Back to Journals » OncoTargets and Therapy » Volume 12
Evaluating ivosidenib for the treatment of relapsed/refractory AML: design, development, and place in therapy
Authors Nassereddine S ![]() , Lap CJ, Tabbara IA
, Lap CJ, Tabbara IA
Received 6 October 2018
Accepted for publication 5 December 2018
Published 28 December 2018 Volume 2019:12 Pages 303—308
DOI https://doi.org/10.2147/OTT.S182443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Samah Nassereddine,1,2 Coen J Lap,2 Imad A Tabbara1,2
1Department of Internal Medicine, The George Washington University School of Medicine, Washington, DC, USA; 2Division of Hematology/Oncology, The George Washington Cancer Center, Washington, DC, USA
Abstract: Improvements in the last decade in understanding the molecular mechanisms underlying acute myeloid leukemia (AML) have emphasized that treatment regimens should be personalized with agents that can selectively target genetic abnormalities if present. Neomorphic mutations in isoform 1 of isocitrate dehydrogenase (IDH1) result in the formation of the oncometabolite R-2-hydroxyglutarate, which drives leukemic transformation by affecting processes such as chromatin remodeling, the cellular defense against oxidative stress and cell survival. Preclinical studies with small molecule inhibitors have validated mutant IDH1 as a molecular target, and a recent Phase 1 clinical trial with the first mutant IDH1 inhibitor ivosidenib has prompted approval by the US Food and Drug Association for the treatment of patients with IDH1-mutated AML in the relapsed and refractory setting due to impressive results. This approval has given a group of patients, that otherwise has a very poor prognosis and limited options, new hope, and it is to be expected that more indications for ivosidenib will follow soon. These developments highlight the potential of precision medicine in AML, with more agents currently under evaluation in clinical trials. Although the first reports have also already emerged describing acquired resistance for these mutant IDH inhibitors, combination treatment might overcome this problem, which could drastically change the treatment landscape of AML over the next few years.
Keywords: ivosidenib, AML, IDH1, relapsed, refractory
Introduction
In the era of precision medicine, physicians are anchoring hope to find a cure for even the most challenging diseases. Acute myeloid leukemia (AML), initially described as “a suppuration of the blood,” remained a black box for several decades due to its complex biology and unpredictable clinical course, which resulted in a stagnation in treatment evolution.1 With the better understanding of the underlying molecular abnormalities driving leukemogenesis, more intense scientific efforts have led to the development of various targeted therapies for AML.2–4 As a result, over the last 2 years, we have witnessed the approval of five new agents gemtuzumab ozogamicin, midostaurin, Vyxeos (liposomal cytarabine-daunorubicin), enasidenib, and ivosidenib, which have expanded the treatment arsenal significantly, especially in the setting of relapsed and refractory disease (R/R AML).
An especially promising development in the treatment of AML appears to be the ability to specifically inhibit the recurrently mutated isoforms of isocitrate dehydrogenase (IDH) with small molecule inhibitors.5 Following successful Phase 2 clinical trials showing efficacy of these drugs for the treatment of IDH-mutated AML, the US Food and Drug Association (FDA) first granted approval in 2017 for Enasidenib (AG221/CC-90007, IDHIFA; Celgene Corp., Summit, NJ, USA) in the setting of R/R AML with a mutated IDH2 and subsequently in 2018 for ivosedinib (AG-120, Tibsovo; Agios Pharmaceuticals, Inc., MA, USA) for the treatment of R/R AML with a mutant IDH1.6,7 The combination of a promising efficacy in clinical trials with a mild side effect profile has generated new hope for a group of patients who otherwise has a dismal outlook. In this review we focus on the most recently approved drug, ivosidenib, and discuss its molecular mechanism of action, clinical development, and current place in the treatment of IDH1-mutated R/R AML.
Exploring the role of mutant IDH1 inhibition in AML
Advances in the last decade in next generation sequencing technologies have revolutionized our understanding of the molecular foundations of AML, by revealing that the majority of cases can be categorized into biological and prognostical distinct subtypes based on the presence or absence of recurrent cytogenetic and molecular abnormalities.2–4 Two recently discovered genes found to be recurrently mutated in AML are two isoforms of a metabolic enzyme named IDH, which catalyzes the oxidative decarboxylation of isocitrate into α-ketoglutarate (αKG) in the tricarboxylic acid cycle in the cytosol and mitochondria of cells.8 Neomorphic mutations in the catalytic site of these isoforms result in the ability to reduce αKG into the oncometabolite R-2-hydroxyglutarate (R-2HG), which affects various cellular processes, including the epigenetic state of the cell, the cellular defense against oxidative stress as well as cell survival9–12 (Figure 1). Although mutated IDH1 has shown oncogenic potential independent of R-2HG, R-2HG appears to have crucial role in leukemogenesis by competitively inhibiting multiple classes of αKG-dependent dioxygenases, which includes the ten-eleven translocation family of 5-methylcytosine hydroxylases and the histone lysine demethylase (KDM) family.9,10 Both the families of enzymes orchestrate chromatin remodeling, either through DNA methylation or by post-translational modification of histone ends, thereby regulating gene expression programs in the cell. Cellular accumulation of R-2HG results in a global dysregulation of gene expression, which in hematopoietic cells has been shown to directly impair cellular differentiation and promote leukemogenesis. Beyond its effect on disrupting global gene expression patterns, two other mechanisms that contribute to transformation have been described as well, including stimulation of the EgIN family of prolyl 4-hydroxylases and direct inhibition of cytochrome c oxidase (COX) in the inner membrane of mitochondria. Stimulation of prolyl 4-hydroxylases results in aberrant HIF-1α signaling, which has been linked to cancer stem cell formation and chemo-resistance of leukemic blasts.11,13 Variously, COX inhibition makes leukemic cells become dependent on anti-apoptotic BCL-2 for cell survival, which is induced as a direct response to increased activation of pro-apoptotic BAK and BAX.12
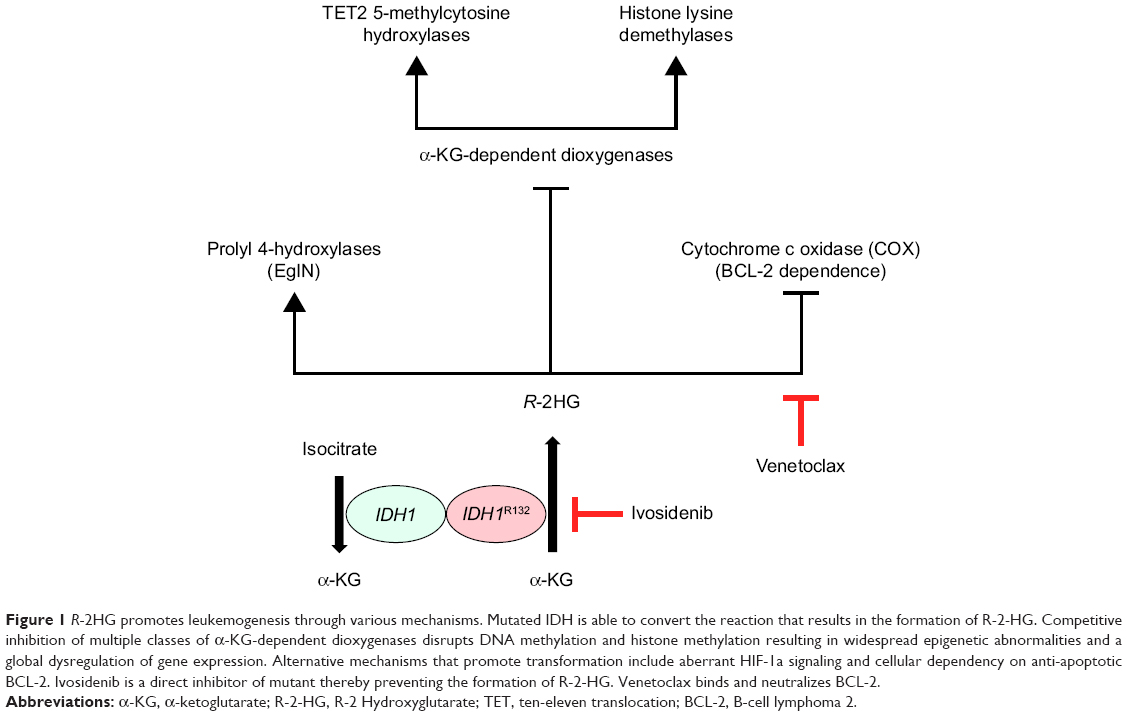 | Figure 1 R-2HG promotes leukemogenesis through various mechanisms. Mutated IDH is able to convert the reaction that results in the formation of R-2-HG. Competitive inhibition of multiple classes of α-KG-dependent dioxygenases disrupts DNA methylation and histone methylation resulting in widespread epigenetic abnormalities and a global dysregulation of gene expression. Alternative mechanisms that promote transformation include aberrant HIF-1a signaling and cellular dependency on anti-apoptotic BCL-2. Ivosidenib is a direct inhibitor of mutant thereby preventing the formation of R-2-HG. Venetoclax binds and neutralizes BCL-2. |
Heterozygous mutations affecting IDH1 in AML are all identified in a catalytic hotspot that codes for an arginine at position 132 (IDH1R132), which results in the replacement of the arginine into a cysteine, histidine, glycine, or serine.2,3 The incidence of mutations in IDH1 varies between studies but is estimated to lie between 6% and 10%, which increases to 16% if only cases with normal cytogenetics are analyzed (CN-AML).3,4,14 Mutations in IDH1 frequently co-occurs with other molecular abnormalities, including mutations in nucleophosmin (NPM1) and Mixed Lineage Leukemia-partial tandem duplications. In addition, mutations in IDH1 have also been identified in other myeloid malignancies, albeit in a lower incidence, including in 3%–12% of patients with myelodysplastic syndrome (MDS) and in 1%–8% of patients with myeloproliferative neoplasms (MPN).15–17 Although mutations in IDH1 are often described as initial driving events in the process of leukemogenesis and have been identified in pre-leukemic hematopoietic stem and progenitor cells, other studies have also shown that acquisition of a mutation in IDH1 is associated with leukemic transformation of MDS and MPN.18,19 While reports are inconsistent, most studies looking into the prognostic significance of recurrent mutations in AML report an adverse prognosis if the sole abnormality that can be detected in CN-AML is a mutation in IDH1. Nevertheless, prognosis appears to be highly correlated by the presence of other mutations, as a co-occurring mutation in NPM1 positively influences the prognosis resulting in an intermediate risk disease.
Extensive preclinical studies have convincingly shown that inhibition of mutant IDH in leukemic cells with either ivosidenib or other mutant IDH inhibitors is able to suppress R-2HG levels, reverse DNA and histone hypermethylation, and, importantly, release the myeloid differentiation block, both in vitro and in vivo.10,20–22 Due to these observations, various small molecule inhibitors have been developed, which are currently evaluated in clinical trials including a pan-mutant IDH1 and IDH2 inhibitor (AG881) as well as inhibitors that specifically target mutant IDH1 (ivosidenib/AG-120, IDH305, FT2102, BAY1436032).23,24 Although some of these inhibitors have additional properties in comparison with ivosidenib, such as the ability to cross the blood–brain barrier for BAY1436032, only ivosidenib has so far received FDA approval for the treatment of IDH1-mutated AML in the R/R setting.
Use of ivosidenib in patients with IDH1-mutated R/R AML
Ivosidenib (AG-120) is a first-in-class, potent and reversible inhibitor, specifically targeting mutant IDH1, which is currently being evaluated in various clinical trials for the treatment of IDH1-mutated AML and MDS (NCT03173248, NCT03471260, NCT03564821, NCT02074839, NCT03503409, NCT02632708, NCT02677922, NCT03013998). This review discusses in more depth the results of a recent trial that resulted in the FDA approval of ivosidenib/AG-120 for the treatment of R/R AML. Conversely, we expect that the results of trials evaluating the treatment of ivosidenib in other settings, including for patients with IDH1-mutated MDS or treatment of ivosidenib in combination with hypomethylating agents, will be published soon.
The recent FDA approval was based on the promising results of a multicenter, open-label, single-arm, dose-escalation, and dose-expansion Phase 1 clinical trial (NCT02074839).25 Although the study is a non-randomized, Phase 1 clinical trial and the end-point did not include quality of life, the results are impressive for a group of patients with an otherwise extremely poor prognosis and limited treatment options. In the study, 179 patients with a diagnosis of R/R AML were included, including 33% of patients with a secondary form of AML that failed prior therapy. About 24% of patients had relapsed after allogeneic stem cell transplantation. Median age was 67 years (range: 18–87 years), and IDH1 mutational status was confirmed by digital PCR. Patients were assigned to receive 500 mg ivosidenib oral daily until disease progression, poor tolerance, or transplant (12% of population). About 30.2% (95% CI: 23.5–37.5) of patients reached the combined endpoint of either a complete remission (CR) or a complete remission with partial hematologic recovery (CRh) with a median time to CR or CRh of 2.7 months (range: 0.9–5.6 months) and a median duration of response of 6.5 months (95% CI: 5.5–11.1). About 21.8% (95% CI: 16.0–28.6) of patients reached a CR with a median time to CR of 2.8 months (range: 0.9–8.3) and a median duration of CR of 9.3 months (95% CI: 5.6–12.5). About 41.6% of patients (95% CI: 31.9–46.7) had an overall response (OR) with a median time to first response of 1.9 months (range: 0.8–4.7 months) and a median duration of response of 6.5 months (95% CI: 4.6–9.3). Plasma R-2HG levels were suppressed in almost all patients, regardless of response. Patients with R/R AML responding with a CR or CRh had a lower mean level of mutant IDH1 variant allele frequency compared to patients who did not meet the combined endpoint. In these patients, R-2HG levels remained stable overtime. This last observation suggests a possibility to predict treatment response. Clearance of mutant IDH1 was seen in 21% of patients. Moreover, preliminary data also showed that patients in CR or CRh achieving a clearance of mutant IDH1 had a longer duration of remission (11.1 months; 95% CI: 6.5 to not known) and longer OS (14.5 months; 95% CI: 13.9 to not known) compared to patients in CR or CRh who did not reach clearance (duration of remission, 6.5 months; 95% CI 4.6–9.3 and OS, 10.2 months; 95% CI: 9.0–12.5). At 56 days of treatment, 37.3% of patients who were dependent at baseline on red blood cell and/or platelet transfusions became transfusion independent and 59.4% of those who were at baseline independent of red blood cell and/or platelet transfusions remained transfusion independent.
Overall, ivosidenib was well tolerated, although most patients experienced side effects (98.9%). The most common adverse events were diarrhea (30.7%), leukocytosis (29.6%), febrile neutropenia (28.5%), nausea (27.9%), and fatigue (25.7%). About 20.7% of patients experienced adverse events of grade 3 or higher, most commonly prolongation of the QT interval (7.8%) and IDH differentiation syndrome (DS) (3.9%). These last two adverse events are of particular interest because they were also observed with the use of enasidenib in IDH2-mutated AML.26 IDH DS has symptoms similar to the DS observed in patients treated with all-trans retinoic acid (ATRA) and/or arsenic trioxide (ATO) for acute promyelocytic leukemia (APL), which can be potentially lethal, and is thought to be a mediated by cytokine release from differentiating myeloid blasts.27 The median time of onset of the DS was 29 days (range: 5–59 days) and 37% of these patients developed leukocytosis. The patients were treated with steroids, diuretics with or without hydroxyurea, resulting in resolution of symptoms in 89% of patients, with no patients experiencing grade 4 or lethal events. QT prolongation resulted in dose interruptions and dose reductions in the study. This requires close monitoring of patients because of the large number of drugs that can cause QT prolongation that may be used by these patients.
Acquired resistance to mutant IDH inhibition
Despite the promising data published about mutant IDH inhibitors, reports have already emerged describing patients who developed resistance to these small molecule inhibitors, resulting in progressive disease with increases in plasma R-2HG concentration and re-occurrence of leukemic blasts. In an initial report, two patients with IDH2-mutated AML were presented, who developed resistance to the mutant IDH2 inhibitor enasidenib as a result of second-site IDH2 mutations (Q316E, I319M) in the wild-type allele (in trans), which cooperated with the gain-of-function mutation (R140Q) on the other allele in inducing resistance either by disrupting the hydrogen bond between the IDH2 dimer and enasidenib or by hindrance of binding of the IDH2 dimer to enasidenib.28 It remains to be investigated if the mechanism underlying the resistance to ivosidenib seen in this patient is similar to the resistance for enasidenib observed in patients with second-site mutations in trans. Furthermore, standard assessment of IDH1 mutational status does not include the evaluation of a potential S280F mutation. It might be interesting to see if a mutation at position S280F is present before the start of ivosidenib therapy (or any other mutant IDH1 inhibitor) in these patients, to see if presence of a S280F mutation can predict resistance to these small molecule inhibitors.
Regardless, whether it is second-site mutations in cis, in trans, or other mechanisms such as isotype switching from mutant IDH1 to mutant IDH2 (or vice versa), as suggested in a recent pre-publication, the occurrence of resistance emphasizes the crucial role that mutant IDH plays as a therapeutic target.29 Importantly, the fact that leukemic cells in these patients can develop resistance, underlines the requirement to identify strategies, including combination treatment, to prevent the occurrence of resistance. In this regard, the selective inhibitor of BCL-2, venetoclax, could be a very interesting approach.30 BCL-2 enhances leukemic cell survival by blunting the activation of mitochondrial apoptosis pathway by directly antagonizing pro-apoptotic proteins BAX and BAK. Based on the molecular mechanism by which IDH mutations promote leukemogenesis, dual treatment of these patients with both ivosidenib and venetoclax might result in synergy or prevent the development of future resistance12 (Figure 1).
Conclusion
Ivosedinib has a promising role for the treatment of R/R AML, especially because the outcome in this group of patients is notoriously poor with CR rates not higher than 10% resulting in a median OS of less than 4 months and a 60-day mortality of 30%. Following last year’s approval of enasidenib for the treatment of IDH2-mutated AML, the recent FDA approval of the mutant IDH1 inhibitor ivosidenib will give clinicians another targeted therapy option, provided that a mutation in IDH1 is present. Not surprisingly, due to the impressive results, ivosidenib is also recommended for induction as well as post-remission maintenance therapy for older patients who are ineligible for intensive remission induction therapy in the most recent National Comprehensive Cancer Network AML guidelines.31
So far, clinical trials have shown impressive data with 21.8% of patients reaching a CR and nearly one in three patients achieving either CR or CRh. While special attention needs to be given to QT prolongation and IDH DS, overall, ivosidenib was well tolerated with few grade 3 (or greater) side effects. Various clinical trials are currently underway in the hope to show benefit of combining ivosidenib with either intensive chemotherapy or hypomethylating agents for the upfront treatment of IDH1-mutated AML, or as maintenance therapy post-transplant. After nearly four decades of little progress in treatment options, the approval of ivosidenib and other targeted therapies in the last 2 years have advanced the field of AML treatment tremendously, forthcoming as a result of a greater appreciation of the heterogeneity of this complex disease. Within the next few years, more agents are expected to be introduced including the BCL-2 inhibitor venetoclax, the cyclin-dependent kinase 9 inhibitor alvocidib and second-generation FLT3 inhibitors quizartinib and gilteritinib.30,32–34 While a cure for AML still requires the combination of various treatment modalities, current advances highlight the potential of precision medicine as well as of other targeted approaches that are in development to treat AML.
Disclosure
The authors report no conflicts of interest in this work.
References
Piller G. Leukaemia – a brief historical review from ancient times to 1950. Br J Haematol. 2001;112(2):282–292. | ||
Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. | ||
Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. | ||
Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. | ||
Nassereddine S, Lap CJ, Haroun F, Tabbara I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. 2017;96(12):1983–1991. | ||
Center for Drug Evaluation and Research Application Number 209606orig1s000 Multi-Discipline Review; 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209606Orig1s000MultidisciplineR.pdf. Accessed March 30, 2018. | ||
Center for Drug Evaluation and Research Application Number 211192Orig1s000 Multi-Discipline Review. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211192Orig1s000MultidisciplineR.pdf. Accessed December 17, 2018. | ||
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966. | ||
Sasaki M, Knobbe CB, Munger JC, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488(7413):656–659. | ||
Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. | ||
Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. | ||
Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. | ||
Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8(4):399–411. | ||
Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood. 2010;116(25):5486–5496. | ||
Kosmider O, Gelsi-Boyer V, Slama L, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24(5):1094–1096. | ||
Pardanani A, Patnaik MM, Lasho TL, et al. Recurrent IDH mutations in high-risk myelodysplastic syndrome or acute myeloid leukemia with isolated del(5q). Leukemia. 2010;24(7):1370–1372. | ||
Tefferi A, Jimma T, Sulai NH, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26(3):475–480. | ||
Corces-Zimmerman MR, Hong W-J, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548–2553. | ||
Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24(7):1015–1023. | ||
Hansen E, Quivoron C, Straley K, Lemieux RM, Popovici-Muller J. AG-120, an oral, selective, first-in-class potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood. 2014;124:3734. | ||
Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S. Discovery of the first potent inhibitors of mutant IDH1 that lowers tumor 2-HG in vivo. ACS Med Chem Lett. 2012;3(10):850–855. | ||
Wang F, Travins J, Delabarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. | ||
Chaturvedi A, Herbst L, Pusch S, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. 2017;31(10):2020–2028. | ||
Dinardo CD, Schimmer AD, Yee KW. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128(22):Abstract 1073. | ||
Dinardo CD, Stein EM, de Botton S, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med. 2018;378(25):2386–2398. | ||
Stein EM, Dinardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. | ||
Birendra KC, Dinardo CD. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant IDH1 inhibitor, AG-120. Clin Lymphoma Myeloma Leuk. 2016;16(8):460–465. | ||
Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559(7712):125–129. | ||
Harding JJ, Lowery MA, Shih AH, Schvartzman JM, Hou S. Isoform switching as a mechanism of acquired resistance to isocitrate dehydrogenase inhibition. BioRxiv. 2018;8(12):1540–1547. | ||
National Comprehensive Cancer Network. NCCN Practice Guidelines in Oncology: Acute Myeloid Leukemia. V.2; 2018. Available from: www.nccn.org/professionals/physician_gls/pdf/aml.pdf. Accessed November 12, 2018. | ||
Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. | ||
Bogenberger J, Whatcott C, Hansen N, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget. 2017;8(63):107206–107222. | ||
Cortes JE, Tallman MS, Schiller GJ, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood. 2018;132(6):598–607. | ||
Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–1075. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.