Back to Journals » OncoTargets and Therapy » Volume 8
Evaluation and comparison of two commercially available targeted next-generation sequencing platforms to assist oncology decision making
Authors Weiss G, Hoff B, Whitehead R, Sangal A, Gingrich S, Penny R, Mallery D, Morris S, Thompson E, Loesch D, Khemka V
Received 1 February 2015
Accepted for publication 24 February 2015
Published 24 April 2015 Volume 2015:8 Pages 959—967
DOI https://doi.org/10.2147/OTT.S81995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Daniele Santini
Glen J Weiss,1 Brandi R Hoff,1 Robert P Whitehead,1 Ashish Sangal,1 Susan A Gingrich,1 Robert J Penny,2 David W Mallery,2 Scott M Morris,2 Eric J Thompson,2 David M Loesch,2 Vivek Khemka1
1Cancer Treatment Centers of America, Western Regional Medical Center, Goodyear, AZ, USA; 2Paradigm Diagnostics, Phoenix, AZ, USA
Background: It is widely acknowledged that there is value in examining cancers for genomic aberrations via next-generation sequencing (NGS). How commercially available NGS platforms compare with each other, and the clinical utility of the reported actionable results, are not well known. During the course of the current study, the Foundation One (F1) test generated data on a combination of somatic mutations, insertion and deletion polymorphisms, chromosomal abnormalities, and deoxyribonucleic acid (DNA) copy number changes at ~250× coverage, while the Paradigm Cancer Diagnostic (PCDx) test generated the same type of data at >5,000× coverage, plus provided messenger RNA (mRNA) expression levels. We sought to compare and evaluate paired formalin-fixed paraffin-embedded tumor tissue using these two platforms.
Methods: Samples from patients with advanced solid tumors were submitted to both the F1 and PCDx vendors for NGS analysis. Turnaround time (TAT) was calculated. Biomarkers were considered clinically actionable if they had a published association with treatment response in humans and were assigned to the following categories: commercially available drug (CA), clinical trial drug (CT), or neither option (hereafter referred to as “None”).
Results: The demographics of the 21 unique patient tumor samples included ten men and eleven women, with a median age of 56 years. Due to insufficient archival tissue from the same collection period, in one case, we used samples from different collections. PCDx reported first results faster than F1 in 20 cases. When received at both vendors on the same day, PCDx reported first results for 14 of 15 cases, with a median TAT of 9 days earlier than F1 (P<0.0001). Categorization of CA compared to CT and none significantly favored PCDx (P=0.012).
Conclusion: In the current analysis, commercially available NGS platforms provided clinically relevant actionable targets (CA or CT) in 47%–67% of diverse cancer types. In the samples analyzed, PCDx significantly outperformed F1 in TAT, and had statistically significant higher clinically relevant actionable targets categorized as CA.
Keywords: next-generation sequencing, cancer, assay, platform, treatment decision making
A letter to the Editor has been recieved and published for this article.
Introduction
During the course of their lifetime, advanced cancer patients will often receive several lines of treatment. With each subsequent line of therapy, there may be diminishing rates of return on durable response.1 At a minimum, the identification of the Achilles’ heel of a tumor to invoke disease stabilization and durable tumor control is what many clinicians are seeking to help their patients. The era of precision medicine and consequent genomic profiling a patient’s tumor to identify a unique treatment option is upon us.2 We and others have reported on use of next-generation sequencing (NGS) technologies in prospective studies.3–7 The field of sequencing technologies has rapidly evolved over the last decade, and there are now benchtop sequencers that allow for high-depth coverage of several megabases in a matter of hours with relatively lost cost and reasonable turnaround time (TAT) for clinical applications.7
There are now a variety of commercially available assays that can be ordered by clinicians. How commercially available NGS platforms compare with each other and the clinical utility of the reported actionable results is not well known. One of the most popular and prevalent is the Foundation One (F1) test (Foundation Medicine, Cambridge, MA, USA), launched in 2011. Paradigm Cancer Diagnostic (PCDx) is a newer commercially available NGS platform (Paradigm, Ann Arbor, MI, USA) launched in 2014. During the time frame of the current analysis, the F1 test generated data on a combination of somatic mutations, insertion and deletion polymorphisms (indels), chromosomal abnormalities, and deoxyribonucleic acid (DNA) copy number changes at ~250× coverage, while the PCDx test generated the same type of data at >5,000× coverage, plus provided messenger ribonucleic acid (mRNA) expression levels. To replicate the experience of real clinical practice, additional validation of actionable targets reported by commercial laboratory tests was not performed by another independent laboratory. We sought to compare and evaluate paired formalin-fixed, paraffin-embedded (FFPE) tumors using these two platforms.
Materials and methods
Ethics statement
After obtaining ethics approval from the Western Institutional Review Board (IRB) under Exemption 4 of Title 45 Code of the US Federal Regulations (CFR) concerning retrospective study of existing data, qualifying NGS reports were collected. Under exemption 4, patient consent is not required and was not obtained for this study, as Title 45 CFR Part 46 does not apply. NGS results were de-identified and the clinical information associated with these specimens is not individually identifiable, such that subjects cannot be identified either directly or indirectly through identifiers linked to the subjects.
Tumor specimens and clinical annotation
FFPE tumor tissues were ordered by the treating physician when deemed clinically appropriate for F1 testing and were submitted for commercial testing according to Foundation Medicine’s commercial laboratory standards.8 Clinically appropriate testing was defined as (i) patient having a stage 4 solid tumor by AJCC staging and (ii) tumor progressed on at least one line of therapy when there exists a standard of care (SOC) treatment or (iii) there is no SOC for that cancer diagnosis. Where feasible, an additional tumor tissue block from the same collection date or with the same pathologic diagnosis from a different collection time point was also sent for PCDx testing.
Methods for PCDx testing
FFPE specimens were received for PCDx testing, and the diagnosis of each case was confirmed on a freshly cut hematoxylin and eosin (HE)-stained slide by a board-certified pathologist (author RJP). Cases were micro/macro dissected when less than 60% tumor cells were present in order to enrich for tumor cells. DNA was extracted from all specimens, and where feasible, ribonucleic acid (RNA) was also extracted. Complementary DNA was created from RNA. A proprietary polymerase chain reaction (PCR)-based method was used to create libraries. All libraries from a given case were simultaneously sequenced on an Ion 318™ chip on the Ion PGM sequencer (Thermo Fisher Scientific, Waltham, MA, USA). mRNA was analyzed for elevated expression at P≤0.001. Copy number variants and alterations were reported. Mutations were compared to a database of mutations, and only those contained within the database were reported. The test was optimized to detect base substitutions with 4% frequency at 99.9% sensitivity and indels with 7% frequency at 99.4% sensitivity. The specificity of mutation assays was optimized to be >99.99% at the patient level, meaning that <0.01% of patient reports will contain a false positive result. A report was generated, reviewed by a board-certified oncologist (author DML) and pathologist (author RJP), signed out, and transmitted to the patient’s physician.
Statistical analysis
Clinical annotation related to the tumor samples analyzed, such as age at the time of NGS testing, sex, histology, and tumor stage at the time of NGS testing were collected and summarized. TAT was calculated as the difference in calendar days between the date the sample was received at the vendor to the date of the first NGS report for that sample. A biomarker was considered clinically actionable if it had a published association with treatment response in humans. The NGS report was assigned the highest ranked category of the following categories based on the list of actionable biomarker(s): commercially available drug (CA) (highest category) > clinical trial drug (CT) > neither option (abbreviated as “None” hereafter) (lowest category). In other words, if there was more than one actionable biomarker in the F1 or PCDx report, the biomarker with the highest category was used in the comparison between the commercial NGS reports.
To be considered CA, one of the DNA actionable biomarkers or mRNA had to have demonstrated clinical efficacy in human cancer prospective trials using the biomarker and a CA that can target that biomarker.9 For PCDx, both ERBB2 and TOP2A mRNA have high (>80%) concordance with protein expression, and were the only mRNA biomarkers included for the CA categorization. For example, ERBB2 is a known biomarker candidate for trastuzumab or lapatinib, and EGFR is a known biomarker candidate for erlotinib or afatinib.
To be considered CT, one of the DNA actionable biomarkers was required to have a drug in clinical development using the biomarker. For example, MET is being explored as a biomarker for MET inhibitors, and PIK3CA is a biomarker being explored for PI3 kinase inhibitors. To be considered None, either KRAS mutation was reported or none of the remaining DNA or mRNA biomarkers was reported to have a drug candidate that had demonstrated clinical efficacy in human cancer prospective trials. A Student’s t-test was applied to TAT results and chi-square and Fisher’s exact tests were used to analyze the categorization of actionable biomarkers. A two-tailed P-value of <0.05 was considered to indicate a significant difference.
Results
Twenty-one cases were identified as meeting the inclusion criteria of paired F1 and PCDx testing. There were ten men and eleven women associated with the tumors analyzed, and these patients had a median age of 56 years (range 35–65 years). The most common cancer types were thoracic (n=7), gastrointestinal (n=4), and genitourinary (n=3) (Table 1). Paired F1 and PCDx results for the 21 unique patient tumors were submitted between March 2014 and September 2014. The results for the samples are provided in Table 2, and the list of targets for these assays is provided in Figures 1 and 2. Due to insufficient archival tissue from the same collection period, for one case, we used tumor tissue samples from different collection periods. All other samples included in this analysis underwent DNA analysis. For PCDx testing, RNA was extracted from all specimens, with the exception of one case for which sufficient tissue was not available. RNA was sequenced to a mean depth of coverage of 21,562×, while DNA copy number and mutation testing were sequenced to a mean depth of 56,085× and 13,656×, respectively.
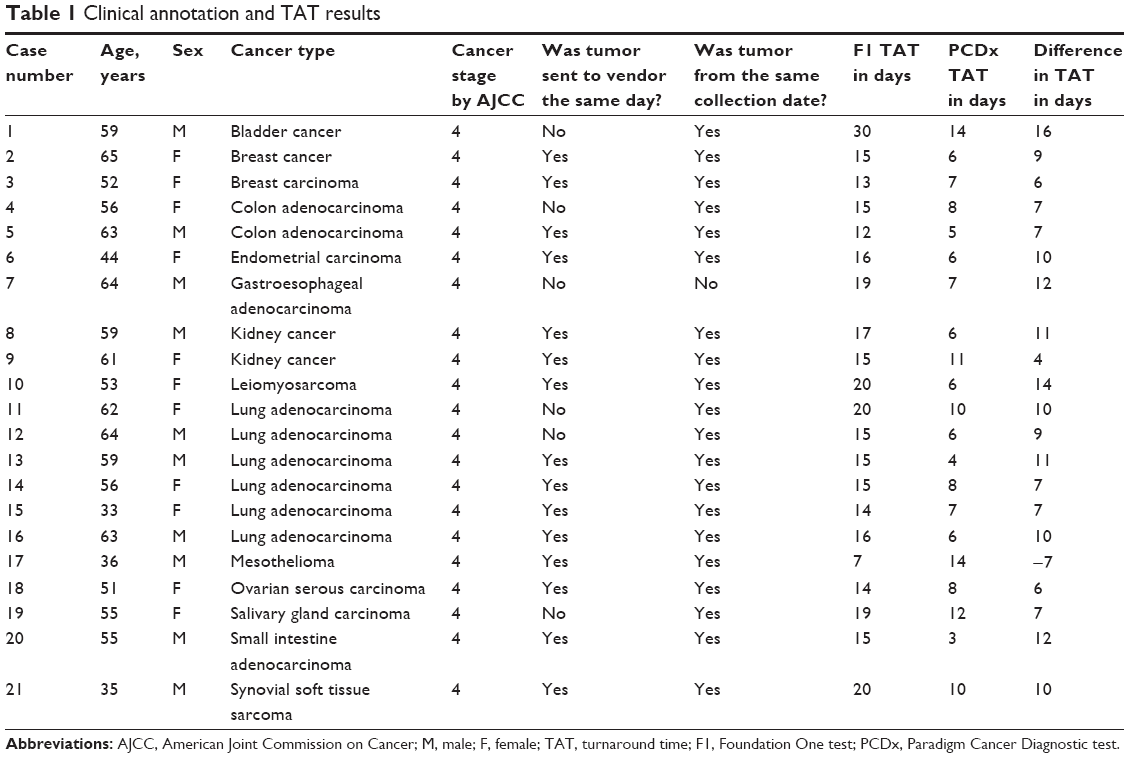 | Table 1 Clinical annotation and TAT results |
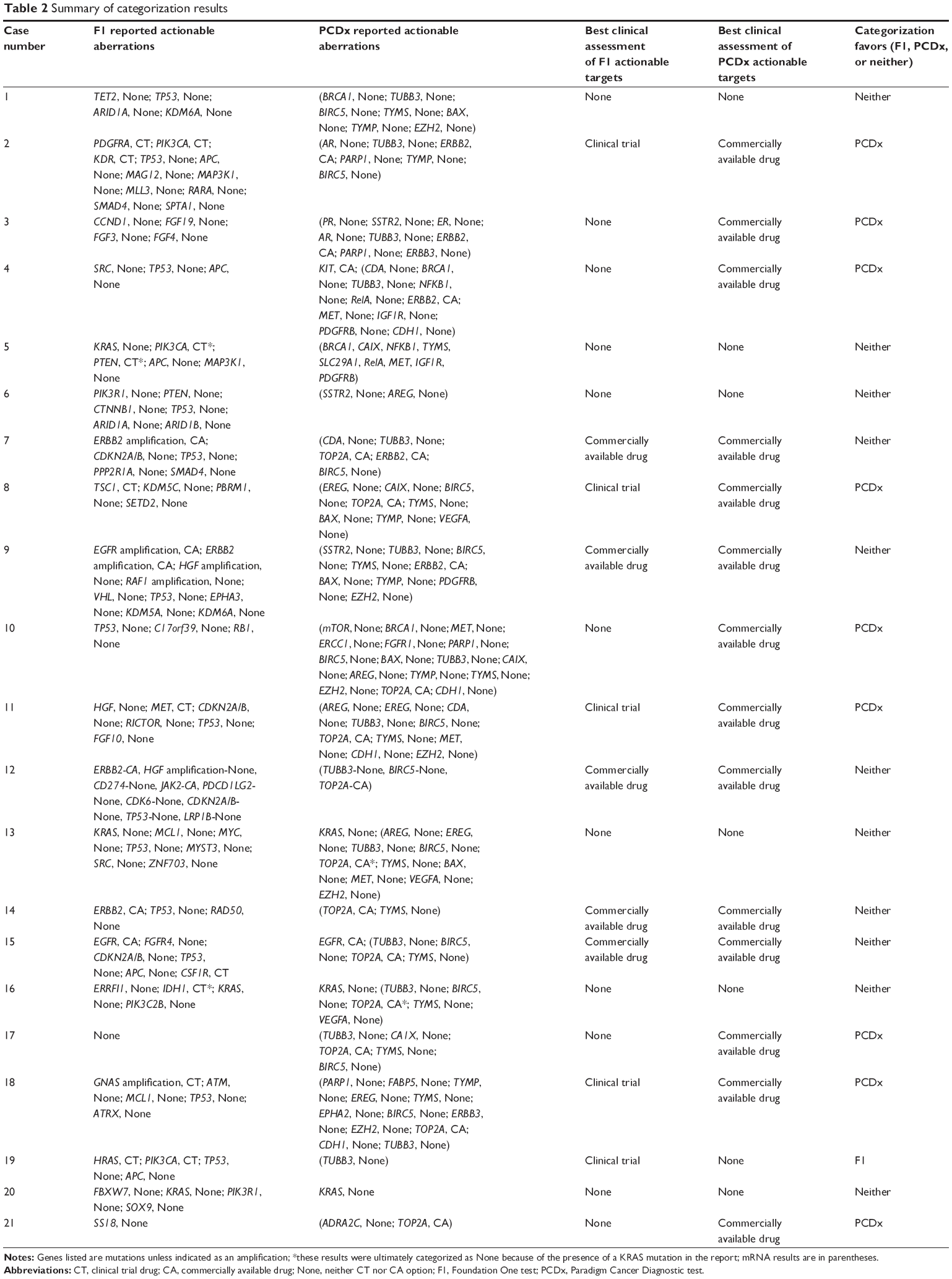 | Table 2 Summary of categorization results |
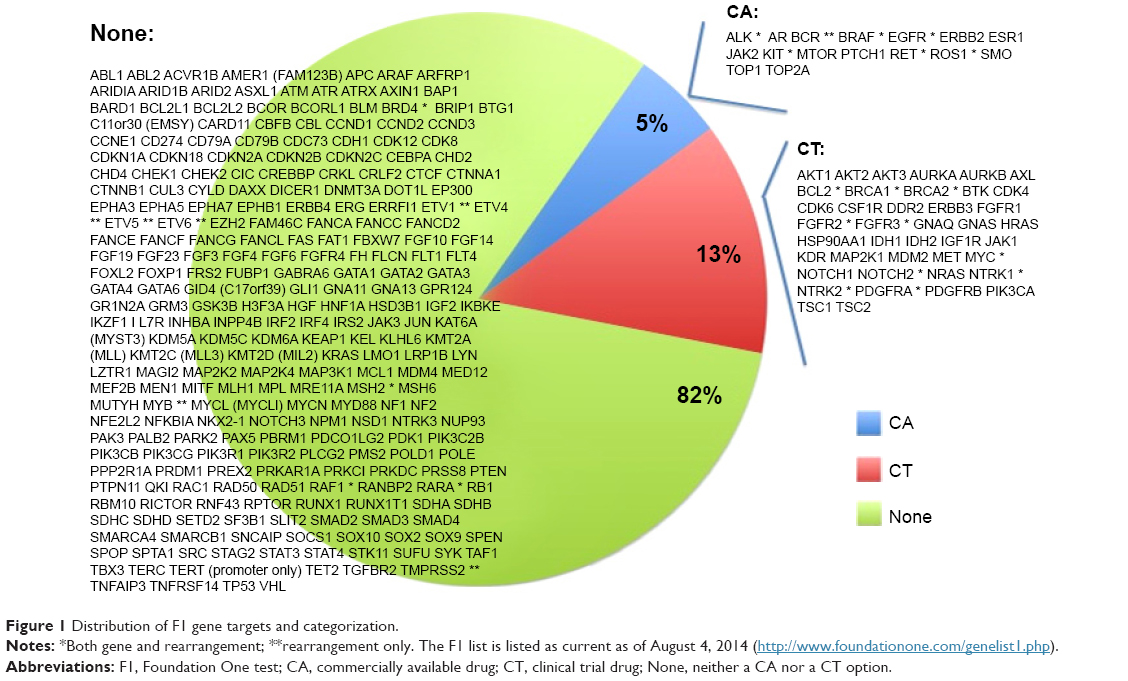 | Figure 1 Distribution of F1 gene targets and categorization. |
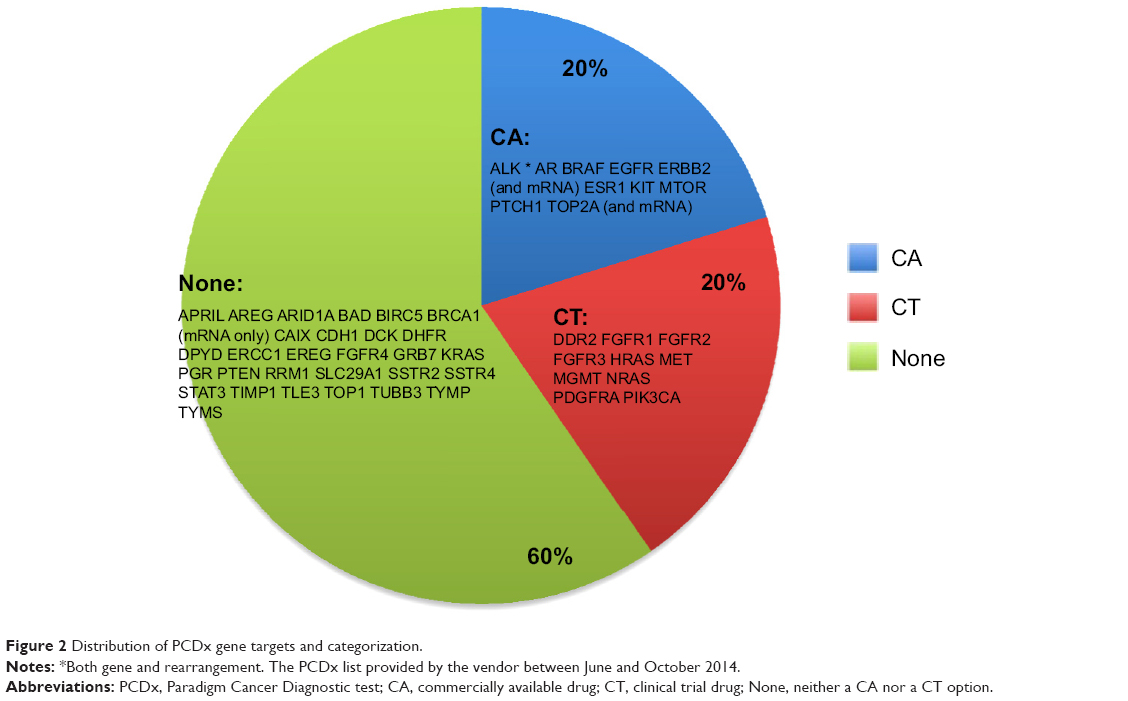 | Figure 2 Distribution of PCDx gene targets and categorization. |
PCDx reported first results faster than F1 in 20 of 21 cases (95.2%), and these TAT were statistically significant (P<0.0001). PCDx had a median TAT of 7 days (range 3–14 days) to report first results compared to F1’s median TAT of 15 days (range 7–30 days). When both vendors received tumor samples on the same day, PCDx reported NGS results first for 14 of 15 cases (93.3%), with a median TAT of 9 days earlier than F1 (range −7 to 14 days). These TAT were also statistically significant (P<0.0001).
PCDx and F1 reported a CA 14 and five times, CT zero and five times, and None seven and eleven times, respectively (P=0.004). Categorization of CA compared to CT and None significantly favored PCDx (P=0.012). PCDx provided higher ranking actionable targets for nine cases vs one case for F1, while the remainder of cases had equivalent ranking for actionable targets. Six paired cases were categorized as None for both NGS platforms. F1 and PCDx platforms yielded eleven and seven cases, respectively, that were categorized as None, resulting in a 47%–67% rate of clinically relevant actionable targets (CA or CT) in diverse cancer types using a commercially available NGS platform.
Discussion
In recent years, there has been a surge of new molecular targeted agents that are available to treating medical oncologists. In parallel, multiplexed molecular tests have arrived on the market, with varying degrees of evidence or summary reports that pose challenges to treating medical oncologists. A key issue in clinical oncology practice is the ability to accurately interpret what is truly clinically meaningful and actionable on a report from a commercially available NGS assay. Recently, 22% of adult cancer physicians from a tertiary-care, National Cancer Institute-designated comprehensive cancer center reported low confidence in their genomic knowledge.10 The majority of cancer care is not delivered at tertiary comprehensive cancer centers, but rather in busy community oncology practices. Treating physicians will have time constraints to read and synthesize NGS assay reports, so there is a need for clear, concise reporting of clinically relevant targets.
In this study, we sought to compare the F1 and PCDx NGS platforms to determine if we could discriminate differences in TAT and clinically relevant actionable biomarkers in tumor tissue from patients with advanced cancers. It should be noted that these comparisons are based on the information provided on the commercial vendors’ reports, the same reports that a treating medical oncologist would be provided. Furthermore, since the purpose of the current study was to compare and evaluate these two NGS platforms, outcome data and prescribed therapeutic agents that may have been based on these reports are not available. As in real clinical practice, when there was more than one tissue block collected at the same time point, multiple blocks were not sent to a commercial vendor to independently run each sample as a unique test with its own unique NGS report. We expected to observe a large degree of overlap on actionable targets from the same individual’s cancer, particularly when tissue collected at the same time point and from the same source was sent to the vendors for analysis.11,12
In the current study, we found that PCDx significantly outperformed F1 in TAT in nearly every paired case by a median time of 9 calendar days. For patients with advanced cancers, particularly those who have recently progressed, the treating physician is trying to make a determination on the next appropriate treatment as soon as possible. The longer the delay in initiating a new treatment for patients with progressing metastatic cancer, the shorter the window of opportunity to alter the disease course is. Recent guidelines for genomic testing to determine first-line therapy in advanced non-small cell lung cancer (NSCLC) called for a 5–10 working day TAT.13 Therefore, in the real world scenario where the use of a NGS testing platform is likely to occur, an even shorter TAT may be more clinically relevant. The results from our present study show the median TAT for F1 is 15 calendar days, while for PCDx, it is 7 calendar days. Considering that shipping the tumor sample to the commercial vendor adds at least 1 calendar day, PCDx’s TAT would fall well within the 5–10 working day TAT to meet the published guidelines for first-line therapy decision making in NSCLC.13
There are two notable discrepancies in the categorization of actionable biomarkers between F1 and PCDx that warrant further elaboration. Case number 5 did not have a KRAS mutation identified on PCDx. During the time frame these analyses were conducted, KRAS exon 4 was not available on the PCDx platform; however, none of the mRNA targets were CA or CT, so the resulting categorization for PCDx was the same as F1. It is important to note that the other three cases with KRAS mutations and the one case with an EGFR mutation were identified on both platforms (cases 13, 16, and 20, and case 15, respectively). In case number 12, while F1 reported a mutation in ERBB2, PCDx did not identify an ERBB2 mutation, as this was not yet available on the PCDx platform at the time of these analyses; it has been added by the vendors since. However, ERBB2 mRNA was not elevated, suggesting that perhaps F1 was likely upstaged with the CA categorization, when in fact there was unlikely protein expression of the ERBB2 biomarker. In this same case, TOP2A mRNA was elevated, so the PCDx result was categorized as CA.
We find in the current study that some commercially available NGS platforms are misleading customers with exceedingly high rates of “actionable targets” that can be offered to their patients based on test results. Barring the drug safety concerns, treatment coverage, and reimbursement issues that arise when prescribing recommended agent(s) off-label either alone or in combination, our study demonstrates that for diverse cancer types, the rate of clinically relevant targets is 47% for F1 and 67% for PCDx. This rate includes both CA and CT targets. The addition of protein expression measurement to an NGS platform would assist in determining clinically relevant targets. Indeed, at least two other vendors are offering immunohistochemistry (IHC) with NGS testing.3,14 Since the analysis of this study has been conducted, PCDx now offers IHC for several actionable biomarkers that would be categorized as CA or CT, including: AR, ER, HER2, MET, MGMT, PTEN, PR, TOP1, TYMS, PD-L1, and PD-1.
At the time of the study, the PCDx test total charge to insurance was US $4,800 per sample vs US $5,800 for the F1 test. The average depth of coverage for PCDx was >5,000× compared with >250× for the F1 test. PCDx reports results based on published clinical literature on drug response. The F1 results are tied most directly to cancer drivers found in the TCGA (The Cancer Genome Atlas), with some of these results also associated with the clinical literature on drug responses. During the study, PCDx interrogated 114 genes (DNA and mRNA combined), whereas the F1 platform tested 315 genes. The fact that PCDx identified clinically relevant actionable alterations more frequently while querying approximately one-third as many genes speaks to the relative utility of the gene sets tested. The interrogation of cancer driver genes does not necessarily have current clinical utility, although some subset of these driver genes is likely to become druggable in the future.
While a potential drawback to our study is that the sample size is potentially too small to draw definite conclusions for a wide range and large number of cancer types and specimens, it does call attention to disparate results on TAT, and to reporting of clinically meaningful targets in commercial vendor NGS assay reports. The results of this study underscore the need to differentiate clinically relevant and meaningful targets with a likelihood of benefit. It is important to distinguish actionable targets associated with CAs vs suggested options that should be tried in the context of a clinical trial vs lack of a reasonable or plausible treatment suggested by NGS analysis. With the integration of additional levels of testing such as proteomics and/or epigenetics, NGS reports can hopefully provide more context and substance that will lead to better outcomes for patients and toward the goal of precision medicine, which is to create better outcomes for patients.
Conclusion
In this analysis, commercially available NGS platforms provided clinically relevant actionable targets (CA or CT) in 47%–67% of diverse cancer types. In the samples analyzed, PCDx significantly outperformed F1 in TAT, and had statistically significant, higher clinically relevant actionable targets categorized as CA.
Acknowledgments
We thank the patients and clinical staff who participated in this research.
Author contributions
All other authors had control of the data and information submitted for publication. All authors contributed toward data analysis, drafting, and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
GJW and SAG have received honoraria from Caris. Paradigm provided Western Regional Medical Center, Inc with PCDx assay testing, and had no role in the control of the data and information submitted for publication. RJP, DWM, SMM, EJT, and DML are current or former employees of Paradigm, a joint venture between the University of Michigan Health System and the International Genomics Consortium. RJP and DWM also have stock in Caris but no current employment relationship with Caris. RJP, DWM, and SMM have stock in Viomics, a blood-based molecular cancer screening company. GJW is a Clinical Oncology Advisor to Viomics. The authors report no other conflicts of interest in this work.
References
Bailey CH, Jameson G, Sima C, et al. Progression-free survival decreases with each subsequent therapy in patients presenting for phase I clinical trials. J Cancer. 2012;3:7–13. | ||
MacConaill LE. Existing and emerging technologies for tumor genomic profiling. J Clin Oncol. 2013;31(15):1815–1824. | ||
Von Hoff DD, Stephenson JJ, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28(33):4877–4883. | ||
Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3(111):111ra121. | ||
Tran B, Brown AMK, Bedard PL, et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer. 2013;132(7):1547–1755. | ||
Weiss GJ, Liang WS, Demeure MJ, et al. A pilot study using next-generation sequencing in advanced cancers: feasibility and challenges. PLoS One. 2013;8(10):e76438. | ||
Servant N, Roméjon J, Gestraud P, et al. Bioinformatics for precision medicine in oncology: principles and application to the SHIVA clinical trial. Front Genet. 2014;5:152. | ||
Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. | ||
US Food and Drug Administration. [Webpage on the Internet]. Hematology/Oncology (Cancer) Approvals & Safety Notifications. Available from http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm. Accessed December 3, 2015. | ||
Gray SW, Hicks-Courant K, Cronin A, Rollins BJ, Weeks JC. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32(13):1317–1323. | ||
Vignot S, Frampton GM, Soria JC, et al. Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J Clin Oncol. 2013;31(17):2167–2172. | ||
Tsongalis GJ, Peterson JD, de Abreu FB, et al. Routine use of the Ion Torrent AmpliSeq™ Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 2014;52(5):707–714. | ||
Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the study of lung cancer/association for molecular pathology guideline. J Clin Oncol. 2014;32(32):3673–3679. | ||
Genomeweb.com [homepage on the Internet]. New York: GenomeWeb; 2015. Available from: https://www.genomeweb.com/clinical-genomics/belgiums-oncodna-sequence-1300-patients-under-breast-international-groups-aurora. Accessed February 21, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.