Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect
Authors Schoedel K, Morrow S, Sellers E
Received 14 March 2014
Accepted for publication 7 May 2014
Published 26 June 2014 Volume 2014:10 Pages 1161—1174
DOI https://doi.org/10.2147/NDT.S30713
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Kerri A Schoedel,1 Sarah A Morrow,2 Edward M Sellers3,4
1Altreos Research Partners, Inc., Toronto, 2Western University, London, 3DL Global Partners, Inc., Toronto, 4University of Toronto, Toronto, Ontario, Canada
Abstract: Pseudobulbar affect (PBA) is a common manifestation of brain pathology associated with many neurological diseases, including amyotrophic lateral sclerosis, Alzheimer’s disease, stroke, multiple sclerosis, Parkinson’s disease, and traumatic brain injury. PBA is defined by involuntary and uncontrollable expressed emotion that is exaggerated and inappropriate, and also incongruent with the underlying emotional state. Dextromethorphan/quinidine (DM/Q) is a combination product indicated for the treatment of PBA. The quinidine component of DM/Q inhibits the cytochrome P450 2D6-mediated metabolic conversion of dextromethorphan to its active metabolite dextrorphan, thereby increasing dextromethorphan systemic bioavailability and driving the pharmacology toward that of the parent drug and away from adverse effects of the dextrorphan metabolite. Three published efficacy and safety studies support the use of DM/Q in the treatment of PBA; significant effects were seen on the primary end point, the Center for Neurologic Study-Lability Scale, as well as secondary efficacy end points and quality of life. While concentration–effect relationships appear relatively weak for efficacy parameters, concentrations of DM/Q may have an impact on safety. Some special safety concerns exist with DM/Q, primarily because of the drug interaction and QT prolongation potential of the quinidine component. However, because concentrations of dextrorphan (which is responsible for many of the parent drug’s side effects) and quinidine are lower than those observed in clinical practice with these drugs administered alone, some of the perceived safety issues may not be as relevant with this low dose combination product. However, since patients with PBA have a variety of other medical problems and are on numerous other medications, they may not tolerate DM/Q adverse effects, or may be at risk for drug interactions. Some caution is warranted when initiating DM/Q treatment, particularly in patients with underlying risk factors for torsade de pointes and in those receiving medications that may interact with DM/Q.
Keywords: clinical pharmacology, Nuedexta, drug interactions, CYP2D6, QTc interval, CNS-LS
Introduction
Pseudobulbar affect (PBA), also called pathological laughing and crying (PLC), is a common manifestation of brain pathology. The terms used to describe this abnormally expressed emotion have varied over the years, including disinhibition, affective lability, emotional dysregulation, and involuntary emotional expression disorder.1 However, PLC and PBA are more specifically defined disorders that distinguish more clearly between mood and affect. PLC is defined as involuntary and uncontrollable expressed emotion (laughing or crying or both) that is exaggerated relative to the stimulus as well as the experienced emotion.2 In contrast, PBA is a broader term in which the emotion is still exaggerated and inappropriate and also incongruent with the underlying emotional state.1 Thus, both involve discordance between mood and the degree of expressed affect.
A defining feature of PBA is that it is due to a brain disorder. It has been associated with many neurological diseases. The exact prevalence has been difficult to determine, partly due to different definitions being used to describe the disorder. The best estimate of PBA prevalence comes from Work et al (2011).3 Her group found that the prevalence in neurological disorders ranges from 9.4%–37.5% depending on the criteria used for diagnosis. PBA is most common in amyotrophic lateral sclerosis (ALS), occurring in up to 49% of patients, and in Alzheimer’s disease (39%).4,5 It also occurs frequently in stroke patients, especially in the first year (11%–34%),6,7 in multiple sclerosis (MS) (10%–29%),8,9 Parkinson’s disease (5%–17%),10–12 and in traumatic brain injury (5%–11%).13,14
Although the underlying pathology of PBA is not fully understood, case reports of PBA with isolated cerebellar, pontine, or thalamic lesions,15–17 combined with postmortem studies, have led to the currently accepted theory. It postulates that the cerebellum modulates the appropriate emotional response to the current social situation and mood based on cortico-pontine-cerebellar circuits. Disruption of these circuits leads to PBA.17
Current treatment options for PBA
Neuropathology of PBA and mode of action of DM/Q
As noted earlier, the basis of PBA appears to be a loss of control (disinhibition) over emotional context and motor response. This disconnection is associated with lesions and altered function in the cortico-limbic-subcorticothalamic-ponto-cerebellar network. In essence, cortical inhibition of upper brain stem is disrupted and releases the bulbar nuclei that control laughing and crying. A variety of studies (electroencephalography, imaging) implicate decreased serotonin and dopamine, glutamate excess, and sigma receptor abnormalities.18 The cerebellum is also thought to play a role since 36% of patients with brain stem and cerebellar atrophy meet criteria for PBA.19 Glutamate is a central neurotransmitter and an agonist of N-methyl-D-aspartate (NMDA) receptors that can have widespread effects. Similarly, sigma 1 receptor agonists are thought to inhibit glutamate’s actions in the brainstem and cerebellum. The goal of treating PBA is to restore control by resetting the neurotransmitter abnormalities: increasing serotonin and dopamine, blunting NMDA activity, and enhancing sigma function in specific networks.
Treatment in the past has focused primarily on the neurotransmitters serotonin or glutamate, thought to be involved in the cortico-limbic pathways or pathways originating in the cerebellum. There are multiple case reports and case series demonstrating improvement or complete resolution of symptoms related to PBA in MS, stroke, traumatic brain injury, and ALS with selective serotonin receptor inhibitors (SSRIs) and other mood stabilizers (Table 1). Only three controlled trials have focused on PBA/PLC specifically; two examined tricyclic antidepressants, the other used SSRIs.20–22 All three demonstrated a significant improvement by decreasing the frequency and severity of the symptoms. These studies are difficult to compare due to varying outcomes and time on treatment as well as different agents used to treat PBA.
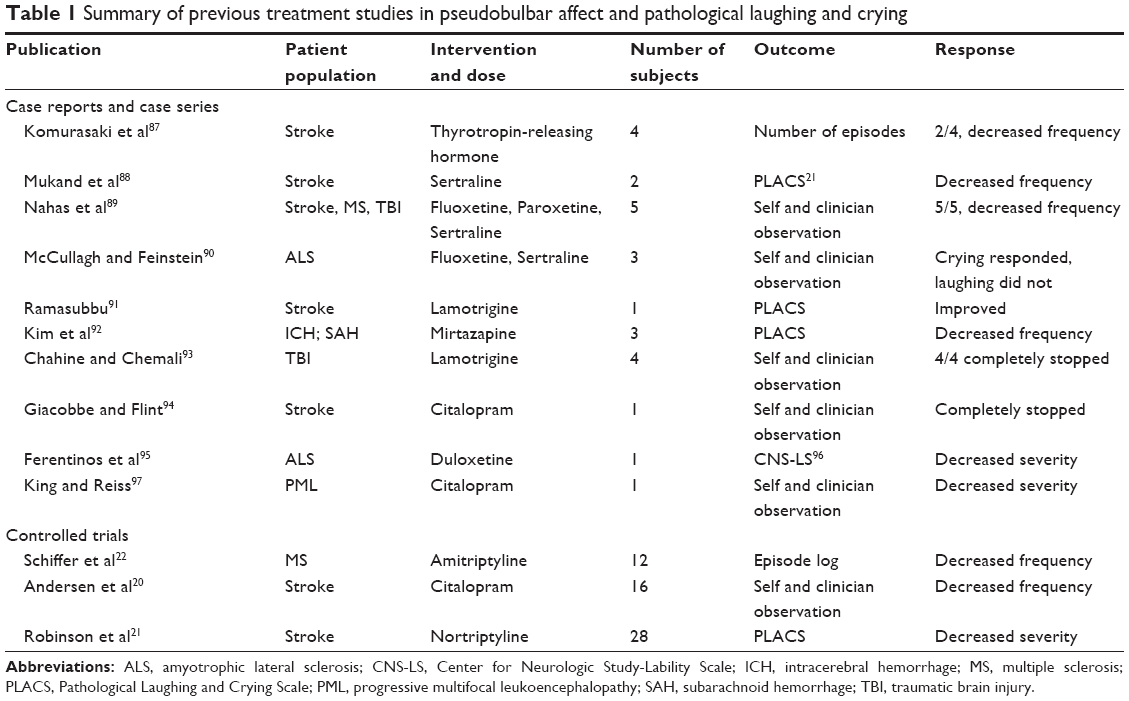 | Table 1 Summary of previous treatment studies in pseudobulbar affect and pathological laughing and crying |
Nuedexta™ (Avarnir Pharmaceuticals, Aliso Viejo, CA, USA) is a combination product containing dextromethorphan hydrobromide and quinidine sulfate. Dextromethorphan/quinidine (DM/Q) was approved by the US Food and Drug Administration in 2010 for the treatment of PBA.23
The mechanism of DM/Q is not fully understood.18 Dextromethorphan is biotransformed to dextrorphan predominantly by cytochrome P450 (CYP) 2D6;24 however, the pharmacologic properties of dextrorphan are very different than those of the parent.25 Dextromethorphan is a relatively selective and specific NMDA receptor antagonist but also acts as a sigma receptor agonist. In contrast, the primary active metabolite of dextromethorphan, dextrorphan, has much lower affinity for sigma receptors and higher affinity for NMDA receptors (Table 2).26–29 In addition, there is evidence that both dextromethorphan and dextrorphan can inhibit serotonin uptake into neurons. Given the relatively high brain:plasma partition ratio of dextromethorphan (estimated as 7–30-fold in rabbits and up to 68-fold in humans),30 it is possible that despite its lower relative NMDA affinity, dextromethorphan could be associated with substantial sigma activation and some NMDA antagonism at concentrations achieved after 30/10 mg of DM/Q (Table 2). This specificity for sigma and lower affinity for NMDA is thought to be an important mechanism in minimizing the prominent NMDA antagonist side effects of dextrorphan (altered perception, hallucinations, dissociation), which limit the ability to increase dextromethorphan doses. Therefore, DM/Q solves the practical problem of increasing dextromethorphan concentrations in order to take advantage of the preferred pharmacologic profile of the parent drug. The DM/Q product achieves this by taking advantage of the very low inhibitory constant of quinidine for CYP2D6, which permits strong inhibition of the enzyme at relatively low quinidine concentrations, thereby increasing dextromethorphan systemic bioavailability and plasma drug concentrations and decreasing those of dextrorphan (see Table 3).31–33 This drives the pharmacology of dextromethorphan toward that of the parent drug and away from the adverse effects of the dextrorphan metabolite.25,34–36 In addition, as discussed later in this review, the relatively low dose of quinidine has important implications for the safety profile of this combination product.
 | Table 2 Summary of dextromethorphan and dextrorphan pharmacodynamics and plasma concentrations |
 | Table 3 Dextromethorphan, dextrorphan, and quinidine human plasma concentrations reported in the literature |
Clinical pharmacology of DM/Q
The clinical pharmacology of dextromethorphan and quinidine are well understood; pharmacokinetic properties of dextromethorphan and quinidine are described in the product label and will not be described here.23 However, the plasma concentrations of dextromethorphan, the dextrorphan active metabolite, and quinidine are important to consider in relation to the efficacy and safety of DM/Q and are briefly summarized below.
Dextromethorphan concentrations with DM/Q
The dextromethorphan plasma concentration data are the most relevant for understanding of the DM/Q safety and efficacy profile since the goal of blocking CYP2D6 is to maximize dextromethorphan concentrations and bioavailability. Dextromethorphan plasma concentrations associated with dosing of DM/Q at steady-state (30/10 mg twice daily [bid]) are in the range of approximately 64–114 ng/mL (Table 3). In contrast, in individuals with normal CYP2D6 activity (extensive metabolizers), dextromethorphan plasma concentrations associated with chronic dosing (for cough suppression) or following dextromethorphan 30 mg alone are typically in the range of 0.5–6 ng/mL (mean of approximately 2 ng/mL) (Table 3). Thus, dextromethorphan exposure with DM/Q is approximately 20-fold higher compared to dextromethorphan administered alone (or even higher based on the literature).23
Quinidine concentrations with DM/Q
The mean peak plasma concentration of quinidine following DM/Q 30/10 mg bid in patients with PBA is approximately 1%–3% of the therapeutic concentrations required for antiarrhythmic efficacy (1,500–5,000 ng/mL).23,37
Efficacy of DM/Q in the treatment of PBA
Clinical efficacy of DM/Q
The treatments for PBA, including the efficacy of DM/Q, have been reviewed previously.38–43 Historically, the most common treatments for PBA have been tricyclic antidepressants, SSRIs, and dopaminergic agents. None of these have been approved for treatment of PBA, and the data supporting their efficacy is weak.40,44
Three controlled DM/Q studies have examined DM/Q doses of 30/30 mg and 30/10 mg bid and 20/10 mg bid (the product is approved in the US at a 20/10 mg dose and in Europe at a 20/10 mg and 30/10 mg dose).45–47 Figure 1 compares the Center for Neurologic Study-Lability Scale (CNS-LS) changes from the three trials, which was the primary dependent variable in these studies.96 The primary and secondary efficacy results from the three randomized controlled trials are summarized in Table 4. Specifically, the US registration trial resulting in approval of 20/10 mg of DM/Q bid showed a 49% reduction in PBA episode rate compared to placebo overall, and on all weekly rates. More DM/Q patients were in remission at 12 weeks compared to placebo (51% versus 29%).39 Episode-free days were more common in the active-treated group. These findings, in addition to the large and consistent effects on the CNS-LS, point to a meaningful clinical improvement in these patients. All of the trials also showed highly significant improvements on other clinically important outcomes, such as decreased number of episodes, quality of life, functioning, and interactions with others, which help to confirm the potential clinical relevance of the treatment effects.
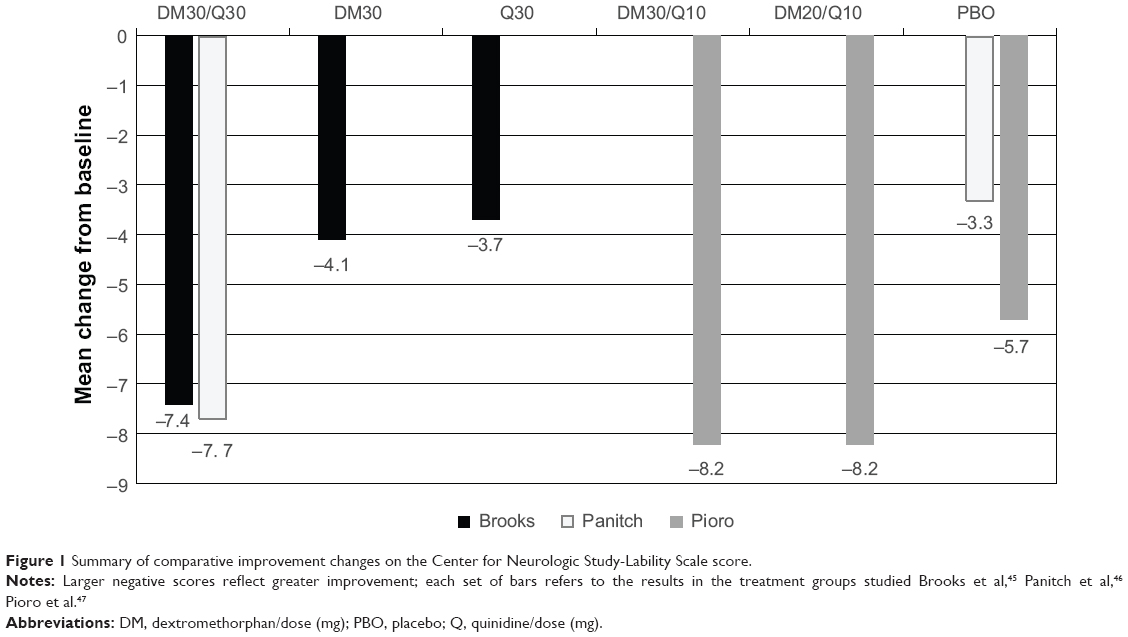 | Figure 1 Summary of comparative improvement changes on the Center for Neurologic Study-Lability Scale score. |
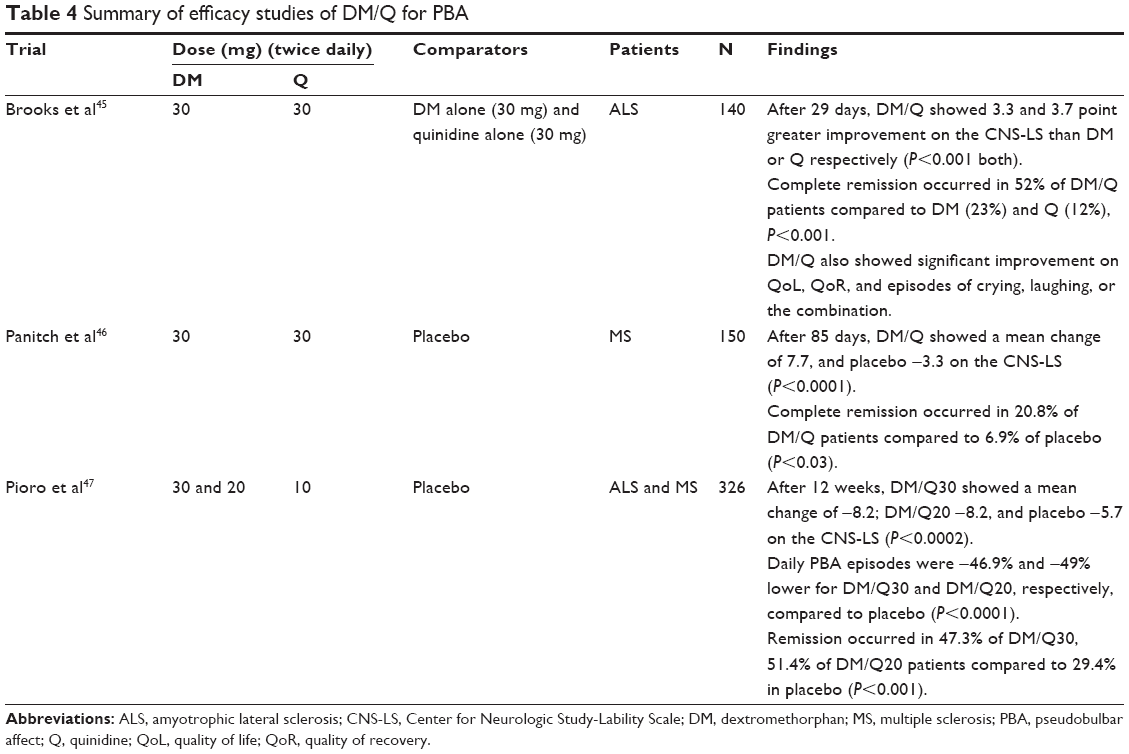 | Table 4 Summary of efficacy studies of DM/Q for PBA |
The Safety, Tolerability, and Efficacy Results (STAR) trial47 was somewhat different to the other two trials because it included the Beck Depression Inventory (BDI-II), the Neuropsychiatric Inventory (NPI), and the SF-36 (Medical Outcomes 36-Item Short Form Health Survey).48–50 On the BDI-II, mean improvement was greater for DM/Q 30/10 mg compared to placebo. No changes were seen on the NPI compared to placebo. On the SF-36, significant improvement occurred for DM/Q 30/10 mg versus placebo on the mental summary score and on its subdomains for social functioning and mental health, with no change in physical summary scores. Comparison of the 30/10 mg and 20/10 mg dose groups also suggested that the 30/10 mg group may have responded a bit earlier on the CNS-LS score. This would be consistent with the BDI-II changes observed. Changes on these secondary measures suggest that DM/Q-related reductions in PBA symptoms, as measured by CNS-LS, were also reflected in improvements in global quality of life and functioning in these patients.
Efficacy in relationship to DM/Q concentrations
Early data derived from case series and case reports suggested DM/Q might be efficacious in the treatment of PBA.51 These early studies used doses of DM/Q of 30/75 mg bid. Across these high dose trials and the more recent studies, is a puzzling lack of obvious relationship to either dextromethorphan or quinidine dose. On the other hand, the STAR trial47 did suggest that the 30/10 mg bid dosing was somewhat more efficacious than the 20/10 mg dose. In one other trial, a weak correlation was found between 12-hour postdose plasma dextromethorphan concentration and CNS-LS score at days 29 and 85 (r= −0.41 to −0.51; P<0.0001).46 However, taken as a whole, Figure 1 suggests that effect sizes were the same irrespective of either quinidine (30 mg or 10 mg) or dextromethorphan dose (30 mg, 20 mg, or 10 mg).
If the proposed mechanism of action is robust, it might be expected that there should be some clearer relationship between dextromethorphan concentration and effect, and/or quinidine concentration and degree of inhibition and resultant dextromethorphan concentrations. Virtually complete inhibition of CYP2D6 occurs with doses of quinidine in excess of 100 mg per day, and as doses of quinidine are lowered, there is a graded lesser degree of inhibition.52 There are several possible explanations why the relationship to dose (or concentration) is apparently weak. First, the extent of quinidine inhibition of CYP2D6 is not constant. The CYP2D6 enzyme exists in many isoforms with differing substrate selectivity and specificity, and these isoforms also show quite variable sensitivity to differing inhibitors.53–56 Hence, quinidine inhibition can be highly variable at any given quinidine concentration. The second possibility is that we do not fully understand the mechanism of action, and dextromethorphan concentrations at the sigma or NMDA receptors are much higher than estimated, and/or the receptor capacity is very low and dextromethorphan has very high intrinsic activity. These data suggest that some patients might respond to even lower doses of DM/Q. However, since the safety of the DM/Q product seems to be quite acceptable, whether it is worth finding the lowest effective dose is unclear. Lowering the doses of quinidine and/or dextromethorphan might come at the “cost” of not being effective for those patients with atypical CYP2D6 or with differing pathophysiology.
Safety considerations with DM/Q
General safety of DM/Q in clinical trials
Table 5 summarizes the most important clinically reported safety data from the three largest clinical trials of PBA.45–47 Excluded from this table are adverse events that were more common in the placebo group and those for which there is no plausible causal relationship (eg, nasopharyngitis). The full data can be obtained in the papers reporting these studies. The most commonly reported adverse events of dizziness, fatigue, nausea, and weakness are consistent with side effects attributable to dextromethorphan and potentially quinidine. A recent 1 year study in 553 patients with PBA (296 completers) receiving DM/Q 30/30 mg bid confirms the apparent safety of DM/Q (Pattee et al, unpublished data, 2014). The most frequently reported treatment emergent adverse events in this study were nausea (11.8%), dizziness (10.5%), headache (9.9%), somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), and dry mouth (5.1%). These adverse events were mainly mild to moderate in severity and generally transient. There were no clinically important cardiac or respiratory adverse events reported.
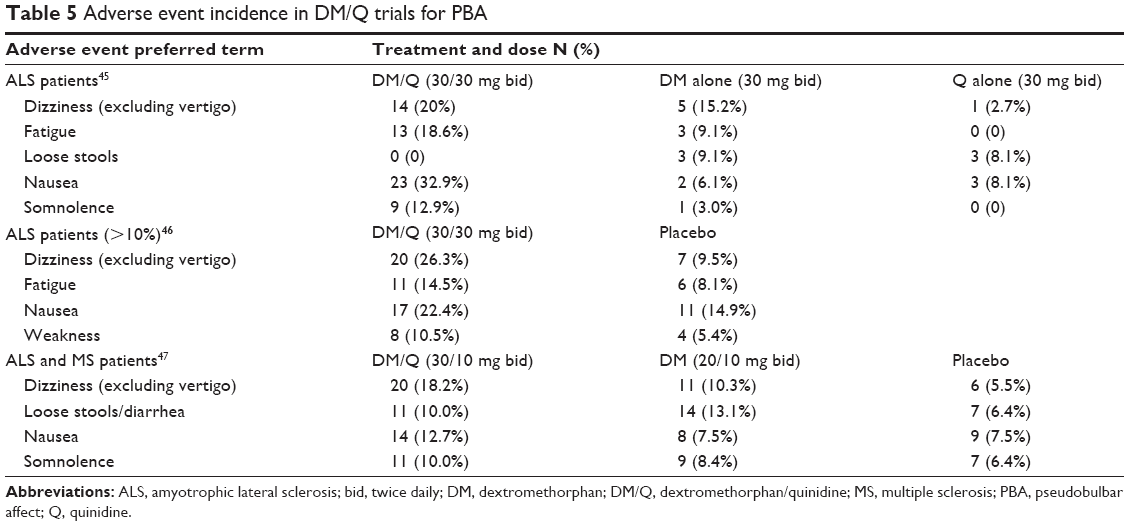 | Table 5 Adverse event incidence in DM/Q trials for PBA |
In other indications, the placebo data in the Panitch et al study46 are of interest because some adverse events were actually less common in the active-treated group (eg, dermatitis, fatigue aggravated, headache, pain in limbs), suggesting a hypothesis that there are dimensions of DM/Q effect that are not being captured by the typical efficacy measures. MS patients often report disabling pain. Since decreased glutaminergic signaling may alleviate pain, the lower reports of pain in limbs are particularly interesting. The study was not powered to detect such a change, nor were patients required to have pain for entry into the trial. These potential other effects of DM/Q merit further study. The discontinuation rates due to adverse events in the MS study were lower than in the ALS study (24%). In the ALS study, discontinuations were mainly due to musculoskeletal complaints that did not occur in the MS patients.
Impact of plasma concentrations on perceived special safety concerns with DM/Q
A summary of plasma dextromethorphan, dextrorphan, and quinidine plasma concentrations is provided in Table 3. Several studies and cases have reported plasma concentrations of dextromethorphan in relation to safety parameters. Peak dextromethorphan concentrations (49–168 ng/mL) in a similar range as those associated with DM/Q (64–114 ng/mL) do not appear to be associated with serious psychomotor/cognitive impairment (an objective measure of sedation) in CYP2D6 poor metabolizers or in extensive metabolizers when coadministered with quinidine, although subjective sedation/dysphoria has been reported at concentrations in the higher end of this range (>100 ng/mL).25,34,35 Peak dextromethorphan concentrations in a higher range (185–233 ng/mL) were tolerable in these subjects but were associated with motor impairment and subjective sedation. In general, when high doses of dextromethorphan are given alone (>100 mg per day), the resultant higher dextromethorphan/dextrorphan concentrations than those associated with DM/Q have been tolerated in epilepsy (up to 150 ng/mL), neurosurgery (up to 193 ng/mL), and Huntington’s disease patients (up to 275 ng/mL) (see Table 3). In neurosurgery patients, doses associated with dextromethorphan concentrations of 400–500 ng/mL were safely administered although these were associated with side effects such as dizziness, ataxia, hallucinations, and nystagmus. These are side effects typical of dextrorphan and likely mediated by this active metabolite.
Other data supporting the general safety of dextromethorphan come from overdoses and poisonings (Table 3). The lowest concentration of dextromethorphan reported in association with an overdose fatality was estimated as 542 ng/mL; however, this was observed in combination with diphenhydramine concentrations approximately five times higher than the therapeutic range. A case of serotonin syndrome (nonfatal) was associated with a concentration of 930 ng/mL, and impaired drivers have been tested with concentrations up into the 695–1,026 ng/mL range, indicating that 542 ng/mL does not necessarily represent the threshold concentration for fatality. Although there are relatively few reports of dextromethorphan overdose fatalities, most were associated with concentrations in the range of ~1,800–5,000 ng/mL. Overall, the plasma concentrations of dextromethorphan and lower relative concentrations of dextrorphan associated with DM/Q administration may confer a fairly robust margin of safety with this product. Although, it is possible that even at these relatively low concentrations, older and/or cognitively-impaired individuals may be more sensitive to some of the sedating/central nervous system side effects of DM/Q, as events such as dizziness and fatigue were observed in clinical trials (Table 5), and some patients (1%) have discontinued DM/Q due to falls.23
Dextromethorphan respiratory effects
Dextromethorphan belongs to the opioid class of compounds that can cause respiratory depression. However, even though dextromethorphan has been shown to dampen compensatory responses to hypoxic or hypercapnic stimuli in animal models, it has no apparent effect on respiration in awake animals breathing normal air.57–58 Typically, these effects were observed at doses ranging from 5–80 mg/kg administered by the subcutaneous route. This route is relevant to DM/Q as it is the route associated with the least amount of metabolite formation. In clinical studies, single oral doses in the range of 30–60 mg dextromethorphan do not alter vital signs parameters, including respiratory rate, even when combined with morphine (30–60 mg), a known respiratory depressant.59 Similarly, multiple dose studies have failed to show hemodynamic or respiratory effects, including a study that used doses up to 400 mg (dextromethorphan plasma concentrations up to 1,514 ng/mL),60 suggesting that respiratory depression or failure, while observed in a small proportion of patients (1%),23 is not a major concern, at least in those patients without underlying respiratory pathology.
Cardiac safety
Quinidine is classified as a type 1A drug with respect to its propensity to cause torsades de pointes (TdP) and to prolong the QT interval.61 Increases in corrected QT (QTc) are consistently seen with free (unbound) quinidine concentrations of 300 nM, but of course, there is variation in sensitivity among patients due to cardiac disease, race, sex, and concomitant medications.61 After 30 mg of quinidine bid, mean (± standard deviation [SD]) quinidine concentrations of 168.4±103 ng/mL have been reported (466 nM based on total drug concentration; free drug concentration estimated to be about 50 nM). This concentration is well below the reported concentrations causing clinically important QTc prolongation, but this does raise the possibility of an effect on QTc in PBA patients treated with DM/Q. Grenier et al62 analyzed 7,446 QT measurements in 82 subjects given DM/Q, created a robust pharmacokinetic-pharmacodynamic model, and found that low average quinidine concentrations (41.3 ng/mL) were associated with small QTc changes (<3 milliseconds). The marketed dose of DM/Q contains 10 mg of quinidine. The product label carries a warning to this effect:
NUEDEXTA causes dose-dependent QTc prolongation. QT prolongation can cause torsades de pointes-type ventricular tachycardia, with the risk increasing as the degree of prolongation increases. When initiating NUEDEXTA in patients at risk of QT prolongation and torsades de pointes, electrocardiographic evaluation of QT interval should be conducted at baseline and 3–4 hours after the first dose. This includes patients concomitantly taking/initiating drugs that prolong the QT interval or that are strong or moderate CYP3A4 inhibitors, and patients with left ventricular hypertrophy or left ventricular dysfunction.23
In the earliest trial in ALS patients comparing DM/Q 30/30 mg bid and dextromethorphan (30 mg bid) and quinidine (30 mg bid) alone, an increase in QTc on day 29 (end of trial) of +7.5 milliseconds was observed compared to quinidine alone, but not dextromethorphan alone.45 In a trial of MS patients with DM/Q 30/30 mg bid, a +7.5 millisecond change in QTc was seen compared to placebo.46 Both these changes would not be regarded as clinically important. The most careful assessment of this matter was done by Pioro et al47 who found a mean change of 3.0 QTc Bazett’s formula (QTcB) and 4.8 QTc Fridericia’s formula (QTcF) milliseconds with DM/Q 30/10 mg; −1.9 (QTcB) and 1.0 (QTcF) with DM/Q 20/10 mg; and +1.6 (QTcB) and 1.0 (QTcF) with placebo. These changes are also very small. The proportion of individuals who had a QTc prolongation of >30–60 milliseconds in a DM/Q (30/10 mg)-treated group was 7.0%/7.2% (QTcB/QTcF), with placebo having a rate of 6.6%/3.5%. These data are difficult to interpret. On one hand, this trial used lower doses of quinidine than the previous two trials and did find a slightly lower change in QTc, suggesting that the quinidine in the DM/Q can have an effect on QTc. On the other hand, the changes are small and were not reported to be associated with any adverse clinical events. The most parsimonious interpretation of these data would be that quinidine can have a physiologic effect on QTc, but this is not normally a clinical concern with DM/Q. Quinidine concentrations associated with TdP cases appear to be primarily within the therapeutic range of quinidine when administered as an antiarrhythmic (1,500–5,000 ng/mL); some cases of TdP had quinidine concentrations “<1,500 ng/mL” but exact concentrations not cited.37,63 However, these patients were on mean quinidine doses >1,500 mg/day, and so presumably still had much higher concentrations of quinidine than following DM/Q administration.
However, as a final word of caution, patients with PBA often have multiple comorbidities and/or are being treated with numerous other medications, hence quinidine is likely to be combined with other drugs that may interact with it. These interactions can include other drugs that block quinidine metabolism or transport by p-glycoprotein, or that have direct effects on QTc or are arrhythmogenic themselves. Table 6 summarizes examples of such potential interactions and suggests the appropriate clinical approach or alternate drug in patients at risk. Other risk factors associated with quinidine-induced TdP include low serum potassium or moderately low potassium levels in association with diuretics, heart block, and unrecognized congenital long QT syndrome.37 These factors should be considered in any patient being started on DM/Q, but should not limit use of the medication in most patients. In some cases, patients at risk for TdP may require substitutions, where possible, or dose reductions and supervised starts where substitutions cannot be made.
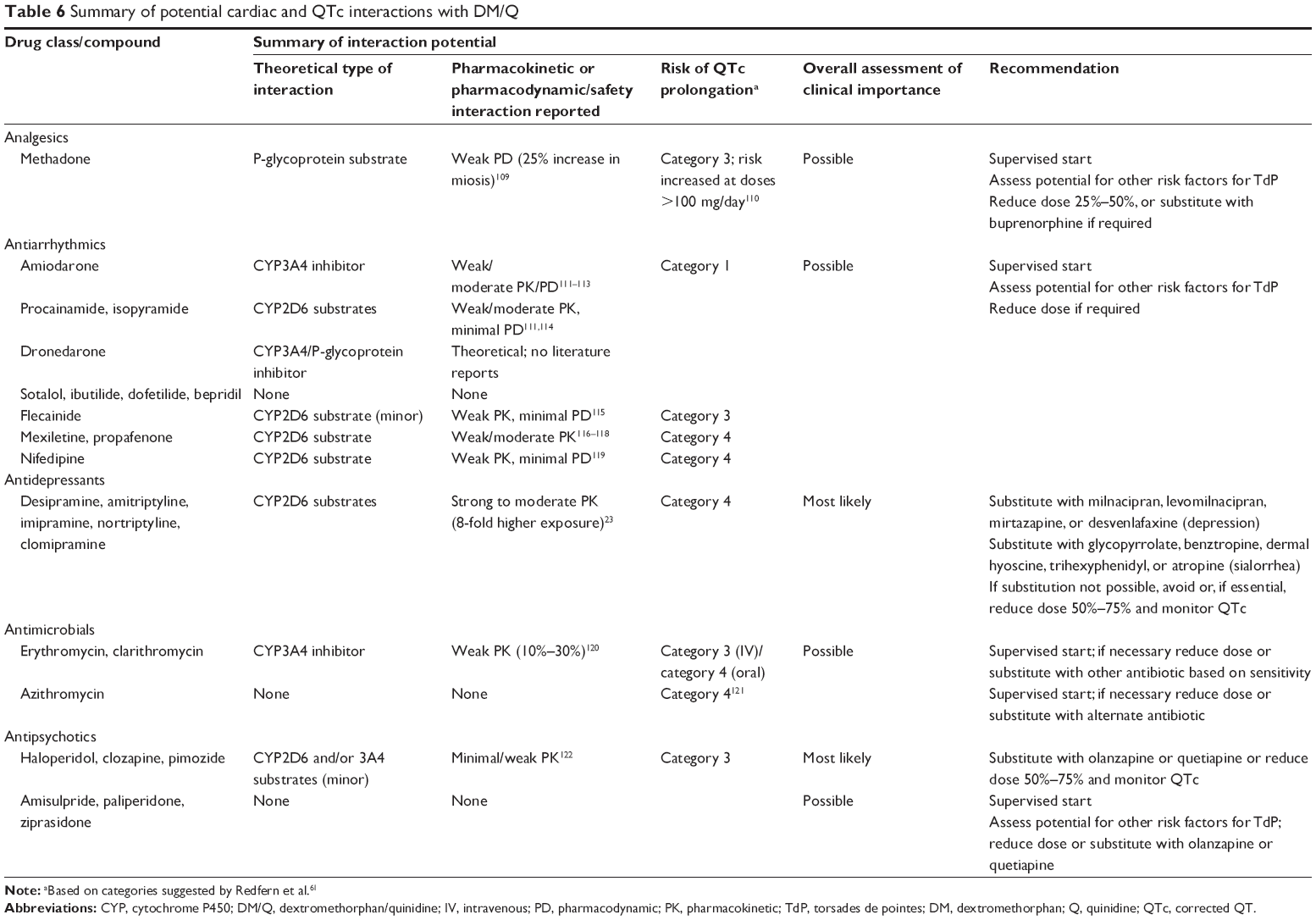 | Table 6 Summary of potential cardiac and QTc interactions with DM/Q |
Other drug interactions
Because the principal mechanism of DMQ involves the inhibition of CYP2D6, an enzyme involved in the metabolism of a number of drugs, there is a possibility of drug interactions. The likelihood of observing clinically relevant drug interactions depends on a number of factors, including the potency of the inhibitor, the concentrations of inhibitor and substrate, the proportion of metabolism of the affected drug that is catalyzed by a particular CYP, and the therapeutic windows of the interacting drugs. There exist many theoretical drug interactions; however, many turn out not to be important in clinical practice for a variety of reasons. Pharmacokinetic changes predicted from in vitro studies may not be as large in patients since most drugs are metabolized by a number of different enzymes, and inhibition of one enzyme may simply lead to shunting of metabolism through a different pathway. As shown in Table 6, some drugs should be avoided, where possible, and substitutions made, primarily due to pharmacokinetic interaction potential combined with some torsadogenic potential, or strong torsadogenic potential alone.
Further, there is a theoretical risk of observing reduced efficacy with certain analgesics that are known CYP3A4/CYP2D6 and/or p-glycoprotein substrates or inhibitors, such as hydrocodone,64 fentanyl,65 morphine,66 and oxycodone.67–69 In most cases, these drugs can be coadministered with DM/Q, as theoretical or pharmacokinetic interactions have not shown much impact on pharmacodynamic effects or analgesia. The exception to this is codeine and possibly tramadol, where reduced efficacy may be observed due to CYP2D6-mediated conversion to the active metabolites.70–74
In terms of antidepressants, those most likely to result in significant interactions include the tricyclic antidepressants, due to the potential for pharmacokinetic interactions as well as additive torsadogenic risk (Table 6). SSRIs may be administered with DM/Q, although supervised starts are recommended due to the potential for pharmacokinetic and/or pharmacodynamic (serotonin syndrome) interactions, and weak potential to prolong the QT interval. This is most relevant for paroxetine,75 fluoxetine,76,77 sertraline,78 and citalopram.79 Mirtazapine interactions with quinidine appear to be relatively weak,80 and this drug is not associated with known torsadogenic potential. In addition, it is routine practice to avoid the use of monoamine oxidase inhibitors with dextromethorphan due to the rare but known potential for serotonin syndrome.81 Substitutions could be made with milnacipran, levomilnacipran, mirtazapine, or desvenlafaxine, as no reported or theoretical interactions are known with these compounds.
There are a number of other drugs that are likely to be coadministered with DM/Q and may also be substrates, inducers, or inhibitors of CYP2D6 or CYP3A4. However, based on the literature, the likelihood of clinically relevant interactions with these drugs is considered low. This includes drugs where pharmacokinetic interactions were weak/minimal or did not result in changes in pharmacodynamic or safety parameters, or those for which interactions would be relevant only at much higher concentrations of DM/Q than those observed clinically. In Parkinson’s disease, this includes bromocriptine, amantadine, biperiden, apormorphine, and sedative-hypnotics such as alprazolam and zolpidem.82,83 For Alzheimer’s disease, this includes galantamine and memantine.84 Donepezil pharmacokinetic/pharmacodynamic interactions are unlikely to be clinically relevant85 although this drug is considered to have weak torsadogenic potential (category 4).61 Although the theoretical potential for interactions exists, other drugs that may be coadministered and are unlikely to result in clinically significant interactions include methylprednisolone, omeprazole, simvastatin/atorvastatin, warfarin, beta-blockers, verapamil, diltiazem, digoxin, and dipyridamole.
As with any drug, the risk for clinically significant interactions increases with the number of coprescribed and potentially interacting medications. An example of this is oxycodone, whereby concomitant use of both CYP2D6 and CYP3A4 inhibitors may increase exposure, where use of CYP2D6 inhibitors alone does not.86 However, this is relevant for any patient, and not only those on DM/Q. The role of DM/Q in inhibition of CYP2D6 requires some routine caution at the beginning of treatment, but as with the QT prolongation potential, should not limit use in patients requiring treatment. Indeed, many strong CYP2D6 inhibitors have been relatively safely used in clinical practice for many years (SSRIs such as paroxetine).
Conclusion
Overall, the published literature supports the efficacy and relatively high margin of safety with DM/Q, and indicates that serious medical events observed at very high doses of dextromethorphan are not expected to occur with DM/Q administered at recommended doses. Because concentrations of the active metabolite dextrorphan (which is responsible for many of the parent drug’s side effects) and quinidine are lower than those observed in clinical practice with these drugs administered alone, some of the perceived safety issues may not be as relevant with this low dose combination product. However, since patients with PBA have a variety of other medical problems and are on numerous other medications, they may not tolerate DM/Q adverse effects or may be at risk for drug interactions. Some caution is warranted at the initiation of DM/Q treatment, particularly in patients with underlying risk factors for TdP and in those receiving medications that may interact with DM/Q. Given that DM/Q is currently the only treatment indicated for PBA, substitutions may be more easily made with the interacting concomitant medications. Where substitutions are not possible, supervised starts and dosage reductions may be needed.
Disclosure
KAS and EMS were employed at the clinical site that performed some of the drug-drug interaction studies with DM/Q and provided consultation to Avanir Pharmaceuticals. EMS has also acted as an independent consultant to Avanir Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
Cummings JL, Arciniegas DB, Brooks BR, et al. Defining and diagnosing involuntary emotional expression disorder. CNS Spectr. 2006;11(6):1–7. | ||
Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1–14; quiz 15. | ||
Work SS, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586–601. | ||
Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand. 1989;80(2):114–117. | ||
Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55–60. | ||
House A, Dennis M, Molyneux A, Warlow C, Hawton K. Emotionalism after stroke. BMJ. 1989;298(6679):991–994. | ||
Kim JS. Pathological laughter and crying in unilateral stroke. Stroke. 1997;28(11):2321. | ||
Feinstein A, Feinstein K, Gray T, O’Connor P. Prevalence and neurobehavioral correlates of pathological laughing and crying in multiple sclerosis. Arch Neurol. 1997;54(9):1116–1121. | ||
Pratt RT. An investigation of the psychiatric aspects of disseminated sclerosis. J Neurol Neurosurg Psychiatry. 1951;14(4):326–335. | ||
Petracca GM, Jorge RE, Ación L, Weintraub D, Robinson RG. Frequency and correlates of involuntary emotional expression disorder in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(4): 406–412. | ||
Siddiqui MS, Fernandez HH, Garvan CW, et al. Inappropriate crying and laughing in Parkinson disease and movement disorders. World J Biol Psychiatry. 2009;10(3):234–240. | ||
Strowd RE, Cartwright MS, Okun MS, Haq I, Siddiqui MS. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382–1387. | ||
Tateno A, Jorge RE, Robinson RG. Pathological laughing and crying following traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2004;16(4):426–434. | ||
Zeilig G, Drubach DA, Katz-Zeilig M, Karatinos J. Pathological laughter and crying in patients with closed traumatic brain injury. Brain Inj. 1996;10(8):591–597. | ||
Elyas AE, Bulters DO, Sparrow OC. Pathological laughter and crying in patients with pontine lesions. J Neurosurg Pediatr. 2011;8(6): 544–547. | ||
Lauterbach EC, Price ST, Spears TE, Jackson JG, Kirsh AD. Serotonin responsive and nonresponsive diurnal depressive mood disorders and pathological affect in thalamic infarct associated with myoclonus and blepharospasm. Biol Psychiatry. 1994;35(7):488–490. | ||
Parvizi J, Coburn KL, Shillcutt SD, Coffey CE, Lauterbach EC, Mendez MF. Neuroanatomy of pathological laughing and crying: a report of the American Neuropsychiatric Association Committee on Research. J Neuropsychiatry Clin Neurosci. 2009;21(1):75–87. | ||
Cummings J, Gilbart J, Andersen G. Pseudobulbar affect. A disabling but under-recognized consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74–81. | ||
Parvizi J, Joseph J, Press DZ, Schmahmann JD. Pathological laughter and crying in patients with multiple system atrophy-cerebellar type. Mov Disord. 2007;22(6):798–803. | ||
Andersen G, Vestergaard K, Riis JO. Citalopram for post-stroke pathological crying. Lancet. 1993;342(8875):837–839. | ||
Robinson RG, Parikh RM, Lipsey JR, Starkstein SE, Price TR. Pathological laughing and crying following stroke: validation of a measurement scale and a double-blind treatment study. Am J Psychiatry. 1993;150(2): 286–293. | ||
Schiffer RB, Herndon RM, Rudick RA. Treatment of pathologic laughing and weeping with amitriptyline. N Engl J Med. 1985;312(23): 1480–1482. | ||
NUEDEXTA® (dextromethorphan hydrobromide and quinidine sulfate) [prescribing information]. Aliso Viejo: Avanir Pharmaceuticals, Inc; 2010. | ||
von Moltke LL, Greenblatt DJ, Grassi JM, et al. Multiple human cytochromes contribute to biotransformation of dextromethorphan in-vitro: role of CYP2C9, CYP2C19, CYP2D6, and CYP3A. J Pharm Pharmacol. 1998;50(9):997–1004. | ||
Zawertailo LA, Tyndale RF, Busto U, Sellers EM. Effect of metabolic blockade on the psychoactive effects of dextromethorphan. Hum Psychopharmacol. 2010;25(1):71–79. | ||
Musacchio JM, Klein M, Canoll PD. Dextromethorphan and sigma ligands: common sites but diverse effects. Life Sci. 1989;45(19): 1721–1732. | ||
Musacchio JM, Klein M, Canoll PD. Dextromethorphan sites, sigma receptors, and the psychotomimetic effects of sigma opiates. Prog Clin Biol Res. 1990;328:13–16. | ||
Tortella FC, Davey R, Pellicano M, Bowery NG. Autoradiographic localization of 3H-dextromethorphan binding sites differs from NMDA. NIDA Res Monogr. 1989;95:548–549. | ||
Tortella FC, Pellicano M, Bowery NG. Dextromethorphan and neuromodulation: old drug coughs up new activities. Trends Pharmacol Sci. 1989;10(12):501–507. | ||
Steinberg GK, George CP, DeLaPaz R, Shibata DK, Gross T. Dextromethorphan protects against cerebral injury following transient focal ischemia in rabbits. Stroke. 1988;19(9):1112–1118. | ||
Otton SV, Crewe HK, Lennard MS, Tucker GT, Woods HF. Use of quinidine inhibition to define the role of the sparteine/debrisoquine cytochrome P450 in metoprolol oxidation by human liver microsomes. J Pharmacol Exp Ther. 1988;247(1):242–247. | ||
Otton SV, Inaba T, Kalow W. Competitive inhibition of sparteine oxidation in human liver by beta-adrenoceptor antagonists and other cardiovascular drugs. Life Sci. 1984;34(1):73–80. | ||
Schadel M, Wu D, Otton SV, Kalow W, Sellers EM. Pharmacokinetics of dextromethorphan and metabolites in humans: influence of the CYP2D6 phenotype and quinidine inhibition. J Clin Psychopharmacol. 1995;15(4):263–269. | ||
Zawertailo L, Busto U, Tyndale R, Sellers E. Dextromethorphan’s active metabolite contributes to its abuse liability. Clin Pharmacol Ther. 1998;63(2):217. | ||
Zawertailo LA, Kaplan HL, Busto UE, Tyndale RF, Sellers EM. Psychotropic effects of dextromethorphan are altered by the CYP2D6 polymorphism: a pilot study. J Clin Psychopharmacol. 1998;18(4):332–337. | ||
Zhang Y, Britto MR, Valderhaug KL, Wedlund PJ, Smith RA. Dextromethorphan: enhancing its systemic availability by way of low-dose quinidine-mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther. 1992;51(6):647–655. | ||
Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J. 1986;111(6):1088–1093. | ||
Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483–489. | ||
Garnock-Jones KP. Dextromethorphan/quinidine: in pseudobulbar affect. CNS Drugs. 2011;25(5):435–445. | ||
Miller A, Pratt H, Schiffer RB. Pseudobulbar affect: the spectrum of clinical presentations, etiologies and treatments. Expert Rev Neurother. 2011;11(7):1077–1088. | ||
Olney N, Rosen H. AVP-923, a combination of dextromethorphan hydrobromide and quinidine sulfate for the treatment of pseudobulbar affect and neuropathic pain. I Drugs. 2010;13(4):254–265. | ||
Rosen H. Dextromethorphan/quinidine sulfate for pseudobulbar affect. Drugs Today (Barc). 2008;44(9):661–668. | ||
Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17(4): 447–454. | ||
Miller A. Pseudobulbar affect in multiple sclerosis: toward the development of innovative therapeutic strategies. J Neurol Sci. 2006;245(1–2): 153–159. | ||
Brooks BR, Thisted RA, Appel SH, et al; AVP-923 ALS Study Group. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63(8):1364–1370. | ||
Panitch HS, Thisted RA, Smith RA, et al; Psuedobulbar Affect in Multiple Sclerosis Study Group. Randomized, controlled trial of dextromethorphan/quinidine for pseudobulbar affect in multiple sclerosis. Ann Neurol. 2006;59(5):780–787. | ||
Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy Results Trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693–702. | ||
Busija L, Pausenberger E, Haines TP, Haymes S, Buchbinder R, Osborne RH. Adult measures of general health and health-related quality of life: Medical Outcomes Study Short Form 36-Item (SF-36) and Short Form 12-Item (SF-12) Health Surveys, Nottingham Health Profile (NHP), Sickness Impact Profile (SIP), Medical Outcomes Study Short Form 6D (SF-6D), Health Utilities Index Mark 3 (HUI3), Quality of Well-Being Scale (QWB), and Assessment of Quality of Life (AQoL). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S383–S412. | ||
Malloy PF, Cummings JL, Coffey CE, et al. Cognitive screening instruments in neuropsychiatry: a report of the Committee on Research of the American Neuropsychiatric Association. J Neuropsychiatry Clin Neurosci. 1997;9:189–197. | ||
Steer RA, Brown GK, Beck AT, Sanderson WC. Mean Beck Depression Inventory-II scores by severity of major depressive episode. Psychol Rep. 2001;88:1075–1076. | ||
Smith RA. Dextromethorphan/quinidine: a novel dextromethorphan product for the treatment of emotional lability. Expert Opin Pharmacother. 2006;7(18):2581–2598. | ||
Pope LE, Khalil MH, Berg JE, Stiles M, Yakatan GJ, Sellers EM. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol. 2004;44(10):1132–1142. | ||
Bogni A, Monshouwer M, Moscone A, et al. Substrate specific metabolism by polymorphic cytochrome P450 2D6 alleles. Toxicol In Vitro. 2005;19(5):621–629. | ||
Droll K, Bruce-Mensah K, Otton SV, Gaedigk A, Sellers EM, Tyndale RF. Comparison of three CYP2D6 probe substrates and genotype in Ghanaians, Chinese and Caucasians. Pharmacogenetics. 1998;8(4):325–333. | ||
Ramamoorthy Y, Tyndale RF, Sellers EM. Cytochrome P450 2D6.1 and cytochrome P450 2D6.10 differ in catalytic activity for multiple substrates. Pharmacogenetics. 2001;11(6):477–487. | ||
Ramamoorthy Y, Yu AM, Suh N, Haining RL, Tyndale RF, Sellers EM. Reduced (+/−)-3,4-methylenedioxymethamphetamine (“Ecstasy”) metabolism with cytochrome P450 2D6 inhibitors and pharmacogenetic variants in vitro. Biochem Pharmacol. 2002;63(12):2111–2119. | ||
Lee SD, Nakano H, Farkas GA. NMDA receptor-mediated modulation of ventilation in obese Zucker rats. Int J Obes Relat Metab Disord. 2001;25(7):997–1004. | ||
Schlenker EH. Dextromethorphan affects ventilation differently in male and female rats. J Appl Physiol (1985). 1996;81(5):1911–1916. | ||
Frymoyer AR, Rowbotham MC, Petersen KL. Placebo-controlled comparison of a morphine/dextromethorphan combination with morphine on experimental pain and hyperalgesia in healthy volunteers. J Pain. 2007;8(1):19–25. | ||
Steinberg GK, Bell TE, Yenari MA. Dose escalation safety and tolerance study of the N-methyl-D-aspartate antagonist dextromethorphan in neurosurgery patients. J Neurosurg. 1996;84(5):860–866. | ||
Redfern WS, Carlsson L, Davis AS, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58(1):32–45. | ||
Grenier J, Lavigne JR, Pope LE. Population Pharmacokinetic Meta-analysis: Inhibition by Quinidine of the First-Pass and Systemic Metabolism of Dextromehtorphan to Dextrorphan. Lincoln, NE: Celerion, Inc; 2010. Available from: http://www.page-meeting.org/pdf_assets/6239-Celerion_Poster_Page2010_PopPK_QDMDX.pdf. Accessed December 28, 2013. | ||
Jenzer HR, Hagemeijer F. Quinidine syncope: torsade de pointes with low quinidine plasma concentrations. Eur J Cardiol. 1976;4(4):447–451. | ||
Otton SV, Schadel M, Cheung SW, Kaplan HL, Busto UE, Sellers EM. CYP2D6 phenotype determines the metabolic conversion of hydrocodone to hydromorphone. Clin Pharmacol Ther. 1993;54(5): 463–472. | ||
Kharasch ED, Hoffer C, Altuntas TG, Whittington D. Quinidine as a probe for the role of p-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44(3):224–233. | ||
Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of P-glycoprotein in the intestinal absorption and clinical effects of morphine. Clin Pharmacol Ther. 2003;74(6):543–554. | ||
Heiskanen T, Olkkola KT, Kalso E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Clin Pharmacol Ther. 1998;64(6):603–611. | ||
Samer CF, Daali Y, Wagner M, et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160(4):919–930. | ||
Samer CF, Daali Y, Wagner M, et al. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br J Pharmacol. 2010;160(4):907–918. | ||
Caraco Y, Sheller J, Wood AJ. Impact of ethnic origin and quinidine coadministration on codeine’s disposition and pharmacodynamic effects. J Pharmacol Exp Ther. 1999;290(1):413–422. | ||
Laugesen S, Enggaard TP, Pedersen RS, Sindrup SH, Brøsen K. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective O-demethylation and reduces the hypoalgesic effect of tramadol. Clin Pharmacol Ther. 2005;77(4):312–323. | ||
Poulsen L, Arendt-Nielsen L, Brøsen K, Sindrup SH. The hypoalgesic effect of tramadol in relation to CYP2D6. Clin Pharmacol Ther. 1996;60(6):636–644. | ||
Sindrup SH, Arendt-Nielsen L, Brøsen K, et al. The effect of quinidine on the analgesic effect of codeine. Eur J Clin Pharmacol. 1992;42(6):587–591. | ||
Stamer UM, Lehnen K, Höthker F, et al. Impact of CYP2D6 genotype on postoperative tramadol analgesia. Pain. 2003;105(1–2):231–238. | ||
Schoedel KA, Pope LE, Sellers EM. Randomized open-label drug-drug interaction trial of dextromethorphan/quinidine and paroxetine in healthy volunteers. Clin Drug Investig. 2012;32(3):157–169. | ||
Achamallah NS. Visual hallucinations after combining fluoxetine and dextromethorphan. Am J Psychiatry. 1992;149(10):1406. | ||
Skop BP, Finkelstein JA, Mareth TR, Magoon MR, Brown TM. The serotonin syndrome associated with paroxetine, an over-the-counter cold remedy, and vascular disease. Am J Emerg Med. 1994;12(6): 642–644. | ||
DeVane CL. Pharmacogenetics and drug metabolism of newer antidepressant agents. J Clin Psychiatry. 1994;55 Suppl:38–45; discussion 46–47. | ||
Sindrup SH, Brøsen K, Hansen MG, Aaes-Jørgensen T, Overø KF, Gram LF. Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther Drug Monit. 1993;15(1):11–17. | ||
Kirchheiner J, Henckel HB, Meineke I, Roots I, Brockmöller J. Impact of the CYP2D6 ultrarapid metabolizer genotype on mirtazapine pharmacokinetics and adverse events in healthy volunteers. J Clin Psychopharmacol. 2004;24(6):647–652. | ||
Sovner R, Wolfe J. Interaction between dextromethorphan and monoamine oxidase inhibitor therapy with isocarboxazid. N Engl J Med. 1988;319(25):1671. | ||
von Moltke LL, Greenblatt DJ, Cotreau-Bibbo MM, Harmatz JS, Shader RI. Inhibitors of alprazolam metabolism in vitro: effect of serotonin-reuptake-inhibitor antidepressants, ketoconazole and quinidine. Br J Clin Pharmacol. 1994;38(1):23–31. | ||
von Moltke LL, Greenblatt DJ, Granda BW, et al. Zolpidem metabolism in vitro: responsible cytochromes, chemical inhibitors, and in vivo correlations. Br J Clin Pharmacol. 1999;48(1):89–97. | ||
Pope LE, Schoedel KA, Bartlett C, Sellers EM. A study of potential pharmacokinetic and pharmacodynamic interactions between dextromethorphan/quinidine and memantine in healthy volunteers. Clin Drug Investig. 2012;32(8):e1–e15. | ||
Varsaldi F, Miglio G, Scordo MG, et al. Impact of the CYP2D6 polymorphism on steady-state plasma concentrations and clinical outcome of donepezil in Alzheimer’s disease patients. Eur J Clin Pharmacol. 2006;62(9):721–726. | ||
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70(1):78–87. | ||
Komurasaki Y, Yokoyama T, Ogura J, Maeda K. Treatment of pathologic emotionality with thyrotropin-releasing hormone. Jpn J Psychiatry Neurol. 1989;43(4):665–668. | ||
Mukand J, Kaplan M, Senno RG, Bishop DS. Pathological crying and laughing: treatment with sertraline. Arch Phys Med Rehabil. 1996;77(12):1309–1311. | ||
Nahas Z, Arlinghaus KA, Kotrla KJ, Clearman RR, George MS. Rapid response of emotional incontinence to selective serotonin reuptake inhibitors. J Neuropsychiatry Clin Neurosci. 1998;10(4):453–455. | ||
McCullagh S, Feinstein A. Treatment of pathological affect: variability of response for laughter and crying. J Neuropsychiatry Clin Neurosci. 2000;12(1):100–102. | ||
Ramasubbu R. Lamotrigine treatment for post-stroke pathological laughing and crying. Clin Neuropharmacol. 2003;26(5):233–235. | ||
Kim SW, Shin IS, Kim JM, Lim SY, Yang SJ, Yoon JS. Mirtazapine treatment for pathological laughing and crying after stroke. Clin Neuropharmacol. 2005;28(5):249–251. | ||
Chahine LM, Chemali Z. Du rire aux larmes: pathological laughing and crying in patients with traumatic brain injury and treatment with lamotrigine. Epilepsy Behav. 2006;8(3):610–615. | ||
Giacobbe P, Flint A. Pharmacological treatment of post-stroke pathological laughing and crying. J Psychiatry Neurosci. 2007;32(5):384. | ||
Ferentinos P, Paparrigopoulos T, Rentzos M, Evdokimidis I. Duloxetine for pathological laughing and crying. Int J Neuropsychopharmacol. 2009;12(10):1429–1430. | ||
Moore SR, Gresham LS, Bromberg MB, Kasarkis EJ, Smith RA. A self report measure of affective lability. J Neurol Neurosurg Psychiatry. 1997;63(1):89–93. | ||
King RR, Reiss JP. Treatment of pseudobulbar affect with citalopram in a patient with progressive multifocal leukoencephalopthy. J Clin Neurosci. 2012;19(1):185–186. | ||
Drugs and human performance fact sheet: dextromethorphan [webpage on the Internet]. Washington, DC: National Highway Traffic Safety Administration; 2014. Available from: http://www.nhtsa.gov/people/injury/research/job185drugs/dextromethorphan.htm. Accessed March 3, 2013. | ||
Kazis A, Kimiskidis V, Niopas I. Pharmacokinetics of dextromethorphan and dextrorphan in epileptic patients. Acta Neurol Scand. 1996; 93(2–3):94–98. | ||
Walker FO, Hunt VP. An open label trial of dextromethorphan in Huntington’s disease. Clin Neuropharmacol. 1989;12(4):322–330. | ||
Logan BK, Goldfogel G, Hamilton R, Kuhlman J. Five deaths resulting from abuse of dextromethorphan sold over the internet. J Anal Toxicol. 2009;33(2):99–103. | ||
Cochems A, Harding P, Liddicoat L. Dextromethorphan in Wisconsin drivers. J Anal Toxicol. 2007;31(4):227–232. | ||
Logan BK. Combined dextromethorphan and chlorpheniramine intoxication in impaired drivers. J Forensic Sci. 2009;54(5):1176–1180. | ||
Ganetsky M, Babu KM, Boyer EW. Serotonin syndrome in dextromethorphan ingestion responsive to propofol therapy. Pediatr Emerg Care. 2007;23(11):829–831. | ||
Kintz P, Mangin P. Toxicological findings in a death involving dextromethorphan and terfenadine. Am J Forensic Med Pathol. 1992; 13(4):351–352. | ||
Rammer L, Holmgren P, Sandler H. Fatal intoxication by dextromethorphan: a report on two cases. Forensic Sci Int. 1988;37(4):233–236. | ||
Gey GO, Levy RH, Pettet G, Fisher L. Quinidine plasma concentration and exertional arrhythmia. Am Heart J. 1975;90(1):19–24. | ||
Benton RE, Sale M, Flockhart DA, Woosley RL. Greater quinidine-induced QTc interval prolongation in women. Clin Pharmacol Ther. 2000;67(4):413–418. | ||
Kharasch ED, Hoffer C, Whittington D. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. Br J Clin Pharmacol. 2004;57(5):600–610. | ||
Florian J, Garnett CE, Nallani SC, Rappaport BA, Throckmorton DC. A modeling and simulation approach to characterize methadone QT prolongation using pooled data from five clinical trials in MMT patients. Clin Pharmacol Ther. 2012;91(4):666–672. | ||
Saal AK, Werner JA, Greene HL, Sears GK, Graham EL. Effect of amiodarone on serum quinidine and procainamide levels. Am J Cardiol. 1984;53(9):1264–1267. | ||
Tartini R, Kappenberger L, Steinbrunn W, Meyer UA. Dangerous interaction between amiodarone and quinidine. Lancet. 1982; 1(8285):1327–1329. | ||
Toivonen L, Kadish A, Morady F. A prospective comparison of class IA, B, and C antiarrhythmic agents in combination with amiodarone in patients with inducible, sustained ventricular tachycardia. Circulation. 1991;84(1):101–108. | ||
Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide pharmacokinetics and pharmacodynamics. Influence of genetic polymorphism. J Pharmacol Exp Ther. 1989; 249(1):134–142. | ||
Birgersdotter UM, Wong W, Turgeon J, Roden DM. Stereoselective genetically-determined interaction between chronic flecainide and quinidine in patients with arrhythmias. Br J Clin Pharmacol. 1992; 33(3):275–280. | ||
Bigger JT. The interaction of mexiletine with other cardiovascular drugs. Am Heart J. 1984;107(5 Pt 2):1079–1085. | ||
Broly F, Vandamme N, Libersa C, Lhermitte M. The metabolism of mexiletine in relation to the debrisoquine/sparteine-type polymorphism of drug oxidation. Br J Clin Pharmacol. 1991;32(4):459–466. | ||
Hii JT, Duff HJ, Burgess ED. Clinical pharmacokinetics of propafenone. Clin Pharmacokinet. 1991;21(1):1–10. | ||
Bailey DG, Freeman DJ, Melendez LJ, Kreeft JH, Edgar B, Carruthers SG. Quinidine interaction with nifedipine and felodipine: pharmacokinetic and pharmacodynamic evaluation. Clin Pharmacol Ther. 1993;53(3): 354–359. | ||
Damkier P, Hansen LL, Brosen K. Effect of diclofenac, disulfiram, itraconazole, grapefruit juice and erythromycin on the pharmacokinetics of quinidine. Br J Clin Pharmacol. 1999;48(6):829–838. | ||
Huang BH, Wu CH, Hsia CP, Yin Chen C. Azithromycin-induced torsade de pointes. Pacing Clin Electrophysiol. 2007;30(12):1579–1582. | ||
Rogers HL, Bhattaram A, Zineh I, et al. CYP2D6 genotype information to guide pimozide treatment in adult and pediatric patients: basis for the U.S. Food and Drug Administration’s new dosing recommendations. J Clin Psychiatry. 2012;73(9):1187–1190. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.