")
Back to Journals » OncoTargets and Therapy » Volume 12
Evaluating tisagenlecleucel and its potential in the treatment of relapsed or refractory diffuse large B cell lymphoma: evidence to date
Authors Zavras PD, Wang Y , Gandhi A, Lontos K , Delgoffe GM
Received 20 February 2019
Accepted for publication 29 April 2019
Published 11 June 2019 Volume 2019:12 Pages 4543—4554
DOI https://doi.org/10.2147/OTT.S177844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
PD Zavras,1 Y Wang,2,3 A Gandhi,4 K Lontos,2,5 GM Delgoffe2,6
1Infectious Disease Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 2Tumor Microenvironment Center, UPMC Hillman Cancer Center, University of Pittsburgh, Pittsburgh, PA, USA; 3School of Medicine, Tsinghua University, Beijing, People’s Republic of China; 4Blood and Marrow Transplant Program, Department of Medicine, Stanford University, Stanford, CA, USA; 5Division of Hematology/Oncology, University of Pittsburgh Medical Center, Pittsburgh, PA, USA; 6Department of Immunology, University of Pittsburgh, Pittsburgh, PA, USA
Abstract: Chimeric antigen receptor (CAR) T cells have changed the treatment landscape of relapsed or refractory diffuse large B cell lymphoma. This review focuses on the biology of tisagenlecleucel and the clinical data that support its use in this setting. In addition, we discuss how it compares to other CAR T products, the financial implications for payers, and ongoing trials.
Keywords: CAR T, tisagenlecleucel, diffuse large B cell lymphoma, DLBCL, refractory DLBCL, lymphoma
Introduction
Non-Hodgkin lymphoma (NHL) has the seventh highest incidence among all cancers and is responsible for 4% of cancer-related deaths in the USA.1 It is the most common hematological malignancy, with 74,680 estimated new cases in the USA in 2018 and 19,910 projected deaths.2 Diffuse large B cell lymphoma (DLBCL) is the most common subtype of NHL, accounting for 30–40% of newly diagnosed NHL cases.2 While about 40% of patients achieve cure with chemoimmunotherapy, patients with refractory disease have a dismal prognosis with a pooled cure rate of 7%, as reported by the SCHOLAR-1 study.3 As a result, the recent US Food and Drug Administration (FDA) approval of two chimeric antigen receptor (CAR) T products4,5 was welcomed as a breakthrough in the scientific community. This review will discuss the mechanism of action of tisagenlecleucel, its role in the therapeutic armamentarium against DLBCL and future directions.
Structure
T lymphocytes play a vital role in adaptive immunity. The highly variable T cell receptor (TCR) expressed on the surface of T cells can recognize its cognate antigen presented by major histocompatibility complex (MHC) molecules and convey the signal for T cell activation, expansion and function, contributing to pathogen clearance. Immunotherapy based on adoptive T cell transfer can mediate tumor regression, including the transfer of tumor-infiltrating lymphocytes or genetically engineered T cells. Among them, CD19-targeting chimeric antigen receptor-modified T cell therapy has shown promising clinical responses in the treatment of relapsed or refractory (r/r) B cell malignancies.6
A CAR combines the antigen-recognition domains from an antibody with the signaling module of a TCR, and provides efficient targeting of tumor cells independently of the MHC, thus overcoming the limitation of MHC expression and MHC identity in typical TCR recognition.7 The first generation CAR design uses CD3ζ alone as the intracellular signaling component, which cannot generate sufficient antitumor response,8,9 partly because of poor survival of modified T cells in patients.10 Later studies demonstrated that fusion of one or more co-stimulatory domains, such as 4-1BB (CD137) and CD28, to CD3ζ can promote proliferation and persistence of CAR T cells while augmenting their antitumor function.11,12
Tisagenlecleucel (CTL019), one of the CD19-directed CAR T therapies, has been FDA-approved for the treatment of r/r DLBCL after two lines of therapy.4 It is a second generation CAR, designed using a 4-1BB co-stimulatory domain. The single-chain variable fragment (scFv) of tisagenlecleucel is derived from the mouse monoclonal antibody FMC63, which can specifically recognize human CD19 in its native conformation. A graphic representation of its structure is presented in Figure 1. Human CD19 is expressed in most B cell malignancies and the expression is restricted within the B cell lineage, making it an attractive target for CAR T cell therapy. The spacer and transmembrane domain of tisagenlecleucel are derived from human CD8α. The spacer connects the scFv with the transmembrane domain, providing flexibility for the optimal binding of the target antigen. The length and composition of the spacer could exert an influence on the effector function during in vivo tumor treatment.13
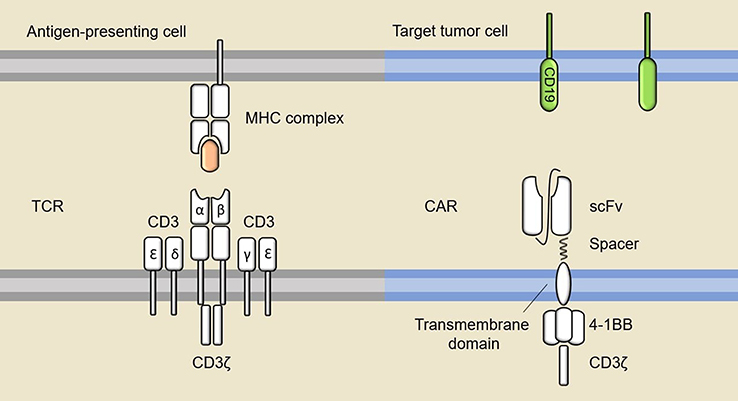 | Figure 1 TCR versus CAR. A typical TCR recognizes peptides presented by MHC molecules on the antigen-presenting cells or tumor cells and convey the signal for T cell activation through CD3. The diagram on the right shows the structure of tisagenlecleucel, which comprises a scFv that can directly bind to target antigen, a transmembrane domain, a co-stimulatory domain and a T cell activation domain.Abbreviations: MHC, major histocompatibility complex; TCR, T cell receptor; CAR, chimeric antigen receptor; scFv, single-chain variable fragment. |
CAR T cells with either a CD28 (axicabtagene ciloleucel) or 4-1BB (tisagenlecleucel) co-stimulatory domain are effective for the treatment of patients with r/r DLBCL.14 However, in animal model-based preclinical research and in clinical trials, the two types of CAR seem to have quite distinct phenotype and behavior, both in vivo and in vitro. Tisagenlecleucel has been reported to persist and remain functional beyond 4 years in some patients, while CD28 CAR T cells are rarely sustained for more than 2 months.15,16 In vitro experiments also suggest that 4-1BB CAR T cells have relatively higher proliferative capacity in culture following stimulation.17,18 In terms of the differentiation status, 4-1BB CAR T cells are more likely to differentiate into central memory T cells with the expression of CD45RO and CCR7, which is associated with sustained antitumor capability. In contrast, CD28 CAR T cells tend to produce a larger proportion of the effector memory cell subset.18
4-1BB co-stimulation has been reported to augment mitochondrial function and biogenesis in CD8+ T cells through the p38–mitogen-activated protein kinase (MAPK) pathway.19 4-1BB CAR T cells correspondingly have an increase in mitochondrial mass, basal oxygen consumption rate, and spare respiratory capacity, indicating they are more reliant on oxidative phosphorylation to generate energy.18 The increased mitochondrial respiratory capacity is also a key characteristic of CD8+ memory T cells. In comparison, CD28 CAR T cells tend to utilize glucose metabolism.18 In terms of signaling, investigators have used liquid chromatography–tandem mass spectrometry (LC-MS/MS) to show that CAR stimulation in both CD28 and 4-1BB CAR induced almost identical protein phosphorylation events. The major difference, however, mainly lies in the kinetics and the strength of signaling. Stimulation through CARs with CD28 co-stimulatory domain alters the phosphorylation states of proteins involved in multiple pathways within a relatively short time and with a larger intensity.20
CAR T cell production
The production of CAR T cells starts with leukapheresis21 and T cell separation using counterflow centrifugal elutriation.22 Further separation of T cell subsets can be achieved through fluorescence-activated cell sorting (FACS) or immunomagnetic methods.23
Transduction of the CAR gene into T cells can be achieved through various methods, the most common of which is the use of a viral vector. Use of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology has also been reported.24 Both lentiviruses and gamma-retroviruses have been used for this purpose, with the former possessing a safer integration site profile,25,26 and thus being preferred in most CAR T cell therapy trials, including CTL019. Before transduction, T cells are activated with beads carrying anti-CD3/CD28 antibodies.27 Then, the vectors are cultured along with the T cells, thereby “injecting” the genetic material responsible for CAR encoding in the form of RNA.28 This CAR encoding information is permanently integrated into the genome of the patient’s cells, as the RNA is reverse-transcribed into DNA. The CAR gene is subsequently translated by the patient’s cells and expressed on the cell surface. The pool of T cells subsequently undergoes expansion, which is usually achieved in a bioreactor. Once the product reaches the target number of cells, it is cryopreserved and transported to the center for infusion into the patient.
Standard of care before CAR T
Upfront chemoimmunotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for DLBCL leads to a cure for approximately 36.5% of the patients older than 60 years after 10 years of follow-up.29 This percentage may actually be higher considering that many patients in this study died from other causes during the long follow-up. Even better results can be observed with younger patients and good-prognosis disease.30 However, many patients either have refractory disease or relapse shortly after they achieve remission. Refractory DLBCL is suggested when the tumor burden has shrunk <50% after first line treatment,31 whereas relapsed DLBCL refers to disease that emerges after a period of complete remission. Patients with r/r DLBCL receive second line chemotherapy, with an overall response rate (ORR) around 40% and complete response (CR) rates around 20%.32 One-third of these patients will subsequently receive autologous stem cell transplantation (ASCT) for consolidation,32 which has been established as the standard of care for this population based on the PARMA trial33 Patients who receive ASCT have a 50% chance of long-term survival without relapse, regardless of the type of response (CR or partial remission [PR]) that they achieved prior to ASCT, as evaluated by computed tomography (CT).34 The response assessed by positron emission tomography (PET) can further refine the prediction of the ASCT outcome, with patients achieving complete metabolic response prior to transplant having an approximately 70% long-term disease-free survival versus 30% for partial responders.35,36 Ultimately, patients who relapse after ASCT have a dismal prognosis, with a median overall survival (OS) of 9.9 months.37 Poor prognostic factors are an International Prognostic Index (IPI) score >2 or relapse within 1 year post-ASCT.37 Patients who did not respond to second line therapy have an even worse prognosis, with median OS of about 4.4 months.38 A follow-up to the CORAL study showed that about 33% of evaluable patients achieved CR with third line regimens,38 but the rest of the patients died soon, probably because of the extent of the disease or accumulating toxicities from chemotherapy. This CR rate is much higher than the one observed in the SCHOLAR-1 study for DLBCL refractory to first or second line chemotherapy or relapse <12 months after ASCT (CR 3–15%).3 This could be partly attributed to the fact that SCHOLAR-1 included patients from observational cohorts and not highly selected patients from clinical trials.
In 1989, Eshhar et al demonstrated the potential of genetically modified T cells in cancer therapy.39 However, it was not until 25 years later that CAR T cells received FDA approval. The first generation anti-CD19 or anti-CD20 CAR T cell administration to refractory DLBCL patients was not very efficacious, probably because of the antitransgene rejection reactions noted in some patients and poor persistence.40 Two clinical trials by Kochenderfer et al were a cornerstone for the establishment of CAR T cell therapy for r/r DLBCL management.41,42 Both trials used constructs with a CD28 co-stimulatory domain. In the first trial, allogeneic anti-CD19 CAR T cells were administered to patients with B cell malignancies who did not respond to or relapsed after allogeneic stem cell transplantation and donor lymphocyte infusions. Three patients exhibited responses.41 In the second trial, patients with r/r DLBCL received conditioning chemotherapy with fludarabine and cyclophosphamide and then an infusion of autologous anti-CD19 CAR T cells. Out of seven patients being treated for r/r DLBCL, four obtained CR, two obtained PR, and one had stable disease after CAR T cell infusion.42 These promising results led to several phase II studies testing different CAR T cell products for r/r DLBCL.
JULIET study
Clinical benefits of CTL019 in human studies were first reported in chronic lymphocytic leukemia (CLL)43 and in acute lymphoblastic leukemia (ALL).44 A single-center phase IIa trial conducted by University of Pennsylvania investigators tested this product in B cell lymphomas. In the interim analysis, six out of 14 patients (43%) with DLBCL achieved CR at 6 months;45 86% of those patients maintained their response at a median follow-up of 28.6 months, thereby demonstrating the durability of treatment.45 This led to a single-arm, multicenter, pivotal phase II trial of CTL019 in adult patients with r/r DLBCL.4 Of the total 165 patients enrolled in the study, 111 (67%) received a single CTL019 infusion (median 3×108 CAR-positive viable T cells) in the inpatient or outpatient setting. Failure to receive cell infusion was primarily due to death, production failure, and/or physician/patient decision to seek other therapy choices during the long period of product delivery (median time from enrollment to infusion was 54 days).4
The median age of patients was 56 years (range 22–76 years) and 76% had stage 3 or 4 disease at the time of enrollment. Nearly all patients (95%) had received at least two and 52% had received at least three prior lines of chemotherapy, whereas 49% had received ASCT.4 Prior to infusion, the majority of patients received lymphodepleting chemotherapy with fludarabine and cyclophosphamide (250 mg/m2/day for 3 days) or bendamustine (90 mg/m2/day for 2 days); seven patients did not receive lymphodepleting chemotherapy, as per the investigator’s decision. From the 111 patients who received CTL019 infusion, the best ORR was 52% (95% CI 41–62%), with 40% CR. At 6 months, CR was 29% and PR was 4%. Four patients with stable disease and 12 patients with PR on imaging at 1 month converted to a CR at a median of 2 months. Patients who exhibited a CR had an estimated probability of 81% of maintaining their response at 1 year. The median OS of the patients who received the infusion was 12 months (95% CI 7 months to not reached). The results of the JULIET study led to FDA approval of tisagenlecleucel for patients with r/r DLBCL (de novo or transformed) after having received two lines of chemotherapy.4
Investigators have tried over the years to identify factors that predict response to CAR T therapy. Studies on patients with ALL and CLL showed that greater expansion and longer persistence of tisagenlecleucel is associated with longer event-free survival.46 However, this was not observed in the JULIET study, where responders and non-responders had similar expansion and persistence of CTL019.4 Response did not depend on CD19 expression either, as the study included patients with no CD19 expression who responded to the infusion. Expression of programmed death-1 (PD-1), lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin and mucin domain-3 (TIM-3) was examined in tumor tissues prior to infusion. None of them was found to be predictive of response but the investigators noted that the five patients with the highest PD-1/PD-L1 interaction score and the 11 patients with the highest number of LAG-3-positive T cells either did not respond or relapsed shortly after their response.
Comparison to other CAR T cells for r/r DLBCL
Two other second generation CAR T cell products have been evaluated for their efficacy against r/r DLBCL. A summary of the differences is shown in Table 1. Their structural difference from tisagenlecleucel lies in the transmembrane and/or co-stimulatory domains, whereas the TCR signaling domain and the anti-CD19 scFv remain the same.4 In contrast to CTL109, axicabtagene ciloleucel (Axi-cel) uses CD28 for both co-stimulatory and transmembrane domains. Lisocabtagene maraleucel (Liso-cel) has IgG4 as a spacer, CD28 for the transmembrane domain, and 4-1BB for the co-stimulatory domain.47 Finally, whereas CTL109 is produced from peripheral blood mononuclear cells enriched for lymphocytes, Liso-cel is produced in a standardized 1:1 CD4+ to CD8+ ratio.
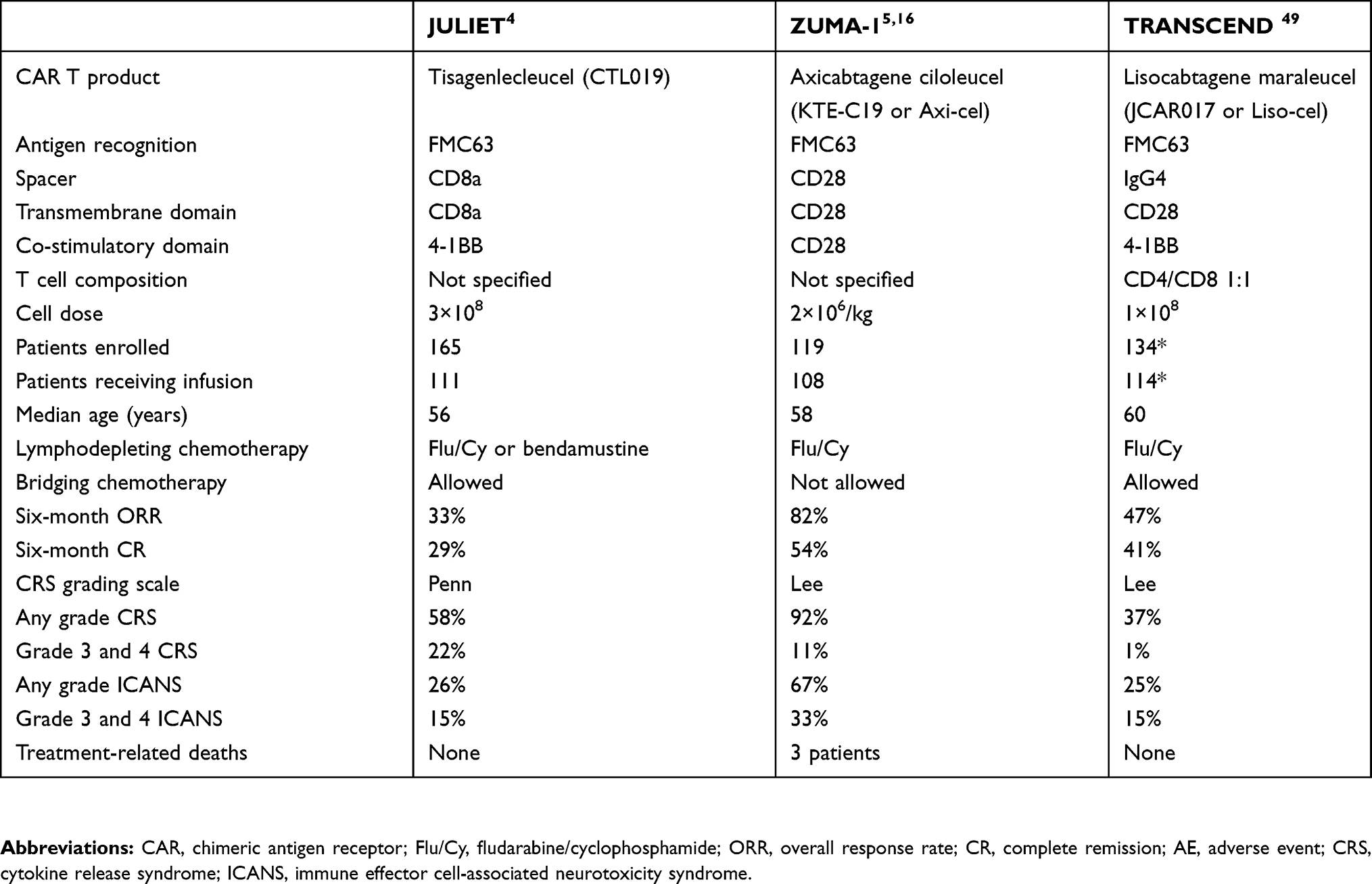 | Table 1 Differences among the three pivotal trials for CAR T cell therapy in aggressive non-Hodgkin lymphoma. The majority of the patients had de novo diffuse large B cell lymphoma |
Axi-cel was the first CAR T cell agent to gain FDA approval for r/r DLBCL, based on the results of the ZUMA-1 study, a phase I/II clinical trial.5 The trial involved patients with r/r DLBCL, primary mediastinal B cell lymphoma and transformed follicular lymphoma who had received at least two prior lines of therapy. Here, the ORR was 83% and the CR 58%.5 Taken together, these results showed a statistically significant 77% decreased risk of early death compared to the standard salvage chemotherapy results of the SCHOLAR-1 study in a retrospective analysis adjusting for multiple variables.48 The major differences between JULIET and ZUMA-1 are the use of cryopreserved apheresis products as well as bridging chemotherapy (in 93% of patients infused) in the former, so as to prevent rapid disease progression.
Liso-cel was investigated through the TRANSCEND NHL 001 study, a phase I trial which has enrolled so far 102 evaluable r/r large B cell lymphoma patients, which, in contrast to ZUMA-1 and JULIET, also included patients with central nervous system (CNS) lymphoma, worse performance status (Eastern Cooperative Oncology Group [ECOG] 2), and relapse post-hematopoietic stem cell transplantation.49 In the CORE population, which included only patients with DLBCL, it showed a 47% ORR and 41% CR rate at 6 months. Liso-cel has already received breakthrough therapy designation from the FDA, and FDA approval is expected in 2019.
Direct comparison among these three products has been difficult for several reasons. First, the eligibility criteria and the patient population differ among the trials. In addition, the numbers of patients who ended up receiving the infusion were not similar, probably owing to manufacturing speed. For example, only 9% of the patients in ZUMA-1 did not receive the infusion versus 33% in the JULIET study.4,16 Thus, patients with more aggressively progressing disease were more likely to be included in the ZUMA-1 trial than in JULIET. Furthermore, bridging chemotherapy between leukapheresis and CAR T cell infusion was allowed in JULIET and TRANSCEND studies, but not in ZUMA-1.4,5,49 The bridging chemotherapy could have decreased the tumor burden, which may have led to less toxicity, as both cytokine release syndrome (CRS) and neurotoxicity have been correlated with tumor burden in ALL studies.50,51 On the other hand, it may have affected the pretreatment inflammatory state of the patient’s body, which is crucial for CAR T cell expansion and persistence. To summarize, direct comparisons are currently infeasible and the final decision on which product to use will most likely depend on the experience that each center has with each product.
Use of tisagenlecleucel in DLBCL after FDA approval
Axi-cel and tisagenlecleucel have both been approved by the FDA for treatment of r/r DLBCL after at least two lines of therapy, including high-grade B cell lymphomas and DLBCL that arises from follicular lymphoma.27,52 Axi-cel has also been approved for r/r primary mediastinal B cell lymphoma. National Comprehensive Cancer Network (NCCN) guidelines have incorporated both as options for DLBCL that responds partially, progresses, or relapses after first salvage therapy including ASCT.53 Most physicians would agree that on progression through first salvage therapy or relapse within 1 year from ASCT, the results of the JULIET study are much improved in comparison to the SCHOLAR-1 study (CR 33% vs 3–15%). However, the role of CAR T after PR to first salvage therapy and relapse more than 1 year after ASCT is contentious. As mentioned above, even the patients who go into ASCT with partial metabolic remission have a 25–30% chance of long-term disease-free survival.35,36 Thus, the choice between ASCT and CTL019 is not straightforward and many people would advocate in favor of ASCT since there are better follow-up data available. In terms of relapse 1 year after ASCT, several small cohorts have been reported with favorable results (median OS >1 year)37,54,55 with either chemotherapy or novel agents, so opting for those modalities before considering CTL019 remains an option.
It is important to highlight that CAR T cells were studied in highly selected patients; patients had ECOG ≤1, no major comorbidities, and no history of CNS pathology. Physicians should remain cautious when extrapolating data from clinical trials to post-FDA approval use in patients, as they can be much sicker, heavily pretreated, and inherently less likely to respond. While there are no “real-world” data available for tisagenlecleucel, we do have the real-world data for Axi-cel.56 Although with a short 30-day follow-up in comparison to the ZUMA-1 report, the ORRs were comparable at 79% versus 82%, and grade 3 or higher CRS 7% versus 13%, with neurotoxicity of 31% in both groups, respectively. Results were comparable within these groups despite differences in patient characteristics, and a long-term follow-up of the real-world group would be reflective of the true benefits. Similarly to the JULIET trial, 56% of patients in the Axi-cel real-world study received bridging chemotherapy between apheresis and cell infusion. In JULIET, the investigators withdrew 23% patients for other reasons, apart from problems in manufacturing.4 The primary reasons were death or physician investigator decision. This decision was mainly driven by fast progression of the disease even though bridging chemotherapy was allowed up to 2 weeks before infusion. This is reflective of the difficulty in managing a progressing patient while waiting for CAR T cell manufacture. It is unlikely that the insurance companies would ever approve the manufacturing and cryopreservation of CAR T cells on first relapse for future use to eliminate the long manufacturing time. However, pharmaceutical companies may elect to provide such programs in the future to boost the use of their products.
Adverse effects
CAR T cell therapy-specific adverse effects have been recognized. The most common ones are CRS, neurotoxicity, hypogammaglobinemia, and prolonged cytopenias. In December 2018, the American Society for Blood and Marrow Transplantation (ASBMT) published consensus guidelines on the criteria used to define and grade CRS as well as neurotoxicities.57
CRS
CRS refers to a hyperinflammatory state that results from a marked increase in serum pro-inflammatory cytokines.58 Patients present with a constellation of symptoms, starting from flu-like symptoms with fever and occasionally progressing to hypoxia and hypotension requiring corrective measures.27 CRS is the most common adverse effect of CAR T cell therapy, with its frequency ranging from approximately 80% to 90% of the patients infused.4,16,59 The onset, duration, and severity of adverse events differ among products. The investigators of the JULIET study reported 3 days as the median time of onset from infusion and 7 days as the median duration of symptoms.4
The grading system of CRS was different in each study. While the CTL019 studies used the Penn grading system,15 the Axi-cel studies used the Lee criteria.60 The differences between the two systems were recently reviewed.61 Based on the ALL studies, the severity of the syndrome highly depends on the tumor burden at the time of CAR T cell infusion.46,62 However, this has not been validated in the lymphoma studies. Teachey et al showed significantly elevated serum interferon-γ (IFNγ), IL-6, IL-8, IL-10, soluble IL-2 receptor-α (sIL2Rα), interferon-γ-inducible protein-10 (IP10), monocyte chemoattractant protein-1 (MCP1), monokine induced by interferon-γ (MIG), and macrophage inflammatory protein-1β (MIP1β) levels among patients with ALL who were treated with CTL019 with CRS grade 4 or higher compared to grades 0–3 CRS.63 C-reactive protein (CRP) can potentially serve as a marker of CRS severity, as patients with higher grade CRS were shown to have significantly more elevated CRP.58,63
The Penn grading scale provides detailed guidance about the treatment algorithm of CRS.61 Mild CRS (grade 1) is treated with supportive care, such as antipyretics and antiemetics. Moderate reaction (grade 2) requires hospitalization for management of CRS-related symptoms, such as fever with neutropenia and the need for intravenous therapies. More severe reaction (grade 3) also prompts hospitalization for management of hypotension with intravenous fluids or low-dose vasopressors, coagulopathy with fresh-frozen plasma, cryoprecipitate, or fibrinogen concentrate, and hypoxia with supplemental oxygen. Life-threatening complications, such as hypotension requiring high-dose vasopressors and hypoxia requiring mechanical ventilation, are classified as grade 4 CRS. Meanwhile, the Lee scale defines grade 2 CRS as hypotension that responds to fluid boluses or a low dose of one vasopressor, with a higher dose required or the addition of a second vasopressor for grade 3 CRS. To date, the mainstay of severe CRS management is IL-6-receptor blockade with the monoclonal antibody tocilizumab. FDA approval was granted based on retrospective analysis of two cohorts, demonstrating response rates 53–69%.64 Tocilizumab is indicated for grade 4 CRS on the Penn scale or grade 2 CRS on the Lee scale based on the FDA-approved package inserts.27,52
Lymphotoxic high-dose steroids (>100 mg daily) have similar efficacy to tocilizumab in reducing CRS symptoms within 1–3 days, but the latter has the advantage of not inhibiting the CAR T cell function, thereby preserving the anti-cancer treatment effect.58 Low-dose corticosteroids, ie, ≤2 mg/kg, and especially when administered early after the onset of symptoms of CRS, on the other hand, did not seem to impact the in vivo expansion of CAR T cells.59,65 However, these data are from the ALL studies. The investigators of ZUMA-1 reported similar response rates between patients who received and those who did not receive steroids.16 In JULIET, 10% of patients received steroids but their response rate was not reported.4 To date, steroids (including methylprednisolone, hydrocortisone, and dexamethasone) are reserved for tocilizumab-refractory CRS.27
The ongoing trial NCT02906371 aims to show whether early administration of tocilizumab prior to CRS development in patients with high tumor burden would be beneficial. IL-6 levels cannot be used to guide pre-emptive therapy, as IL-6 does not significantly rise before the development of CRS and, thus, cannot be a trustworthy marker of subsequent CRS development and the need for tocilizumab.63 The direct IL-6 inhibitor siltuximab is also being used for management of CRS refractory to tocilizumab and steroids by some groups.66 Preclinical data support the theory that IL-1 blockade could also be highly effective in treating CRS,67,68 and clinical trials are likely to start using anakinra (an IL-1-receptor antagonist) in the near future.
Neurotoxicity
Although neurological manifestations can occur in the spectrum of CRS, CAR T cell-related neurotoxicity is distinct from CRS and is termed CAR T cell-related encephalopathy syndrome (CRES).69 More recently, it was named immune effector cell-associated neurotoxicity syndrome (ICANS) by the ASBMT.57 Symptoms range from mild impairment, confusion, aphasia, disorientation, impaired handwriting, tremors, and somnolence, to more severe impairment, eg, seizures, motor weakness, obtundation, increased intracranial pressure (ICP), and cerebral edema.69 Transient neurological symptoms have ranged between 23% and 67% in phase I and II clinical trials for DLBCL.4,5,49 All trials used the Common Terminology Criteria for Adverse Events (CTCAE) grading. ICANS can occur simultaneously with CRS or after CRS has subsided.69 The pathophysiology of ICANS has not been fully elucidated but it seems that high levels of cytokines, endothelial activation, and increased blood–brain barrier permeability play a role.50,51
ICANS is usually fully reversible, even though fatal cases have been reported.5 However, the JULIET trial did not report any deaths related to CTL019.4 Management of ICANS depends on the grade; however, all patients will need assessment for increased ICP with fundoscopy, whereas neuroimaging is reserved for higher grade ICANS.69 The strategy was different among different products but both trials used tocilizumab if CRS was concurrent with ICANS.4,5 Steroids for ICANS were more frequently used with Axi-cel, whereas in JULIET only two patients received steroids because of persistence of ICANS after CRS had resolved.4 This difference was probably related to the higher rates of neurotoxicity that were observed in the ZUMA-1 trial. Additional treatment with anti-epileptics and hyperventilation/hyperosmolar therapy may be required for the management of seizures and cerebral edema, respectively.69 Although not standard of care, levetiracetam is being used for seizure prophylaxis in high-risk patients or in patients receiving a product that is known to have a high incidence of ICANS, such as Axi-cel.
Hypogammaglobulinemia
B cell depletion by CD19 CAR T cells invariably leads to hypogammaglobinemia, which can be managed with immunoglobulin replacement.43,70 Of note, the B cell count can be used as a pharmacodynamic measure of CTL019 functional persistence in the blood, and B cell aplasia is associated with the duration of remission in children and young adults with ALL.44 However, in ZUMA-1 75% of patients who had ongoing responses at 2 years exhibited B cell recovery, while some of them started recovering their B cells at 9 months.5 These data suggest that sustained responses in lymphoma patients do not require long-term persistence of CAR T cells.
Pancytopenia
Cytopenias are seen almost universally in patients who receive CAR T cell infusions. Clinicians attribute them mostly to the lymphodepleting chemotherapy but often they persist for much longer. For example, in the JULIET study, 41% of the treated patients had grade 3/4 thrombocytopenia and/or grade 3/4 neutropenia 28 days from the time of infusion. Whereas all grade 3/4 neutropenias had resolved at 3 months from the infusion, 38% of grade 3/4 thrombocytopenias persisted. Preclinical data from studies on mice suggest that CAR T cells impact bone marrow progenitors regardless of the CAR T target, which is suggestive of toxicity secondary to the cytokine-rich microenvironment after the cell infusion.71 Investigators from the National Institutes of Health noted that in an anti-CD22 CAR T cell trial, the cytopenias were present despite evidence of trilineage hematopoiesis in the bone marrow, which differs from the chemotherapy-induced myelosuppression.72 In addition, the patients‘ neutrophils improved rapidly upon the administration of granulocyte colony-stimulating factor, which again argues in favor of an alternative mechanism of neutropenia (ie, myelokathexis) rather than myelosuppression.
Toxicity comparison among different CAR T cell agents
Toxicities were reported in all lymphoma trials with CAR T cells. However, there are some differences in the incidence of these toxicities among the trials. CTL019 led to CRS of any grade in 58% of patients enrolled in the JULIET study; 22% developed CRS grade ≥3; tocilizumab was administered in 14% of patients.4 ICANS was diagnosed in 26% of patients, whereas 15% had grade 3 or 4 ICANS.4 In ZUMA-1, 92% of the patients developed CRS (11% had CRS grade ≥3 and 43% received tocilizumab), and 67% developed neurotoxicity (33% had ICANS grade 3 or 4).5,16 For Liso-cel, 37% of patients on the updated FULL cohort of the TRANSCEND study developed CRS (only 1% had CRS grade ≥3, whereas 21% received tocilizumab), and 23% developed neurotoxicity (13% had ICANS grade 3 or 4).49 Direct comparison of side effects reported in these trials should be approached with caution given the differences in trial and product design, baseline characteristics, type of lymphodepleting chemotherapy, bridging chemotherapy, and different grading scales. Future studies should be designed using the harmonized definitions and criteria for CRS and neurotoxicity as published in the 2018 ASBMT consensus guidelines.57
Financial implications
CTL019 was first approved for refractory B cell ALL, and the US$475,000 cost stirred up discussions among insurance companies, policy-makers, physicians and patients.73 However, after the FDA approval for r/r DLBCL, Novartis matched the price of Gilead’s Axi-cell at $373,000 only for that indication.74 This is only the cost for treatment acquisition. Severe adverse reactions, such as neurotoxicity and CRS, which may require anti-IL-6 factor administration, ie, tocilizumab, can add an additional cost of up to $200,000.75 This cost has to be compared to the cost of treating an r/r DLBCL patient in the pre-CAR T cell era. The total lifetime cost of r/r DLBCL patient management can approach $600,000–750,000 per patient, including first and second line treatments, as well as the cost of care for adverse events.76 Around 80% of this cost is attributable to the acquisition of third line and subsequent treatment, and care for adverse events from these therapies.76 The Institute for Clinical and Economic Review (ICER) issued a report on the cost-effectiveness of CAR T therapies.77 According to the report, CTL019 and Axi-cel are cost-effective for all approved indications. The committee did not have available outcomes of CTL019 on DLBCL at that time-point, but they reported $45,871 cost per quality-adjusted life year (QALY) gained for CTL019 in ALL and $136,078 per QALY gained for Axi-cel in r/r DLBCL. These costs meet the generally accepted threshold of $50,000–150,000 per QALY gained. These numbers may change as we obtain more information on long-term outcomes, as shown in cost-effectiveness studies of CAR T cells in ALL.78,79 It is important to note that the ICER report is solely targeted to the US market. For example, the UK-based National Institute for Health and Care Excellence initially deemed CTL019 too expensive for the UK National Health Service80 and approved it only after the company offered the product at a confidential discounted price.81
The first year after the FDA approval of CTL019 for ALL was difficult for hospitals as there was no billing code for administration of CAR T cells, creating significant financial strain on the institutions. On August 17, 2018, the Center for Medicare Services (CMS) finally added new ICD-10 procedure codes for the outpatient administration of CAR T cells and modified the existing diagnosis-related group (DRG) for autologous bone-marrow transplantation to include CAR T administration.82 Despite the positive steps, the American Society of Hematology and the ASBMT raised significant concerns as these changes were created to cover outpatient administration of CAR T cells, while the majority of the hospitals in the USA have not yet acquired the experience to perform the infusions in the outpatient setting.83 Apart from that, based on CMS’s 3-day payment rule, if patients who receive infusion of CAR T cells in the outpatient setting develop side effects that need inpatient care within 72 hours of treatment, the cost of CAR T administration is transferred to the DRG that the hospital will use to cover the patient’s hospitalization.84 As more CAR T products for a variety of indications are likely to gain FDA approval in the coming years, these controversies are expected to intensify.
Future potential
CTL019 has proven to be a revolution in the treatment of hematological malignancies. The scientific community is striving to further improve the outcomes. At the time of writing in 2019, there are five active trials using CTL019 in DLBCL. A summary of the trials is shown in Table 2. First, NCT03570892 (BELINDA) is a randomized, open-label, phase III trial comparing CTL019 to first salvage in patients with DLBCL who were refractory or relapsed after rituximab- and anthracycline-containing front-line chemoimmunotherapy. CTL019 is not yet indicated for the first salvage setting and this trial will challenge the standard of care. One of the trials was initiated after investigators at the University of Pennsylvania observed a response to anti-PD-1 therapy in a patient with DLBCL who progressed after CTL019 infusion.85 NCT02650999 is enrolling patients with DLBCL who relapse or fail to respond to CTL019 infusion. Patients receive pembrolizumab and the primary outcome is safety. Preliminary results have been presented and indicate that pembrolizumab is active in this setting.86 NCT03630159 (PORTIA) is a similar trial which aims to determine the efficacy of the combined use of pembrolizumab along with CTL019 in patients with r/r DLBCL. The FDA approval of CTL019 for r/r DLBCL was confined to adults. NCT03610724 (BIANCA) will establish the safety and efficacy of CTL019 in pediatric NHL patients, while NCT02374333 will help the scientific community to understand the role of reinfusion in r/r DLBCL after cell therapy.
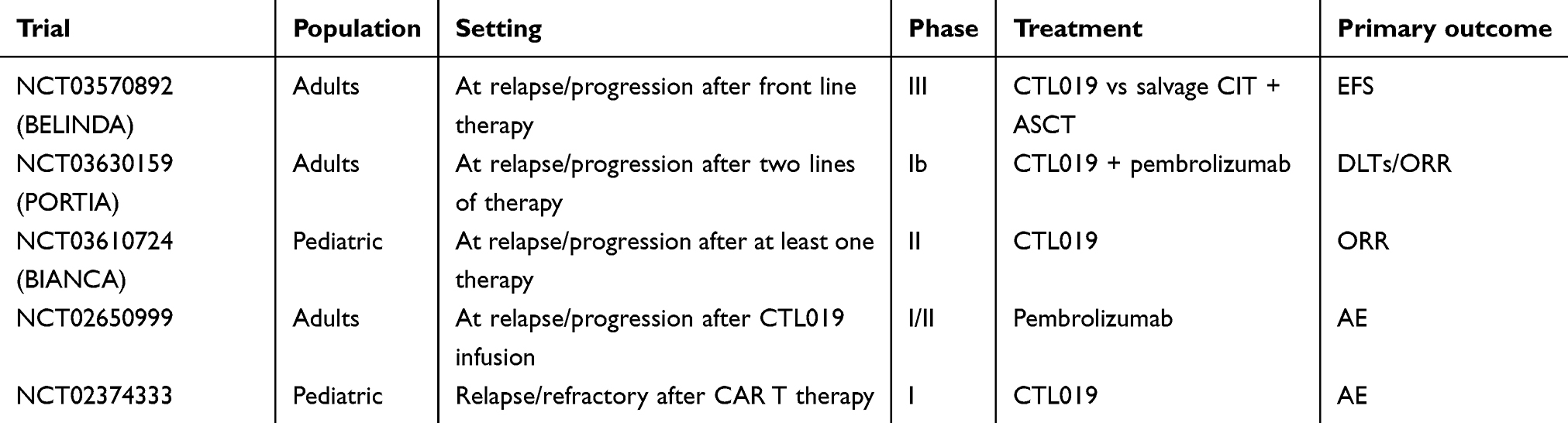 | Table 2 Ongoing trials with CTL019 in diffuse large B cell lymphoma |
Acknowledgments
This work was supported by the Alliance for Cancer Gene Therapy (funds to GMD), Stand Up To Cancer–American Association for Cancer Research (SU2C-AACR-IRG-04-16 to GMD), and a National Institutes of Health Director’s New Innovator Award (DP2AI136598 to GMD).
Disclosure
Dr PDZ has received research funding from Merck & Co., and grants from Merck, outside the submitted work. Dr GMD reports grants from Bluebirdbio, grants and personal fees from TTMS Inc, and grants from Pfizer and TCR2 Therapeutics, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. SEER cancer stat facts: non-hodgkin lymphoma. Available from:
3. Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–1808. doi:10.1182/blood-2017-03-769620
4. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
5. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20(1):31–42.
6. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi:10.1038/nature22395
7. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (New York, NY). 2018;359(6382):1361–1365. doi:10.1126/science.aar6711
8. Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653–1659.
9. Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000;96(5):1999–2001.
10. Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115.
11. Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi:10.1172/JCI46110
12. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi:10.1056/NEJMoa1103849
13. Hudecek M, Sommermeyer D, Kosasih PL, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi:10.1158/2326-6066.CIR-14-0127
14. Chow VA, Shadman M, Gopal AK. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood. 2018;132(8):777–781. doi:10.1182/blood-2018-04-839217
15. Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi:10.1126/scitranslmed.aad3106
16. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
17. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004
18. Kawalekar OU, O’connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(3):712. doi:10.1016/j.immuni.2016.02.023
19. Menk AV, Scharping NE, Rivadeneira DB, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215(4):1091–1100. doi:10.1084/jem.20171068
20. Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11:544. doi:10.1126/scisignal.aat6753
21. Smith JW. Apheresis techniques and cellular immunomodulation. Ther Apheresis. 1997;1(3):203–206.
22. Powell DJ
23. Riddell SR, Sommermeyer D, Berger C, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer Jl (Sudbury, Mass). 2014;20(2):141–144. doi:10.1097/PPO.0000000000000036
24. Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi:10.1038/nature21405
25. McGarrity GJ, Hoyah G, Winemiller A, et al. Patient monitoring and follow-up in lentiviral clinical trials. J Gene Med. 2013;15(2):78–82. doi:10.1002/jgm.2691
26. Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119(4):964–975. doi:10.1172/JCI37630
27. Kymriah FDA package insert. Available from:
28. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. doi:10.1016/j.omtm.2016.12.006
29. Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the groupe d‘Etudes des Lymphomes de l‘Adulte. Blood. 2010;116(12):2040–2045. doi:10.1182/blood-2010-03-276246
30. Pfreundschuh M, Kuhnt E, Trumper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera international trial (MInT) group. Lancet Oncol. 2011;12(11):1013–1022. doi:10.1016/S1470-2045(11)70235-2
31. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the lugano classification. J Clin Oncol. 2014;32(27):3059–3068. doi:10.1200/JCO.2013.54.8800
32. van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: the ORCHARRD study. J Clin Oncol. 2017;35(5):544–551. doi:10.1200/JCO.2016.69.0198
33. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin‘s lymphoma. N Engl J Med. 1995;333(23):1540–1545. doi:10.1056/NEJM199512073332305
34. Gisselbrecht C, Schmitz N, Mounier N, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30(36):4462–4469. doi:10.1200/JCO.2012.41.9416
35. Schot BW, Zijlstra JM, Sluiter WJ, et al. Early FDG-PET assessment in combination with clinical risk scores determines prognosis in recurring lymphoma. Blood. 2007;109(2):486–491. doi:10.1182/blood-2005-11-006957
36. Redondo AM, Valcarcel D, Gonzalez-Rodriguez AP, et al. Bendamustine as part of conditioning of autologous stem cell transplantation in patients with aggressive lymphoma: a phase 2 study from the GELTAMO group. Br J Haematol. 2019;184(5):797–807. doi:10.1111/bjh.15713
37. Nagle SJ, Woo K, Schuster SJ, et al. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am J Hematol. 2013;88(10):890–894. doi:10.1002/ajh.23524
38. Van Den Neste E, Schmitz N, Mounier N, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the international CORAL study. Bone Marrow Transplant. 2016;51(1):51–57. doi:10.1038/bmt.2015.213
39. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028.
40. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–1256. doi:10.1016/j.bbmt.2010.03.014
41. Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129. doi:10.1182/blood-2012-12-471029
42. Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi:10.1200/JCO.2014.56.2025
43. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi:10.1126/scitranslmed.3002842
44. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
45. Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi:10.1056/NEJMoa1708566
46. Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130(21):2317–2325. doi:10.1182/blood-2017-06-786129
47. Havard R, Stephens DM. Anti-CD19 chimeric antigen receptor T cell therapies: harnessing the power of the immune system to fight diffuse large B cell lymphoma. Curr Hematol Malig Rep. 2018;13:534–542. doi:10.1007/s11899-018-0482-6
48. Neelapu S, Locke F, Bartlett N, et al. SCHOLAR-1 versus ZUMA-1: a standardized comparison of outcomes in patients (Pts) with refractory, aggressive non-hodgkin lymphoma (rNHL). Clin Lymphoma Myeloma Leukemia. 2017;17:S362–S363. doi:10.1016/j.clml.2017.07.197
49.
50. Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958–971. doi:10.1158/2159-8290.CD-17-1319
51. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–1419. doi:10.1158/2159-8290.CD-17-0698
52. Yescarta FDA package insert. Available from:
53. NCCN guidelines B-cell lymphomas version 1.2019. Available from:
54. Van Den Neste E, Schmitz N, Mounier N, et al. Outcomes of diffuse large B-cell lymphoma patients relapsing after autologous stem cell transplantation: an analysis of patients included in the CORAL study. Bone Marrow Transplant. 2017;52(2):216–221. doi:10.1038/bmt.2016.213
55. Hunter BD, Herr M, Meacham PJ, et al. Late relapses after high-dose chemotherapy and autologous stem cell transplantation in patients with diffuse large B-cell lymphoma in the rituximab era. Clin Lymphoma Myeloma Leuk. 2017;17(3):145–151. Doi:10.1016/j.clml.2016.11.001
56. Nastoupil LJ, Spiegel MD, Ghobadi JY, et al. Axicabtagene Ciloleucel (Axi-Cel) CD19 Chimeric Antigen Receptor (CAR) T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma: Real World Experience. San Diego, CA: ASH Annual Meeting; 2018.
57. Lee DW, Santomasso BD, Locke FL, et al. ASBMT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638
58. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi:10.1126/scitranslmed.3008226
59. Gardner R, Leger KJ, Annesley CE, et al. Decreased rates of severe CRS seen with early intervention strategies for CD19 CAR-T cell toxicity management. Blood. 2016;128(22):586. doi:10.1182/blood-2016-06-724161
60. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
61. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11(1):35. doi:10.1186/s13045-018-0571-y
62. Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138. doi:10.1126/scitranslmed.3005930
63. Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor t-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–679. doi:10.1158/2159-8290.CD-16-0040
64. Le RQ, Li L, Yuan W, et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor t cell-induced severe or life-threatening cytokine release syndrome. Oncologist. 2018;23(8):943–947. doi:10.1634/theoncologist.2018-0028
65. Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018. doi:10.1158/1078-0432.CCR-18-0758
66. Teachey DT, Bishop MR, Maloney DG, Grupp SA. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit ‘ALL’. Nat Rev Clin Oncol. 2018;15(4):218. doi:10.1038/nrclinonc.2018.19
67. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–738. doi:10.1038/s41591-018-0041-7
68. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
69. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148
70. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–4023. doi:10.1182/blood-2014-12-580068
71. Sauter CT, Chien CD, Shen F, Tasian SK, Fry TJ. Evaluating on-target toxicity of hematopoietic-targeting cars demonstrates target-nonspecific suppression of marrow progenitors. Blood. 2016;128(22):3357.
72. Shalabi H, Shah NN, Fry TJ, Yates B, Delbrook C. Chimeric antigen receptor induced cytopenia differs from chemotherapy induced myelosuppression. Blood. 2017;130(Suppl 1):5048.
73. Bach PB, Giralt SA, Saltz LB. FDA approval of tisagenlecleucel: promise and complexities of a $475000 cancer drug. Jama. 2017;318(19):1861–1862. doi:10.1001/jama.2017.15218
74. Novartis matches gilead on price in new CAR-T use.
75. Chabannon C, Kuball J, McGrath E, et al. CAR-T cells: the narrow path between hope and bankruptcy? Bone Marrow Transplant. 2017;52(12):1588–1589. doi:10.1038/bmt.2017.241
76. Garcia J, Snyder S, Gitlin M. Estimating the lifetime costs in adult patients with relapsed/refractory diffuse large B-cell lymphoma in the United States. Value Health. 2018;21:S27. doi:10.1016/j.jval.2018.04.172
77.
78. Whittington MD, McQueen RB, Ollendorf DA, et al. Long-term survival and value of chimeric antigen receptor T-cell therapy for pediatric patients with relapsed or refractory leukemia. JAMA Pediatr. 2018;172(12):1161–1168. doi:10.1001/jamapediatrics.2018.2530
79. Lin JK, Lerman BJ, Barnes JI, et al. Cost effectiveness of chimeric antigen receptor T-cell therapy in relapsed or refractory pediatric B-cell acute lymphoblastic leukemia. J Clin Oncol. 2018;JCO2018790642.
80. NICE encourages further discussions on Kymriah for adult lymphoma. [Published September 19, 2018]. Available from:
81. NICE recommends another revolutionary CAR T-cell therapy for adults with lymphoma. [Published February 1, 2019]. Available from:
82. CMS finalizes CAR T-cell therapy inpatient payments. Available from:
83.
84. Implementation of new statutory provision pertaining to Medicare 3-day (1-day) payment window policy - outpatient services treated as inpatient. Available from:
85. Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245
86. Chong ES, Nasta SD, Landsburg DJ, et al. Sequential Anti-CD19 Directed Chimeric Antigen Receptor Modified T-Cell Therapy (CART19) and PD-1 Blockade with Pembrolizumab in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphomas. San Diego, CA: ASH Annual Meeting; 2018.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.