2002;300(3):952–957. doi:10.1124/jpet.300.3.952
6. Abedi H, Zachary I. Signalling mechanisms in the regulation of vascular cell migration. Cardiovasc Res. 1995;30(4):544–556. doi:10.1016/S0008-6363(95)00092-5
7. Schini-Kerth VB, Bassus S, Fisslthaler B, Kirchmaier CM, Busse R. Aggregating human platelets stimulate the expression of thrombin receptors in cultured vascular smooth muscle cells via the release of transforming growth factor-beta1 and platelet-derived growth factorAB. Circulation. 1997;96(11):3888–3896. doi:10.1161/01.CIR.96.11.3888
8. Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood. 1990;75(3):550–554. doi:10.1182/blood.V75.3.550.550
9. Neumann FJ, Marx N, Gawaz M, et al. Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation. 1997;95(10):2387–2394. doi:10.1161/01.CIR.95.10.2387
10. Ott I, Neumann FJ, Gawaz M, Schmitt M, Schomig A. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. 1996;94(6):1239–1246. doi:10.1161/01.CIR.94.6.1239
11. Mathew JP, Rinder CS, Tracey JB, et al. Acadesine inhibits neutrophil CD11b up-regulation in vitro and during in vivo cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1995;109(3):448–456. doi:10.1016/S0022-5223(95)70275-X
12. Gawaz MP. Das Blutplättchen: Physiologie, Pathophysiologie, Membranrezeptoren, antithrombozytäre Wirkstoffe und antithrombozytäre Therapie bei koronarer Herzerkrankung. Stuttgart [u.a.]: Thieme; 1999.
13. Friis-Moller N, Weber R, Reiss P, et al. Cardiovascular disease risk factors in HIV patients--association with antiretroviral therapy. Results from the DAD study. Aids. 2003;17(8):1179–1193. doi:10.1097/00002030-200305230-00010
14. Friis-Moller N, Sabin C, Weber R, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. 2003;349(21):1993–2003.
15. Desvarieux M, Boccara F, Meynard JL, et al. Infection duration and inflammatory imbalance are associated with atherosclerotic risk in HIV-infected never-smokers independent of antiretroviral therapy. Aids. 2013;27(16):2603–2614. doi:10.1097/QAD.0b013e3283634819
16. Kristoffersen US, Kofoed K, Kronborg G, Benfield T, Kjaer A, Lebech AM. Changes in biomarkers of cardiovascular risk after a switch to abacavir in HIV-1-infected individuals receiving combination antiretroviral therapy. HIV Med. 2009;10(10):627–633. doi:10.1111/j.1468-1293.2009.00733.x
17. Falcinelli E, Francisci D, Belfiori B, et al. In vivo platelet activation and platelet hyperreactivity in abacavir-treated HIV-infected patients. Thromb Haemost. 2013;110(08):349–357. doi:10.1160/TH12-07-0504
18. Nordell AD, McKenna M, Borges AH, et al. Severity of cardiovascular disease outcomes among patients with HIV is related to markers of inflammation and coagulation. J Am Heart Assoc. 2014;3(3):e000844. doi:10.1161/JAHA.114.000844
19. Westhorpe CL, Schneider HG, Dunne M, et al. C-reactive protein as a predictor of cardiovascular risk in HIV-infected individuals. Sex Health. 2014;11(6):580–582. doi:10.1071/SH14130
20. Lacerda HR, Falcao Mda C, de Albuquerque VM, et al. Association of inflammatory cytokines and endothelial adhesion molecules with immunological, virological, and cardiometabolic disease in HIV-infected individuals. J Interferon Cytokine Res. 2014;34(5):385–393. doi:10.1089/jir.2013.0029
21. Moyle G, Hardy H, Uy J, et al. Changes in biomarkers in HIV-1-infected treatment-naive patients treated with tenofovir DF/emtricitabine plus atazanavir/ritonavir or lopinavir/ritonavir for 96 weeks: the CASTLE biomarker substudy. Antivir Ther. 2014;19(7):693–699. doi:10.3851/IMP2778
22. Rasmussen TA, Tolstrup M, Melchjorsen J, et al. Evaluation of cardiovascular biomarkers in HIV-infected patients switching to abacavir or tenofovir based therapy. BMC Infect Dis. 2011;11:267. doi:10.1186/1471-2334-11-267
23. Duprez DA, Neuhaus J, Kuller LH, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One. 2012;7(9):e44454. doi:10.1371/journal.pone.0044454
24. Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e203. doi:10.1371/journal.pmed.0050203
25. Ikeda H, Nakayama H, Oda T, et al. Neutrophil activation after percutaneous transluminal coronary angioplasty. Am Heart J. 1994;128(6):1091–1098. doi:10.1016/0002-8703(94)90738-2
26. Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23(1):17–25. doi:10.1161/01.ATV.0000046238.23903.FC
27. Hemker HC, Al Dieri R, Beguin S. Thrombin generation assays: accruing clinical relevance. Curr Opin Hematol. 2004;11(3):170–175. doi:10.1097/01.moh.0000130314.33410.d7
28. Baglin T. The measurement and application of thrombin generation. Br J Haematol. 2005;130(5):653–661. doi:10.1111/j.1365-2141.2005.05612.x
29. von Hentig N, Forster AK, Kuczka K, et al. Platelet-leucocyte adhesion markers before and after the initiation of antiretroviral therapy with HIV protease inhibitors. J Antimicrob Chemother. 2008;62(5):1118–1121. doi:10.1093/jac/dkn333
30. Angdujar I, Collado-Diaz V, Ordem S, et al. Prothrombotic effects of abacavir in an in-vivo model.
31. Wohl DA, Arnoczy G, Fichtenbaum CJ, et al. Comparison of cardiovascular disease risk markers in HIV-infected patients receiving abacavir and tenofovir: the nucleoside inflammation, coagulation and endothelial function (NICE) study. Antivir Ther. 2014;19(2):141–147. doi:10.3851/IMP2681
32. Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Activated and unactivated platelet adhesion to monocytes and neutrophils. Blood. 1991;78(7):1760–1769. doi:10.1182/blood.V78.7.1760.1760
33. Schmitz G, Rothe G, Ruf A, et al. European Working Group on Clinical Cell Analysis: consensus protocol for the flow cytometric characterisation of platelet function. Thromb Haemost. 1998;79(5):885–896. doi:10.1055/s-0037-1615088
34. Barbaro G. Vascular injury, hypertension and coronary artery disease in human immunodeficiency virus infection. Clin Ter. 2008;159(1):51–55.
35. McComsey GA, O’Riordan M, Hazen SL, et al. Increased carotid intima media thickness and cardiac biomarkers in HIV infected children. Aids. 2007;21(8):921–927. doi:10.1097/QAD.0b013e328133f29c
36. Masia M, Padilla S, Bernal E, et al. Influence of antiretroviral therapy on oxidative stress and cardiovascular risk: a prospective cross-sectional study in HIV-infected patients. Clin Ther. 2007;29(7):1448–1455. doi:10.1016/j.clinthera.2007.07.025
37. Wang X, Chai H, Yao Q, Chen C. Molecular mechanisms of HIV protease inhibitor-induced endothelial dysfunction. J Acquir Immune Defic Syndr. 2007;44(5):493–499. doi:10.1097/QAI.0b013e3180322542
38. Cotter BR. Endothelial dysfunction in HIV infection. Curr HIV/AIDS Rep. 2006;3(3):126–131. doi:10.1007/BF02696656
39. Barbaro G, Barbarini G. Highly active antiretroviral therapy-associated metabolic syndrome and cardiovascular risk. Chemotherapy. 2006;52(4):161–165. doi:10.1159/000093034
40. Spieker LE, Karadag B, Binggeli C, Corti R. Rapid progression of atherosclerotic coronary artery disease in patients with human immunodeficiency virus infection. Heart Vessels. 2005;20(4):171–174. doi:10.1007/s00380-004-0790-8
41. Zhou H, Pandak WM, Lyall V, Natarajan R, Hylemon PB. HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol. 2005;68(3):690–700. doi:10.1124/mol.105.012898
42. Carr A. Cardiovascular risk factors in HIV-infected patients. J Acquir Immune Defic Syndr. 2003;34(Suppl 1):S73–78. doi:10.1097/00126334-200309011-00011
43. Baker J, Sharma S, Grund B; Group ISS. The effect of ART on inflammation, coagulation, and vascular injury in START.
44. Baker J, Sharma S, Grund B; Group ISS. Association of inflammation and coagulation with clinical risk in the START trial.
45. Ryom L, Lunsgren J, el-Sadr W, et al. Associations between cardiovascular disease and contemporarily used protease inhibitors.
46. Ryom L, Lundgren JD, El-Sadr W, et al. Cardiovascular disease and use of contemporary protease inhibitors: the D:A:D international prospective multicohort study. Lancet HIV. 2018;5(6):e291–e300. doi:10.1016/S2352-3018(18)30043-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.