")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Evaluation of drug interactions between fimasartan and rosuvastatin after single and multiple doses in healthy Caucasians
Authors Lee J , Rhee SJ, Lee S , Yu KS
Received 2 July 2017
Accepted for publication 3 January 2018
Published 6 April 2018 Volume 2018:12 Pages 787—794
DOI https://doi.org/10.2147/DDDT.S145339
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Sukesh Voruganti
Jieon Lee, Su-jin Rhee, SeungHwan Lee, Kyung-Sang Yu
Department of Clinical Pharmacology and Therapeutics, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Republic of Korea
Objectives: As hypercholesterolemia is often accompanied by hypertension, statins are usually prescribed with angiotensin receptor blockers in clinical practice. This study was performed to evaluate the pharmacokinetics and safety of fimasartan and rosuvastatin when coadministered or administered alone as a single dose or as multiple doses to healthy Caucasians.
Methods: Thirty-six subjects were enrolled into an open-labeled, randomized, 6-sequence, 3-period, 3-way crossover study, and randomly received fimasartan (120 mg), rosuvastatin (20 mg) or both. Blood samples for pharmacokinetics were collected up to 48 hours for fimasartan and 72 hours for rosuvastatin after the last dosing and plasma concentrations of study drugs were determined by liquid chromatography-tandem mass spectrometry. Maximum plasma concentration (Cmax), area under the concentration-time curve (AUC) from 0 to the last measurable time (AUClast), maximum plasma concentration at steady state (Cmax,ss) and AUC to the end of the dosing period at steady state (AUCτ,ss) were estimated using a non-compartmental method. Safety and tolerability were evaluated throughout the study.
Results: Thirty subjects completed the study. After single dose administration, the geometric mean ratio (GMR) and 90% confidence intervals (CIs) of fimasartan with or without rosuvastatin were 0.95 (0.80–1.14) and 0.98 (0.91–1.07) for Cmax and AUClast, respectively. The corresponding values for rosuvastatin with or without fimasartan were 1.32 (1.16–1.50) and 0.97 (0.89–1.05), respectively. After administration of multiple doses, the GMRs (90% CIs) for Cmax,ss and AUCτ,ss of fimasartan with or without rosuvastatin were 0.94 (0.74–1.20) and 1.07 (0.90–1.16), respectively. The corresponding values for rosuvastatin with or without fimasartan were 1.16 (1.02–1.32) and 0.86 (0.79–0.94), respectively. A total of 74 adverse events (AEs) were reported and incidences of AEs did not increase significantly with co-administration.
Conclusion: Co-administration of fimasartan and rosuvastatin did not result in clinically relevant changes in the systemic exposure of fimasartan or rosuvastatin after single and multiple administrations, and they were well tolerated.
Keywords: fimasartan, rosuvastatin, drug interaction, clinical trial, angiotensin II receptor antagonist, healthy subjects
Introduction
Cardiovascular diseases (CVD) are now the leading cause of death globally.1,2 There are several risk factors for CVD, such as high blood pressure, diabetes, cholesterol levels, and obesity.3 Among those factors, hypertension and hypercholesterolemia are the 2 major contributing risk factors for CVD and many studies reported that it is important to control both factors to reduce morbidity and mortality.4,5
In clinical practice, statins are commonly prescribed in combination with angiotensin receptor blockers (ARBs).6–8 The co-administration of statins with ARBs was demonstrated to effectively reduce cardiovascular risk and enhance the inhibitory effect of ARBs on oxidative stress.9 The renin–angiotensin–aldosterone system (RAAS) is an important regulator of cardiovascular function and fluid-electrolyte homeostasis. The excessive activation of angiotensin II leads to hypertension, atherosclerosis, coronary artery disease, myocardial infarction, congestive heart failure, and nephropathy.10
Fimasartan (Kanarb®, Boryung Pharmaceutical Co. Ltd., Seoul, Korea) is a non-peptide ARB with a selective type I receptor blockade effect, which inhibits vascular contraction by acting on the kidney RAAS.11 It has been approved for the treatment of mild-to-moderate hypertension in South Korea in 2010.12 Unlike peptide angiotensin II receptor antagonist, such as saralasin, fimasartan does not show a partial agonistic action, making it more effective as an angiotensin II type I receptor-specific antagonist.11,13
Rosuvastatin (Crestor®, AstraZeneca, Cambridge, UK) is a synthetic lipid-lowering agent, one of the drug class of statins.14 It is used as adjunctive therapy to diet to treat high total cholesterol, low-density cholesterol, and triglycerides and to increase high-density cholesterol concentrations in patients with primary hyperlipidemia or mixed dyslipidemia.5 The dose range for orally administered rosuvastatin is 5–40 mg once daily, and the usual starting dose is between 10 and 20 mg.15
This study was conducted to evaluate the pharmacokinetics (PKs), safety, and tolerability of 120 mg fimasartan and 20 mg rosuvastatin when coadministered or administered alone as a single dose or as multiple doses to Caucasian healthy male subjects.
Materials and methods
Study subjects
Eligible subjects were Caucasian (white) male adults aged between 19 and 55 years whose body mass index was in the range of 18.5–32.0 kg/m2. Subjects were included if they were in good health based on medical history, physical examination, laboratory profiles, vital signs or 12-lead electrocardiography (ECGs), and urinary drug screening that was obtained within 4 weeks of the first administration of the study drug. All subjects understood the study procedures in the informed consent form and provided written informed consent voluntarily before entering the study.
Subjects were not enrolled in the study if there was evidence or a history of any of the following: mentally or legally incapacitated or significant emotional problems; clinically significant medical or psychiatric condition or disease; history of any illness that might have confounded the results of study, including hereditary disease/genetic disorder, gastrointestinal disorder, major injury, muscle problems; abnormal laboratory profiles for creatinine phosphokinase, aspartate aminotransferase, and alanine aminotransferase history of alcoholism or drug abuse; the use of any prescription medication during the 14 days before the first dosing; and participation in clinical trials of any drug within 28 days prior to this study. All subjects agreed to use contraception during the study.
Study design and procedures
This was an open-label, randomized, 6-sequence, 3-period, 3-way crossover study. Before starting subject enrollment, this study was registered in a public trial registry, ClinicalTrials.gov (http://www.clinicaltrials.gov; identifier number NCT02704702). The study was conducted in Celerion (Lincoln, NE, USA). All study documents were reviewed and approved by Chesapeake Research Review, Inc. (CRRI) before study initiation. The CRRI Institutional Review Board is organized and operates in compliance with US Food and Drug Administration regulations and guidelines resulting from the International Conference on Harmonisation and Good Clinical Practice.16
On the first day (day 1) of period 1, all eligible subjects were randomly allocated to 1 of 6 treatment sequences to receive 120 mg fimasartan alone, 20 mg rosuvastatin alone, or both 120 mg fimasartan and 20 mg rosuvastatin. The doses of fimasartan and rosuvastatin were chosen as they were the doses used in previous studies and well tolerated in healthy subjects from previous studies.17–19 In addition, the dose for rosuvastatin is within the limits of the dosing regimens.20 A single dose was administered on day 1, followed by the multiple-dose regimen once daily for a week starting on day 4 (Figure 1). All study drugs were administered orally following an overnight fast of at least 10 hours with ~240 mL of water. There was a washout period of at least 7 days between the last dose in one period and the first dose in the subsequent period. The washout period between doses was considered sufficient to prevent carryover effects of the treatments. In addition, a 6-sequence design was chosen to limit the sequence and period effects.
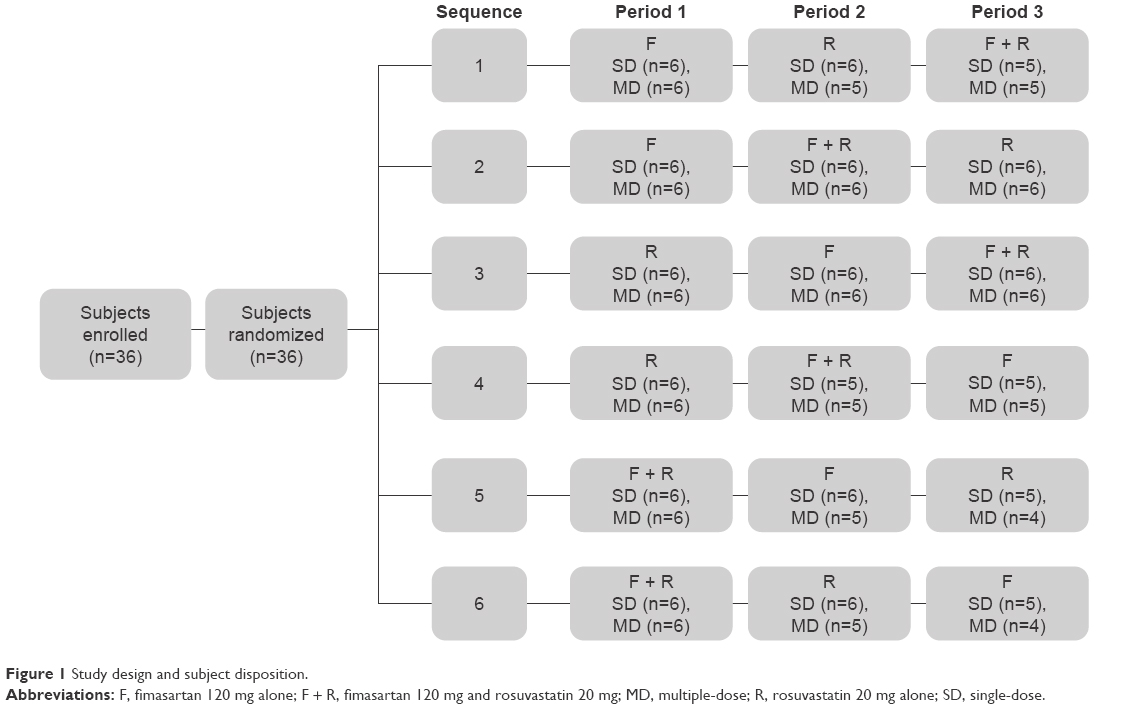 | Figure 1 Study design and subject disposition. |
Determination of fimasartan and rosuvastatin plasma concentrations
For all subjects, blood samples for the determination of rosuvastain and fimasartan plasma concentration levels were collected in blood collection tubes containing di-potassium ethylenediaminetetraacetic acid at the scheduled time points; pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36 and 48 hours after last dosing for fimasartan, pre-dose and 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 24, 48 and 72 hours after last dosing for rosuvastatin, pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 24, 36 and 48 hours after last dosing of coadministered fimasartan and rosuvastatin.
Following blood collection, samples were cooled in an ice bath and centrifuged (approximately at 3,000 rpm for 7 minutes) under refrigeration at a temperature of ~4°C as soon as possible. Within 90 minutes of collection, plasma samples were divided into 2 aliquots and stored in suitably labeled tubes at −80°C±15°C, pending assay.
Plasma concentrations of fimasartan and rosuvastatin were subjected to a liquid–liquid extraction using methyl tertiary-butyl ether, before using high-performance liquid chromatography-tandem mass spectrometry for analysis.21 For analysis of fimasartan plasma concentration, the mobile phase consisted of 0.1% formic acid in 5 mM ammonium acetate and 0.1% formic acid in acetonitrile. For rosuvastatin, the mobile phase consisted of acetonitrile and 0.1% formic acid in 5 mM ammonium acetate (50:50, vol/vol). These methods were validated with respect to accuracy, precision, linearity, sensitivity, and specificity at Celerion. The lower limit of quantitation for plasma fimasartan and rosuvastatin were 2 and 0.2 ng/mL, respectively.21 Fimasartan is freely soluble in methanol and dimethyl sulfoxide (DMSO), sparingly soluble in water, slightly soluble in acetone and acetonitrile. Rosuvastatin is slightly soluble in water, marginally soluble in ethanol, soluble in DMSO (100 mg/mL).
PK analysis
Fimasartan and rosuvastatin PK analyses were performed using a non-compartmental analysis, implemented in Phoenix® WinNonlin® (Version 6.3., Certara, St Louis, MO, USA). The area under the plasma concentration-time curve from zero to the last measurable time point (AUClast) and area under the concentration-time curve during a dosing interval for tau from time 0 to 24 hours at steady state were calculated using the linear trapezoidal with linear interpolation method. A steady-state analysis was performed on the ln-transformed predose Ctrough concentrations on days 8, 9, and 10 using Helmert contrasts. Steady state was concluded at the time point where no more statistical difference (α=5%) was observed from 2 comparisons; 1) Ctrough day 8 vs mean of (Ctrough day 9 and Ctrough day 10), 2) Ctrough day 9 vs Ctrough day 10. The area under the plasma concentration-time curve from time zero to infinite time (AUCinf) was calculated as the sum of the AUClast and the last quantifiable concentration divided by the slope of the final decline portion of the individual log-linear concentration-time curve. The maximum plasma concentration (Cmax), the time to reach the maximum plasma concentration (Tmax) and maximum observed concentration at steady state; maximum concentration between dose time and dose time adding tau (at Tmax) (maximum plasma drug concentration at steady state [Cmax,ss]) were taken directly from the observed values. The apparent clearance was calculated by dividing the dose by the AUC.
Safety and tolerability assessment
Safety and tolerability were assessed in every subject who received at least 1 or more doses of study drugs. Safety and tolerability were evaluated by 12-lead ECGs, vital signs, clinical laboratory tests, and all types of adverse events (AEs). Summary statistics for the 12-lead ECGs, vital signs, and clinical laboratory safety tests were computed and provided, as deemed clinically appropriate. Treatment-emergent AEs (TEAEs) were coded using the Medical Dictionary for Regulatory Activities (Version 19.0) and were summarized by treatment for the number of subjects reporting the AE and the number of AEs reported.
Statistical analysis
All statistical analyses were conducted using SAS (version 9.3., SAS Institute Inc., Cary, NC, USA), and the level of significance was set at 0.05. Continuous variables were summarized using the number of observations (n), arithmetic mean, SD, median, minimum, and maximum. Frequency counts were reported for categorical data when appropriate. The effect of administration on the PK parameters was assessed using the geometric mean ratio (GMR) of combination treatment to fimasartan or rosuvastatin alone for AUClast, AUCinf, and Cmax after a single dose, AUC to the end of the dosing period at steady state (AUCτ,ss), and Cmax,ss after multiple doses. To this end, an analysis of variance (ANOVA) was performed on the log-transformed AUClast, AUCinf, Cmax, AUCτ,ss, and Cmax,ss and the model included sequence, treatment, and period as fixed effects, and subject nested within sequence as a random effect.
Each ANOVA included calculation of least-squares means (LSMs), the difference between treatment LSM, and the standard error associated with this difference. Ratios of LSM were calculated using the exponentiation of the difference between treatment LSMs from the analyses on the ln-transformed parameters. These ratios were expressed as a percentage relative to the reference treatments. For safety and tolerability assessment, the number of AEs and subjects who had AEs was counted by treatment group. The results of vital signs, ECGs, and laboratory tests were also reviewed.
Results
Study subjects
A total of 36 subjects entered the study and were randomized to study treatment, 6 of whom dropped out prior to completion of study, 3 subjects were excluded due to AEs (1 subject in sequence 4 who suffered an accident discontinued the study; 1 subject in sequence 4 was discontinued from the study after developing moderate epididymo-orchitis, the other subject in sequence 4 discontinued the study after experiencing ECG changes consistent with mild paroxysmal atrial flutter); 2 subjects in sequence 6 discontinued due to positive drug or alcohol lab tests; 1 subject in sequence 1 discontinued due to violation of procedural rules. Every subject was a healthy male Caucasian (white). The mean ± SD for age, weight, and height was 36.4±10.5 years, 84.6±10.0 kg, and 176.6±7.5 cm, respectively, which were not significantly different among sequence groups. All subjects who were enrolled in the study were included in the PK analyses.
PK assessments
Effect of rosuvastatin on the PK of fimasartan
As shown in Figure 2A, fimasartan PK profiles did not significantly differ when fimasartan was administered alone and coadministered with rosuvastatin for a given dosing regimen (both single dose and multiple doses). The total exposure to fimasartan was comparable; the GMR (90% confidence interval [CI]) of Cmax and AUClast for fimasartan with and without rosuvastatin were 0.95 (0.80–1.14) and 0.98 (0.91–1.07), respectively (Table 1). After administration of multiple doses, Cmax,ss and AUCτ,ss of fimasartan with or without rosuvastatin were 0.94 (0.74–1.20) and 1.07 (0.90–1.16), respectively (Table 2).
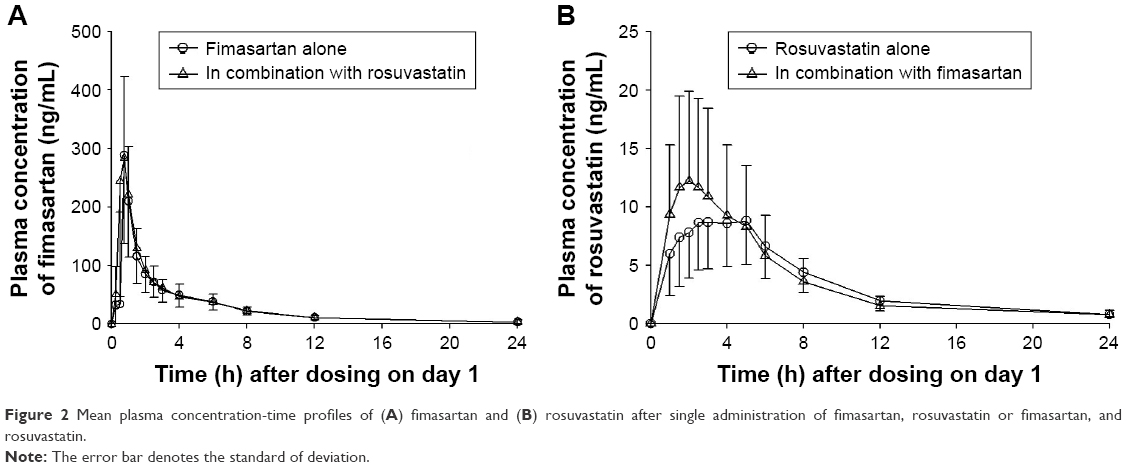 | Figure 2 Mean plasma concentration-time profiles of (A) fimasartan and (B) rosuvastatin after single administration of fimasartan, rosuvastatin or fimasartan, and rosuvastatin. |
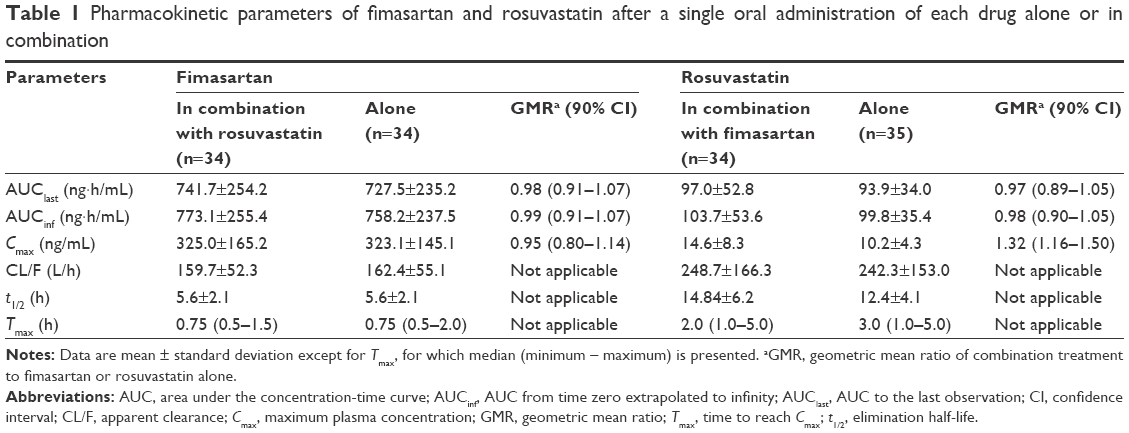 | Table 1 Pharmacokinetic parameters of fimasartan and rosuvastatin after a single oral administration of each drug alone or in combination |
 | Table 2 Pharmacokinetic parameters of fimasartan and rosuvastatin after multiple oral administrations of each drug alone or in combination |
Fimasartan exposure accumulated by 20% following multiple administration of fimasartan and by 30% following multiple doses of fimasartan and rosuvastatin, compared with administration of single doses of each treatment; accumulation ratio (Rac) (AUCτ,ss/AUC0–24) was 1.3 and 1.1, respectively (Figure 3). The Cmax was 1.3-fold greater following multiple doses compared with a single dose. For both treatments, peak mean concentrations were reached by 30–45 minutes post-dose and mean concentrations declined in the same fashion post-peak and remained detectable in all subjects at 12 hours after drug administration.
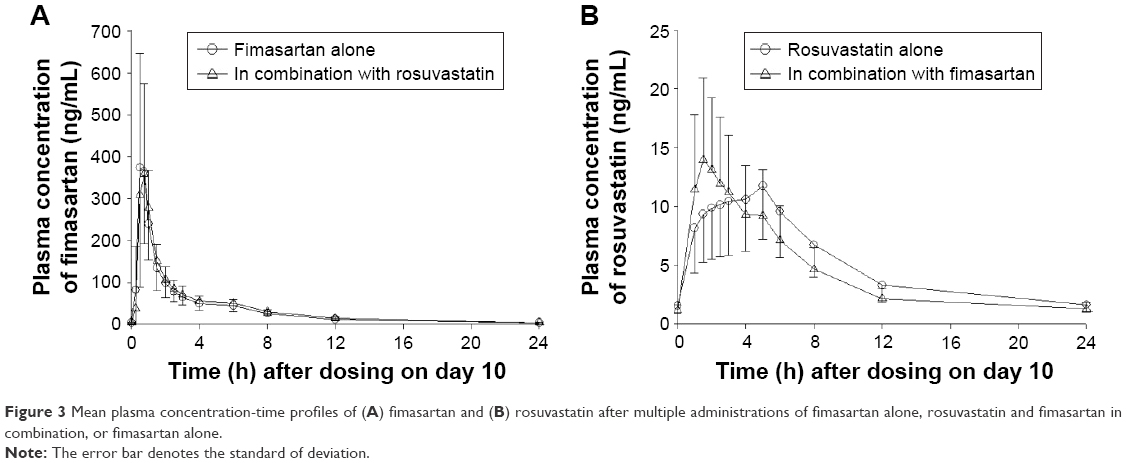 | Figure 3 Mean plasma concentration-time profiles of (A) fimasartan and (B) rosuvastatin after multiple administrations of fimasartan alone, rosuvastatin and fimasartan in combination, or fimasartan alone. |
Effect of fimasartan on the PK of rosuvastatin
No pronounced differences were found between the AUCs of rosuvastatin following single doses, when administered alone or coadministered with fimasartan (Tables 1 and 2); except concentration-time profiles displayed quicker rosuvastatin absorption, leading to higher peaks for the coadministration regimens. Following a single dose, the Cmax of rosuvastatin was ~1.3-fold greater and occurred ~1 hour earlier (median Tmax was 2.0 hours vs 3.0 hours) after coadministered with fimasartan compared with when it was administered alone (Figure 2B). Following multiple doses, the Cmax of rosuvastatin was ~1.3-fold greater and occurred 3 hours earlier (1.5 hours vs 4.5 hours), following coadministration with fimasartan compared with rosuvastatin alone (Figure 3). Rosuvastatin concentrations accumulated by ~54% (RAUC of 1.5) and 36% (RAUC of 1.4) following multiple doses of rosuvastatin, and fimasartan and rosuvastatin, compared with administration of single doses of either treatment.
Tolerability and safety assessments
All the 36 subjects who were enrolled in the study were included in the tolerability and safety analyses, 3 of them discontinued due to TEAEs; including 1 subject with a serious AE of an injury accident and 2 subjects with AEs, including orchitis and atrial flutter. All 3 of these AEs were considered unrelated to study treatment. There were no deaths in this study.
Overall, a total of 74 TEAEs were reported by 18 (50%) subjects. In terms of severity, 68 TEAEs were mild, 1 was moderate, and 5 were severe. Among those TEAEs, 11 (5 dizziness, 2 dizziness postural, 1 headache, 1 nervousness, 1 fatigue, and 1 nausea) were judged to be related to fimasartan, 3 to both fimasartan and rosuvastatin (2 headache, 1 postural dizziness), and 60 unlikely related or unrelated to either drug. No remarkable findings were found in the other safety assessments such as vital signs, ECGs, clinical laboratory, or physical examinations.
Discussion
The aim of this study was to evaluate the PK, safety, and tolerability of fimasartan and rosuvastatin when coadministered or administered alone, as a single dose or as multiple doses to healthy Caucasian male subjects. Considering that statins are commonly prescribed in combination with ARBs to effectively reduce cardiovascular risk, developing combination drugs, including fimasartan and rosuvastatin drugs, would be helpful by increasing patient adherence and medication compliance or by increasing efficacy.18,22–24 The doses of fimasartan and rosuvastatin were chosen as they were the doses used in previous studies and well tolerated by healthy subjects (not published). In addition, the dose for rosuvastatin is within the limits of the dosing regimens of the full prescribing information. From our study, we have shown that fimasartan and rosuvastatin do not exert a significant drug interaction in Caucasian subjects.
From a previous drug–drug interaction study of fimasartan and rosuvastatin in healthy male Korean subjects, no clinically significant difference of coadministration of fimasartan and rosuvastatin was found on the extent of systemic exposure (eg, AUC) or maximum exposure (eg, Cmax) to both fimasartan and rosuvastatin at same dosage (120 and 20 mg for 7 days, respectively).21 In this study, there was no significant effect of coadministration of rosuvastatin and fimasartan on the extent of exposure to fimasartan or rosuvastatin, except a slight decrease and increase by 14% in AUCτ,ss and 16% in Cmax,ss for rosuvastatin following multiple doses of both drugs.
Rosuvastatin is mostly eliminated via bile (70%). It is selectively transported from blood to liver by organic anion transporting polypeptide 1B1 (OATP1B1) which is encoded solute carrier organic anion transporter family, member 1B1 gene.25 The OATP1B1 plays an important role for hepatobiliary elimination of fimasartan together with OATP2B1, and OATP1B3. Considering fimasartan and rosuvastatin are both substrates of OATP1B1, inhibition of OATP1B1 by fimasartan may explain the increase in rosuvastatin maximum exposure due to lower hepatic uptake and efflux of rosuvastatin as observed in the present study.26
Birmingham et al have reported that plasma exposure to rosuvastatin was significantly higher in Asian than Caucasian populations who live in the same environment.27 Their study has concluded that genetic variations of ABCG2 can partially explain the difference.27 In the current study, when comparing the observed PK profiles from our study in healthy fasted male Caucasians with the profiles in the previous study in healthy fasted male Korean subjects, Caucasians showed higher systemic exposure of rosuvastatin without or with fimasartan than Korean subjects with same dosage regimen probably due to different environment, such as diet.28 Our comparison is limited since the study environments were different and the genotype of patients was not assessed (eg, OATP1B1).
Regarding the safety and tolerability assessment results, 120 mg of fimasartan and 20 mg of rosuvastatin were well tolerated when administered together or alone. In this study, the most frequently reported TEAEs throughout the study were headache and dizziness experienced by 7 (19.4%) and 3 (8.0%) subjects, respectively, probably due to transient blood pressure lowering effect. These AEs have also been reported in previous clinical trials in healthy Korean subjects.12,21 Overall, coadministration of rosuvastatin and fimasartan did not increase the incidence of TEAEs.
In conclusion, coadministration of fimasartan and rosuvastatin did not cause a significant drug interaction in terms of drug systemic exposure, safety, and tolerability in the healthy male Caucasian subjects. Single and multiple doses of 120 mg fimasartan, administered alone and in combination with 20 mg rosuvastatin were well tolerated by healthy male Caucasian subjects.
Acknowledgments
This study was supported by Boryung Pharmaceutical Co. Ltd (Seoul, Republic of Korea). KCRN Research (Germantown, MD, USA) was involved in study initiation and study monitoring. Celerion (Lincoln, NE, USA) was involved in data collection, data analysis, and writing the study reports. The funding body had no role in data interpretation and the writing of the manuscript. Jieon Lee is currently a recipient of an ORISE stipend from the US Food and Drug Administration. Jieon Lee’s contribution to the manuscript was based on their prior employment, and the current manuscript does not reflect any position of the US Food and Drug Administration or the US government.
Disclosure
The authors report no conflicts of interest in this work.
References
Hobbs FD. Cardiovascular disease: different strategies for primary and secondary prevention? Heart. 2004;90(10):1217–1223. | ||
GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–171. | ||
de Ruijter W, Westendorp RG, Assendelft WJ, et al. Use of Framingham risk score and new biomarkers to predict cardiovascular mortality in older people: population based observational cohort study. BMJ. 2009;338:a3083. | ||
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evalution, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. | ||
McTaggart F, Jones P. Effects of statins on high-density lipoproteins: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther. 2008;22(4):321–338. | ||
Duru OK, Turk N, Ettner SL, et al. Adherence to Metformin, Statins, and ACE/ARBs Within the Diabetes Health Plan (DHP). J Gen Intern Med. 2015;30(11):1645–1650. | ||
Kim-Mitsuyama S. Combination therapy of angiotensin receptor blocker with statin or thiazolidinediones as promising therapeutic strategy for metabolic syndrome and atherosclerosis. Hypertens Res. 2009;32(8):639–640. | ||
Lee J, Lee H, Jang K, Lim KS, Shin D, Yu KS. Evaluation of the pharmacokinetic and pharmacodynamic drug interactions between cilnidipine and valsartan, in healthy volunteers. Drug Des Devel Ther. 2014;8:1781–1788. | ||
Koh KK, Han SH, Oh PC, Shin EK, Quon MJ. Combination therapy for treatment or prevention of atherosclerosis: focus on the lipid-RAAS interaction. Atherosclerosis. 2010;209(2):307–313. | ||
Ma TK, Kam KK, Yan BP, Lam YY. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br J Pharmacol. 2010;160(6):1273–1292. | ||
Kim JH, Lee JH, Paik SH, Kim JH, Chi YH. Fimasartan, a novel angiotensin II receptor antagonist. Arch Pharm Res. 2012;35(7):1123–1126. | ||
Chi YH, Lee H, Paik SH, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of fimasartan following single and repeated oral administration in the fasted and fed states in healthy subjects. Am J Cardiovasc Drugs. 2011;11(5):335–346. | ||
Tsang SW, Ip SP, Wong TP, Che CT, Leung PS. Differential effects of saralasin and ramiprilat, the inhibitors of renin-angiotensin system, on cerulein-induced acute pancreatitis. Regul Pept. 2003;111(1–3):47–53. | ||
Aggarwal RK, Showkathali R. Rosuvastatin calcium in acute coronary syndromes. Expert Opin Pharmacother. 2013;14(9):1215–1227. | ||
Quirk J, Thornton M, Kirkpatrick P. Rosuvastatin calcium. Nat Rev Drug Discov. 2003;2(10):769–770. | ||
Morris K. Revising the Declaration of Helsinki. Lancet. 2013;381(9881):1889–1890. | ||
Cardona-Munoz EG, Lopez-Alvarado A, Conde-Carmona I, et al. Safety and efficacy of fimasartan in Mexican patients with grade 1–2 essential hypertension. Arch Cardiol Mex. 2017;87(4):316–325. | ||
Rhee MY, Ahn T, Chang K, et al. The efficacy and safety of co-administration of fimasartan and rosuvastatin to patients with hypertension and dyslipidemia. BMC Pharmacol Toxicol. 2017;18(1):2. | ||
Lee HY, Oh BH. Fimasartan: a new angiotensin receptor blocker. Drugs. 2016;76(10):1015–1022. | ||
Li Y, Jiang X, Lan K, Jiang Q. Comparative single- and multiple-dose pharmacokinetics of rosuvastatin following oral administration in Chinese volunteers. Eur J Drug Metab Pharmacokinet. 2009;34(3–4):221–227. | ||
Kang WY, Kim EH, Seong SJ, et al. Pharmacokinetic drug interaction study using fimasartan and rosuvastatin in healthy volunteers. Int J Clin Pharmacol Ther. 2016;54(12):992–1003. | ||
Shenfield GM. Fixed combination drug therapy. Drugs. 1982;23(6):462–480. | ||
Canbakan B. Rational approaches to the treatment of hypertension: drug therapy-monotherapy, combination, or fixed-dose combination? Kidney Int Suppl (2011). 2013;3(4):349–351. | ||
Montecucco F, Mach F. Statins, ACE inhibitors and ARBs in cardiovascular disease. Best Pract Res Clin Endocrinol Metab. 2009;23(3):389–400. | ||
Hua WJ, Hua WX, Fang HJ. The role of OATP1B1 and BCRP in pharmacokinetics and DDI of novel statins. Cardiovasc Ther. 2012;30(5):e234–e241. | ||
Shin KH, Kim TE, Kim SE, et al. The effect of the newly developed angiotensin receptor II antagonist fimasartan on the pharmacokinetics of atorvastatin in relation to OATP1B1 in healthy male volunteers. J Cardiovasc Pharmacol. 2011;58(5):492–499. | ||
Birmingham BK, Bujac SR, Elsby R, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur J Clin Pharmacol. 2015;71(3):329–340. | ||
Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther. 2008;84(3):417–423. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.