")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Eteplirsen in the treatment of Duchenne muscular dystrophy
Authors Lim KRQ , Maruyama R, Yokota T
Received 30 November 2016
Accepted for publication 9 February 2017
Published 28 February 2017 Volume 2017:11 Pages 533—545
DOI https://doi.org/10.2147/DDDT.S97635
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Kenji Rowel Q Lim,1 Rika Maruyama,1 Toshifumi Yokota1,2
1Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, 2The Friends of Garrett Cumming Research & Muscular Dystrophy Canada, HM Toupin Neurological Science Research Chair, Edmonton, AB, Canada
Abstract: Duchenne muscular dystrophy is a fatal neuromuscular disorder affecting around one in 3,500–5,000 male births that is characterized by progressive muscular deterioration. It is inherited in an X-linked recessive fashion and is caused by loss-of-function mutations in the DMD gene coding for dystrophin, a cytoskeletal protein that stabilizes the plasma membrane of muscle fibers. In September 2016, the US Food and Drug Administration granted accelerated approval for eteplirsen (or Exondys 51), a drug that acts to promote dystrophin production by restoring the translational reading frame of DMD through specific skipping of exon 51 in defective gene variants. Eteplirsen is applicable for approximately 14% of patients with DMD mutations. This article extensively reviews and discusses the available information on eteplirsen to date, focusing on pharmacological, efficacy, safety, and tolerability data from preclinical and clinical trials. Issues faced by eteplirsen, particularly those relating to its efficacy, will be identified. Finally, the place of eteplirsen and exon skipping as a general therapeutic strategy in Duchenne muscular dystrophy treatment will be discussed.
Keywords: Duchenne muscular dystrophy, eteplirsen, Exondys 51, exon-skipping therapy, phosphorodiamidate morpholino oligomers
Duchenne muscular dystrophy (DMD): introduction and management issues in treatment
DMD is a fatal X-linked recessive neuromuscular disorder characterized by progressive muscle weakening and wasting.1 It affects around one in 3,500–5,000 males born worldwide.2,3 The disorder progresses rapidly, with boys losing ambulation by 12 years of age or earlier; death often occurs within the 20s, usually due to respiratory or cardiac complications.4,5 DMD is caused by mutations in the DMD gene coding for dystrophin,1,6 a membrane-associated protein that links cytoskeletal actin in muscle fibers with the surrounding extracellular matrix by forming a network with sarcolemmal glycoproteins (otherwise known as the dystrophin-associated glycoprotein complex [DAGC]).7–9 This linkage strengthens muscle structure during stressful contraction/relaxation cycles;10 recent studies, however, indicate that dystrophin also has nonmechanical roles.11 Dystrophin has four domains: an N-terminal domain for binding actin, a rod domain mainly for structural flexibility, a cysteine-rich domain for facilitating protein–protein interactions, and a C-terminal domain for binding DAGC proteins at the sarcolemma.9,12 Dystrophin loss predisposes muscle fibers to mechanical damage, leading to muscle degeneration.
DMD is considered the longest gene in humans, spanning 2.4 Mb in chromosomal region Xp21 with 79 exons and producing a 14 kb transcript.13,14 Due to its length, it is highly susceptible to mutations. Furthermore, certain regions of DMD are mutation hotspots.15,16 Approximately 60% of DMD cases are due to deletions of at least one exon in DMD,4,12 ~6% to duplications,17 and the rest to small mutations. In most cases, these disrupt the DMD reading frame or introduce a premature stop codon, both of which cease dystrophin production.
At present, most practices for DMD treatment are palliative at best, aimed at managing problems with ambulation, respiration, and cardiac health that are typical of DMD.4,5 Of these, corticosteroid treatment has been found to be the overall most effective option for patients. Improved muscular strength, prolonged ambulation, and better respiratory function were observed in patients treated with the corticosteroids prednisolone/prednisone or deflazacort in separate long-term clinical trials.18,19 However, these improvements were temporary – disease progression was only delayed – and treatment was associated with a number of side effects (eg, weight gain, bone fractures, cataracts).
There is thus a push toward the development of curative therapies for DMD. To date, a number of cell- and gene-based strategies have been explored, with varying degrees of success.5,12 Cell-based strategies involve transplantation of healthy myoblasts into patients, and as such are handicapped by issues of immune rejection and poor systemic delivery and viability of transplanted cells. Likewise, conventional gene-based strategies aiming to deliver functional copies of DMD in patients have turned out problematic, mostly due to poor delivery (owing to the large, complex structure of the gene)13,14 and the activation of an immune response in cases when a viral vector is used.
Novel strategies without these problems of safety and efficacy are currently emerging.12 One promising strategy is exon skipping, which attempts to fix the defective DMD gene through the use of nucleic acid-based drugs.20,21 This approach has shown much promise and has spurred the development of numerous pharmaceuticals, one of which is eteplirsen, also known as Exondys 51 or AVI-4658.
Developed by Sarepta Therapeutics (Cambridge, MA, USA), eteplirsen was approved by the US Food and Drug Administration (FDA) in September 2016, making it the first and currently only FDA-approved drug for DMD.22 Eteplirsen was granted accelerated approval on the basis of surrogate end-point results showing that it was able to increase dystrophin levels in patients.22,23 While the drug is now accessible to patients, an additional clinical trial is still required by the FDA to demonstrate strong evidence of clinical benefit. This review discusses the pharmacology of eteplirsen, findings from clinical trials on its efficacy and safety, and issues faced by the drug in its course to definitive approval. The review ends by highlighting the implications of eteplirsen on the DMD community and the DMD-therapy scene as a whole.
Clinical pharmacology of eteplirsen
Mechanism of action: exon skipping
Not all DMD deletions result in out-of-frame mutations: some lead to in-frame mutations, generating variants able to produce functional albeit truncated versions of dystrophin. This kind of deletion occurs in patients with Becker MD (BMD), a milder dystrophinopathy compared to DMD.24 The genetic difference between DMD and BMD presents an important observation: the nature of the deletion determines the severity of the disorder. This led to the realization that making a deletion less harmful by turning an out-of-frame to an in-frame mutation should alter the DMD phenotype to that of the less severe BMD.
It is with this underlying principle that exon skipping was developed as a therapeutic strategy for DMD. In this approach, the translational reading frame of a gene is restored using synthetic nucleic acid analogs called antisense oligonucleotides (AOs) to interfere with pre-messenger RNA (mRNA) splicing20,25 (Figure 1). AOs are employed to bind target complementary sequences in the pre-mRNA, which influence the splicing machinery to exclude an exon (or exons) from the final transcript. The therapeutic potential of the method was first demonstrated in 1993, where correct splicing of the human β-globin gene was successfully restored in vitro through the use of a 2′O-methyl RNA AO.26 Since then, the strategy has grown to use a wide array of AO chemistries for the treatment of various disorders.25
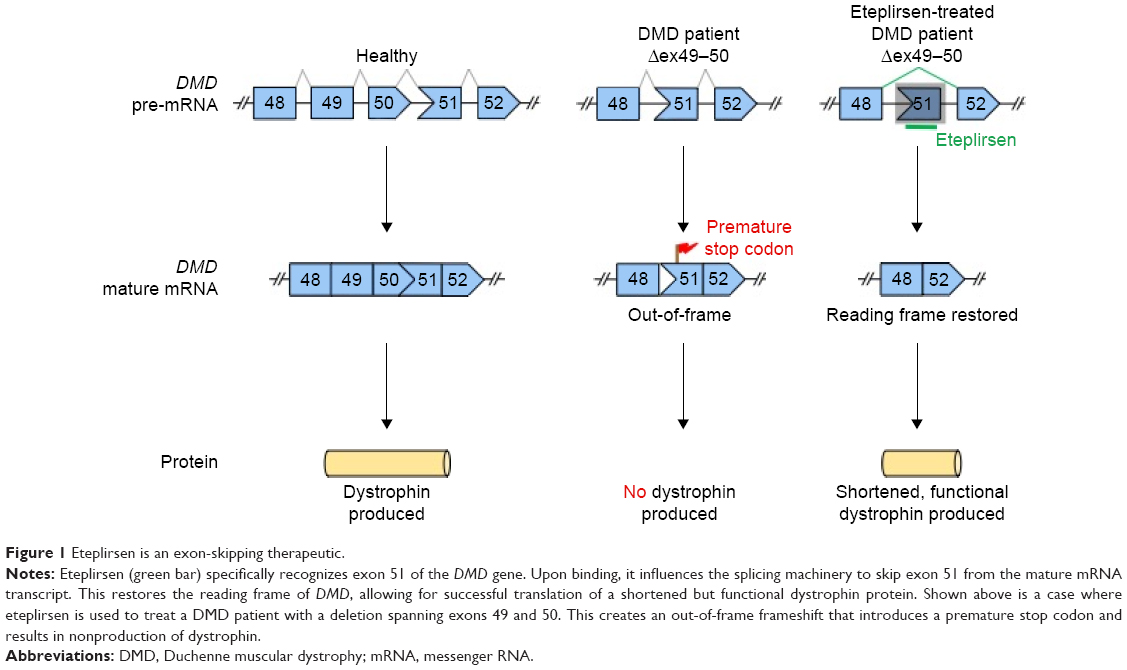 | Figure 1 Eteplirsen is an exon-skipping therapeutic. |
Eteplirsen is a 30-nucleotide phosphorodiamidate morpholino oligomer (PMO) type of AO20,27 (Figure 2) with the sequence CTCCAACATCAAGGAAGATGGCATTTCT.28 In contrast to regular RNA or DNA, PMO bases are attached to a morpholine moiety, and subunits are connected via phosphorodiamidate linkages that are neutrally charged at physiological pH.20,29 Eteplirsen hybridizes to exon 51 of DMD (codes for part of hinge 3 within the rod domain)30 and causes it to be skipped during splicing;20 this corrects the translational reading frame, resulting in the production of shortened functional dystrophin proteins (Figure 1). A related DMD therapeutic, drisapersen (BioMarin, San Rafael, CA, USA), is also an AO-based drug with the same mechanism of action.20 It differs from eteplirsen in that it is an 18-mer 2′O-methyl phosphorothioate type of AO, which is negatively charged (Figure 2). The FDA rejected drisapersen in early 2016, due to safety issues associated with the use of the drug and insufficient evidence of clinical utility.31
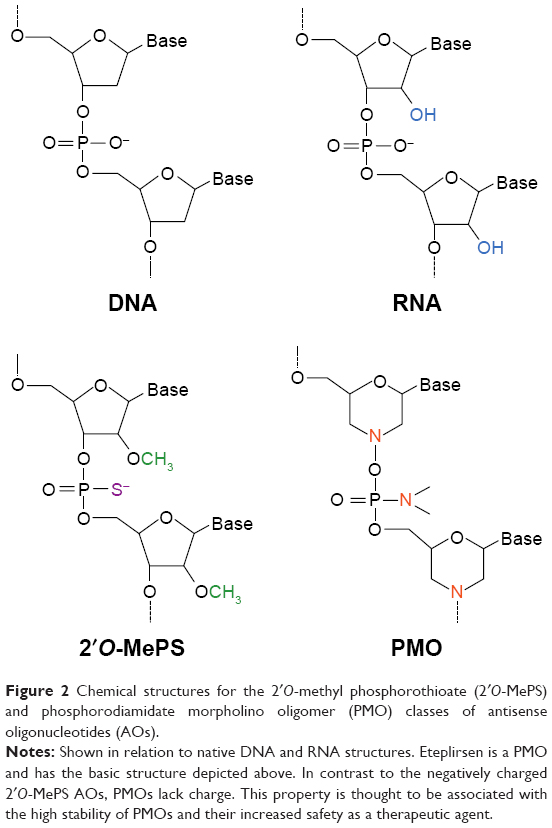 | Figure 2 Chemical structures for the 2′O-methyl phosphorothioate (2′O-MePS) and phosphorodiamidate morpholino oligomer (PMO) classes of antisense oligonucleotides (AOs). |
Eteplirsen is beneficial for DMD patients with deletions ending at exon 50 and starting at exon 52.12 This covers ~20.5% of DMD patients with deletion mutations, or 14% of all DMD patients.32 This is the largest group of patients to which single exon skipping is applicable, making exon 51 a reasonable therapeutic target. Also, in vivo efficacy of DMD exon 51 skipping using PMOs has been demonstrated in the mdx52 dystrophic mouse model,33 further making it a good target choice.
Pharmacokinetics
Table 1 lists all clinical trials on eteplirsen to date, together with the respective study details. The pharmacokinetic properties of eteplirsen were studied in two trials: NCT00844597 (Cirak et al)34 and NCT01396239 (Mendell et al).35 The former was an open-label Phase I/II dose-escalation study, with eteplirsen administered to DMD patients (19 patients total) as an intravenous (IV) infusion over a dose range of 0.5–20 mg/kg/week for 12 weeks. The latter was a double-blind Phase II placebo-controlled study that involved treating DMD patients with eteplirsen at 30 mg/kg/week or 50 mg/kg/week doses for 24 weeks. Each cohort, including the placebo-treated cohort, consisted of four patients, for a total of 12 enrolled patients. Inclusion/exclusion criteria for these studies are shown in Table 2. Besides these sources, pharmacokinetic information on eteplirsen is also available via its drug label.36
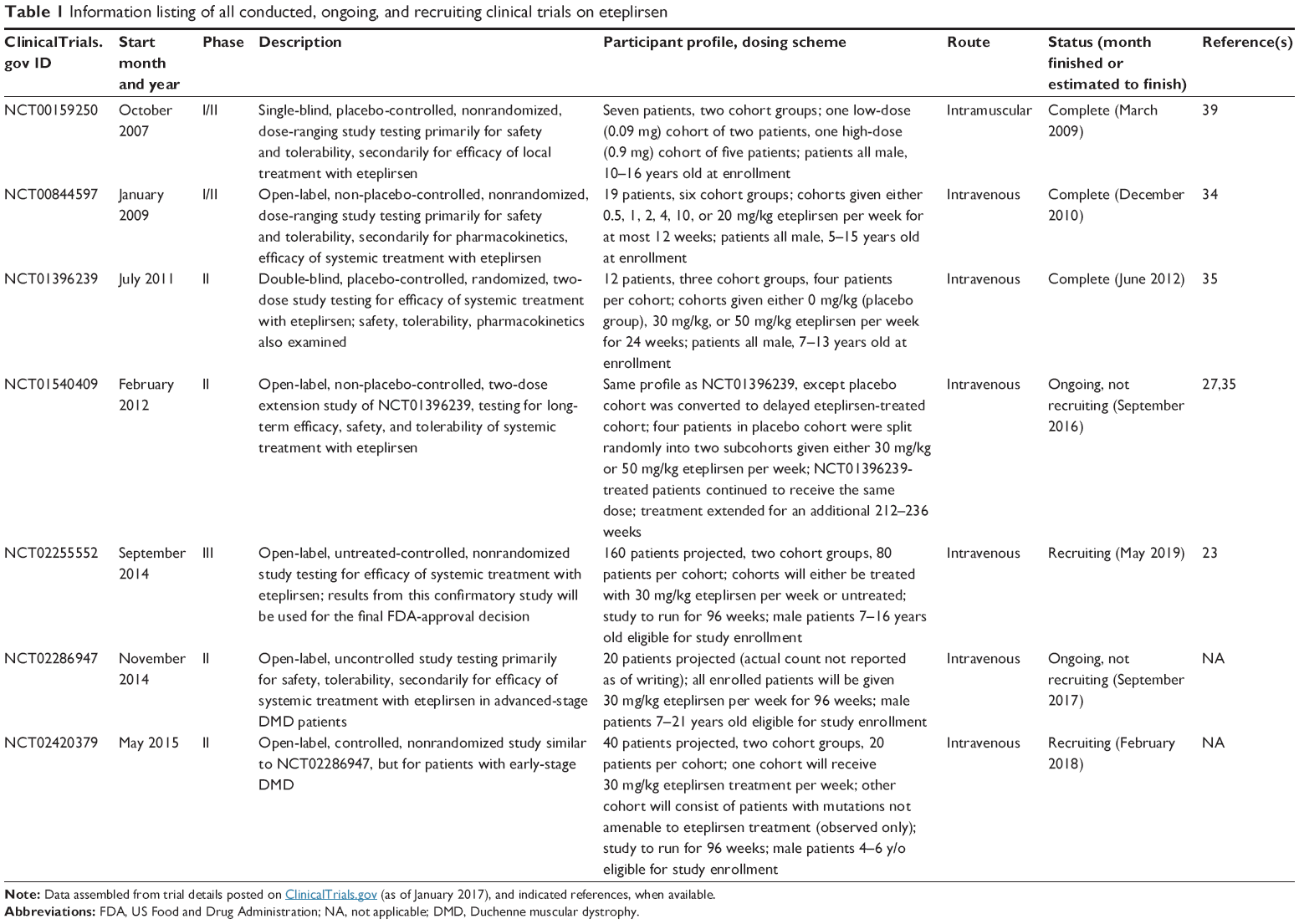 | Table 1 Information listing of all conducted, ongoing, and recruiting clinical trials on eteplirsen |
 | Table 2 Major inclusion/exclusion criteria for NCT00844597, NCT01396239/NCT01540409, and NCT02255552 |
Eteplirsen had a volume of distribution of 450–981 mL/kg in the dose-escalation study.34 At the recommended dose of 30 mg/kg/week, the mean apparent volume of distribution was 600 mL/kg.36 Specific tissue-distribution data for eteplirsen are not available in the current literature. However, studies have shown that PMOs do exhibit broad tissue distribution,37 with one study showing PMO uptake in six different muscle groups in mdx mice.38 Note though that PMO distribution to muscle and most tissues, other than in the kidney and liver, is poor; this is because PMOs are neutral, water-soluble molecules, and are thus more favorably cleared from the circulation than other drugs.37,38 On a different note, metabolism of eteplirsen was not found to occur in the liver;36 this is consistent with PMOs being unamenable to metabolic action.39
Cirak et al34 showed that eteplirsen had a plasma half-life of 1.62–3.6 hours within the 0.5–20.0 mg/kg dose range. In Mendell et al,35 after 12 weeks of treatment with single IV infusion doses of 30 mg/kg or 50 mg/kg eteplirsen, mean plasma half-lives found were 3.3 hours and 3.2 hours, respectively. No drug accumulation was observed between doses for dosing schemes of 0.5 mg/kg/week to 50 mg/kg/week.34–36 Total plasma clearance was 233–615 mL/h/kg over the dose range examined in Cirak et al;34 at a 30 mg/kg/week dose, total clearance was 339 mL/h/kg after 12 weeks of treatment.36 The kidneys are responsible for most of this clearance, with the extent of renal clearance increasing with dose. Mendell et al35 showed that 65%–70% of total clearance was attributable to renal clearance. It was observed that this magnitude of clearance occurred within the first 24 hours after drug administration.36
Efficacy of eteplirsen
Much of the data on the efficacy of eteplirsen as an IV administered drug for DMD treatment comes from four trials: NCT00844597, NCT01396239, NCT01540409, and NCT02255552 (Table 1). An earlier trial39 also produced efficacy data, but it involved an intramuscular route of administration. NCT0154040927 is an extension of NCT01396239, with two modifications: masking was changed to open-label, and patients in the placebo-treated cohort were switched to receive eteplirsen treatment. The four patients in the placebo-treated cohort were split, with two patients receiving either 30 mg/kg/week or 50 mg/kg/week eteplirsen. Matched historical controls from Italian and Belgian databases were used for the study.
NCT02255552 is the confirmatory study required by the FDA to support the clinical benefit of the drug.22,23 It is an ongoing, recruiting Phase III trial. Inclusion/exclusion criteria for this study are shown in Table 2. It is an open-label study planned to consist of two 80-patient cohorts: an eteplirsen-treated and an untreated cohort. Patients in the former cohort will be subjected to 30 mg/kg/week of eteplirsen for 96 weeks.40 The reasons behind the choice of dose were not discussed in the published clinical trial literature. Efficacy will be assessed 48 weeks posttreatment; treatment will go on to 96 weeks for longitudinal evaluation. In deciding whether to grant accelerated approval for the drug, the FDA requested preliminary efficacy data from 13 patients enrolled in the trial 48 weeks posttreatment.23
The efficacy of eteplirsen in these studies was assessed by dystrophin amounts produced as a result of treatment (a surrogate end point) and by treatment effect on patient ambulation (a clinical end point). Data from immunohistochemistry (IHC) and Western blotting (WB), which were employed to determine dystrophin protein localization and levels in patient muscle with antibodies against DMD rod-domain regions, served as outcome measures for the surrogate end point.34,35 Results from the 6-minute walk test (6MWT), which measures the distance a patient can independently walk in 6 minutes, mostly formed the basis for the clinical end point.35
NCT00844597 findings
Eteplirsen treatment was found to improve dystrophin expression in seven of 19 patients: six given 10–20 mg/kg/week and one given 2 mg/kg/week. Dystrophin fluorescence intensity from semiquantitative IHC showed a significant average increase to 16.4% from 8.9% of healthy controls posttreatment (P=0.0287). Three patients responded particularly well, showing posttreatment increases in dystrophin expression to 7.7%, 17%, and 18% of healthy controls in WB. Considering all results, however, there was an inconsistency of effect. The other four positive responders did not give appreciable results as the three patients mentioned, and the assays gave results that did not always agree with each other. Overall, dystrophin expression was not found to increase with dose. Result variability was ascribed to genetic background differences and random events surrounding eteplirsen uptake. The investigators suggested performing an extended clinical trial, with the argument that clinical benefit would only be observable upon prolonged treatment.
NCT01396239/NCT01540409 and NCT02255552 findings
NCT01396239/NCT01540409 was an extended clinical trial that responded to the shortfall of NCT00844597 with regard to study length. The results from these studies (summarized in Table 3) mostly formed the basis of the FDA decision.23 Patients treated with 30 mg/kg/week of eteplirsen had a significant mean 22.9% increase in dystrophin-positive fibers via IHC 24 weeks posttreatment compared to pretreatment values (P≤0.002); such an increase was not observed in the placebo-treated cohort. This rose significantly to a mean 51.7% at 48 weeks posttreatment (P≤0.001). Significant increases in dystrophin-positive fibers were also observed in the 50 mg/kg/week and delayed eteplirsen-treated cohorts. Because the FDA found the IHC method questionable, additional testing was conducted on 11 patient biopsies from all cohorts at 180 weeks posttreatment with an improved protocol.23 Dystrophin-positive fiber counts were observed at 17.4% on average, casting further doubt on results obtained earlier in the trial.
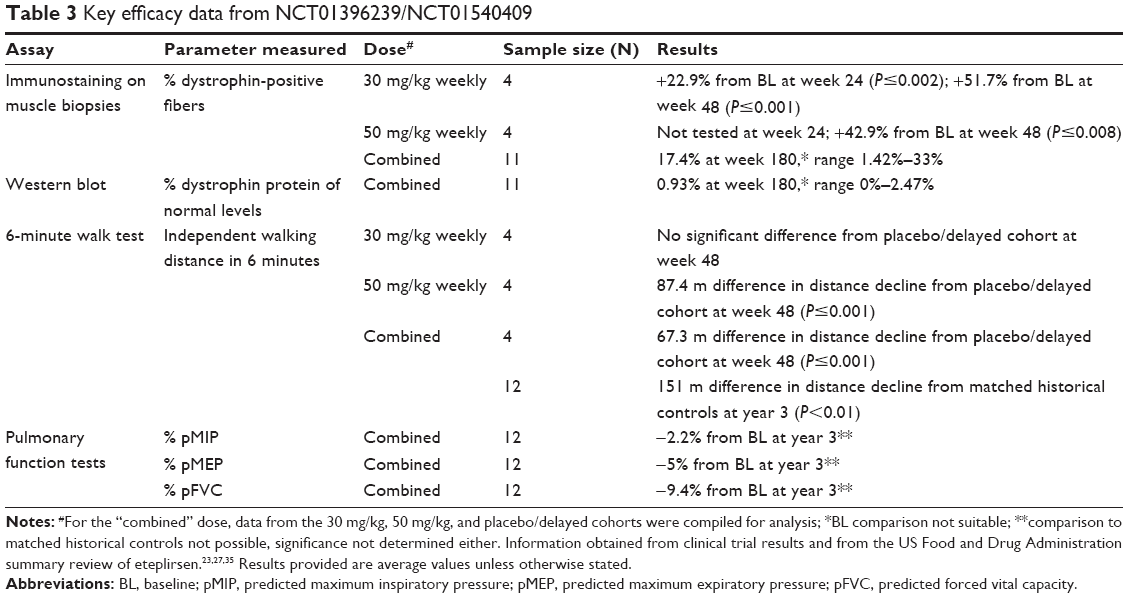 | Table 3 Key efficacy data from NCT01396239/NCT01540409 |
WB-based quantification of dystrophin expression from 11 of 12 patient muscle biopsies after 180 weeks of treatment in NCT01396239/NCT01540409 revealed that eteplirsen-treated patients (combined data from 30 mg/kg/week- and 50 mg/kg/week-treatment cohorts) had 0.93% of dystrophin levels observed in healthy individuals. A similar method was performed for 13 patients in NCT02255552, 48 weeks into the 30 mg/kg/week-treatment regimen, and a statistically significant mean increase in dystrophin levels was observed at 0.22%–0.32% of normal levels (the lower percentage obtained may partly have been because patients were treated for a shorter time). As at least 10% of normal dystrophin amounts are predicted to translate into clinical benefit in patients, there has been much dispute23,31 as to whether the dystrophin levels observed are “reasonably likely to predict clinical benefit”.23
Functional assays conducted in NCT01396239/NCT01540409 include the 6MWT to test ambulation and measurement of forced vital capacity and maximum inspiratory and expiratory pressures to test pulmonary function. Results from the 6MWT showed that 30 mg/kg/week eteplirsen did not appear to provide clinical benefit compared to placebo/delayed-treatment controls 24 and 48 weeks posttreatment. On the other hand, treatment with 50 mg/kg/week of eteplirsen for 48 weeks showed a significant difference in the 6MWT compared to the placebo/delayed-treatment control group (P≤0.001). Comparison of the combined eteplirsen-treated cohort to historical controls after 3 years of treatment showed a significant 151 m mean difference between the groups in the 6MWT (P<0.01).
In this study, eteplirsen treatment was observed at most only to delay disease progression in terms of ambulatory ability as measured by the 6MWT. In fact, 3 years into the study, two of 12 patients lost ambulation. Although it was argued that this was a considerable improvement compared to historical controls (where six of 13 patients lost ambulation in the same period of time), the action of eteplirsen still cannot be deemed sufficient to satisfy the clinical end point of the trial, as also concluded by the FDA.23
On the other hand, eteplirsen was observed to affect pulmonary function positively in patients (Table 3). Compared to natural history data – as pulmonary function was not tested in historical controls – treatment markedly slowed progressive decline in forced vital capacity and maximum inspiratory and expiratory pressure predicted percentages. Again, however, it would seem that eteplirsen had more of a delaying rather than an improving effect on these parameters.
Issues and challenges
Eteplirsen is facing two major issues in proving its efficacy as a DMD therapeutic. One is its lack of apparent efficacy, and the other related issue is clinical trial design, particularly for NCT01396239/NCT01540409. The unsatisfactory performance of eteplirsen in NCT01396239/NCT01540409 may be ascribed in part to its chemistry. A main challenge of using PMOs for treatment is to increase target-tissue uptake, as PMOs exhibit rapid clearance due to their neutral nature.37 Without altering its chemistry, eteplirsen uptake can be improved by increasing either its dose or administration frequency. Key efficacy results in NCT01396239/NCT01540409 were mostly presented with combined data from both the 30 mg/kg/week and 50 mg/kg/week cohorts (Table 3), and so the effect of an increased dose in this case cannot be clearly established; dosing frequency was not studied in any of the clinical trials. As also suggested by the FDA,23 it may prove helpful to see how increasing both parameters can improve efficacy. Additionally, uptake can be improved by administering eteplirsen with hexoses. Research has shown that PMO administration in a formulation containing glucose and fructose is eight times more effective in improving dystrophin production through exon skipping than PMOs administered in saline.41
Another key question is whether the antisense sequence used for eteplirsen was an optimal choice. The efficacy of exon skipping at different target positions of an exon typically varies more than 20-fold.42 Although screening efforts to identify the best target positions for exon 51 skipping were made, they relied highly on nonquantitative reverse-transcription polymerase chain-reaction methods from nonimmortalized, nonclonal primary DMD muscle cells,28 which can produce very high background signals. Currently available DMD myoblast cell models immortalized by introduction of the telomerase catalytic subunit (hTERT) and CDK443 can proliferate and differentiate well enough to produce a large amount of dystrophin after exon skipping that can be quantified by WB.42 Optimization of the sequence using this cell-based model could potentially improve the efficacy of exon skipping.
On a related note, dystrophin produced from in-frame DMD deletions starting/ending at exon 51 was found to be more associated with DMD than BMD patients.44,45 As such, another possible reason for the poor efficacy observed for eteplirsen might be that the truncated dystrophin produced from the skipped transcript was not as functional as initially hoped. However, as the functionality of truncated dystrophin variants resulting from exon skipping has not yet been studied in depth, this assertion remains a possibility at best.
As mentioned, another issue is clinical trial design. NCT01396239/NCT01540409 is mainly dealing with four issues: absence of good controls, sample heterogeneity, inadequate sample size, and the use of a limited selection of outcome measures. While the use of historical controls is somewhat justified in NCT01540409 – with investigators selecting the most matched controls – there remain external factors that can make conclusions on efficacy difficult.23 For instance, not all historical control patients seem to meet the inclusion/exclusion criteria for NCT01396239/NCT01540409 (Table 2). As such, control values used for assessing treatment effect may not be entirely accurate.
Cohort heterogeneity is another complicating factor. The patients who participated in NCT01396239/NCT01540409 had different ages, mutation types, and baseline characteristics (eg, 6MWT baseline results).35 Given the natural history of DMD, age plays a critical role in disease progression5 and could affect how well a patient responds to treatment. Different DMD-mutation genotypes have also been shown to lead to DMD-phenotype variability.46 No details were given on the corticosteroid treatments received by participants; however, it is likely that these varied among patients and may confound efficacy results. The challenge, therefore, would be to design a trial that can handle the natural phenotypic variability associated with DMD,46 constructing as homogeneous a cohort as possible with the available participants. Among other things, this should help set a representative baseline and improve the reliability of efficacy tests.
Another issue is the low sample size used in NCT01396239/NCT01540409. Due to the said variability among patients and the difficulty of obtaining statistically useful results with a limited number of patients, the efficacy of eteplirsen may not have been duly represented in the study. While the following trial, NCT02255552, has addressed this by planning to enroll 160 patients (Table 1), other steps can be taken to increase sample size further. One way would be to consider enrolling nonambulant patients into trials, as the majority of DMD patients are nonambulant.47 This prevents patients from dropping out due to ambulation loss, provides the opportunity for nonambulant patients to participate in trials, and potentially facilitates the creation of more homogeneous cohorts.47,48 This would, however, entail the development and/or use of appropriate outcome measures to assess efficacy.
On the topic of outcome measures, not all those used in NCT01396239/NCT01540409 were sufficient to demonstrate efficacy, specifically with regard to muscle function. While the 6MWT is a standard end point for assessing the clinical utility of DMD therapeutics,49 it is limited, as it applies only to ambulant patients and has been shown to be motivation-dependent.50 It is suggested to explore other methods for assessing muscle function, such as the Performance of Upper Limb scale, which grades the ability of patients to perform 22 different daily tasks using their upper muscles,48 and myometry, which quantitatively assesses muscular strength, eg, hand grip and knee extension, through a variety of tests.51 Not only are these more inclusive to nonambulant patients, they will also determine how treatment affects specific muscle groups.
Finally, there is the issue with the reproducibility and reliability of the methods done for the surrogate end point in NCT01396239/NCT01540409. IHC and WB are standard procedures for quantifying effects on dystrophin expression; however, investigations by the FDA have shown that the methods used for these techniques were questionable.23 Consequently, any correlation between results from these tests and observed effects on muscle function in patients cannot be reliably made. After being subjected to rigorous examination, these methods likely have been improved and validated according to FDA standards for NCT02255552. Nevertheless, it would be helpful to use an additional highly reproducible outcome measure, such as the quantification of muscle–fat conversion through magnetic resonance imaging and spectroscopy,47,52,53 to further strengthen the surrogate end point.
Safety and tolerability of eteplirsen
The chemical nature of PMOs presents certain advantages in terms of safety. This safety is, for the most part, due to the fact that PMOs lack charge29 (Figure 2). It is thought that this makes PMOs largely incapable of interacting with proteins like nucleases, whose affinity to their natural targets (DNA, RNA) is highly dependent on the presence of a negative charge.29,54 As a result, PMOs are not subject to nuclease-mediated degradation and are highly stable in cellular environments. This improved stability adds to their safety as a therapeutic, as unwanted incorporation of individual PMO subunits into the genetic material of a patient is made virtually unlikely.29
Notably, this insusceptibility to protein interaction also renders PMOs unable to sufficiently bind and activate Toll-like receptors, a class of receptors responsible for producing an innate immune response against pathogenic material.25,54 Upon activation, Toll-like receptors initiate signaling cascades that lead in turn to the activation of transcription factors belonging to the nuclear factor kappa B (NFκB), AP1, and IRF families; these families collectively stimulate the production of pro-inflammatory cytokines and type I IFNs that induce inflammation.55 The independence of PMOs from the RNase H degradative pathway also adds to safety, as it promotes specificity of antisense activity.29,54 Because the RNase H system is not used, unwanted degradation of nontarget (possibly important) transcripts is avoided.
Preclinical trials
Preclinical studies on eteplirsen have been done on cynomolgus monkeys and mdx mice. One of these involved the subcutaneous or IV administration of eteplirsen up to the 320 mg/kg maximum dose (clinically, this translates to 100 mg/kg in humans) in cynomolgus monkeys.56 This study found that eteplirsen was well tolerated using either administration route at the highest dose, with no observable adverse effects on cardiovascular, respiratory, neurological, or renal parameters. Genotoxicity assays were also done through the bacterial reverse-mutation assay with Salmonella typhimurium and Escherichia coli tester strains, the chromosome-aberration assay with CHO cells, and the bone marrow-micronucleus test with ICR mice. No toxic effects were found in these assays with eteplirsen compared to corresponding controls up to the maximum tested doses of 5,000 μg/plate, 5,000 μg/mL, or 2,000 mg/kg, respectively. Safety results in this study paved the way for the initiation of NCT00844597, the Phase I/II dose-ranging study with eteplirsen (Table 1).
Subsequent research in cynomolgus monkeys,57 in an effort to support NCT00844597, tested the toxicological effect of repeated IV bolus dosing of eteplirsen at 5–320 mg/kg/week with untreated controls for 12 weeks. As found previously, eteplirsen was well tolerated, with no observable adverse effects. Histological observations from kidney samples revealed instances of basophilic granules/tubules and tubular vacuolation that became more prevalent with dose, but these spontaneously reversed with time during recovery and did not affect measured serum chemistry parameters or renal function. This histological occurrence has been attributed to the renal accumulation of the drug, likely as a result of clearance activity. Studies in mdx mice58 with up to 960 mg/kg/week of IV-administered eteplirsen produced similar results, including the observed histological changes in kidneys.
Clinical trials
Eteplirsen was well tolerated with no adverse effects in NCT00844597, where the maximum dose administered as an IV infusion was 20 mg/kg/week for 12 weeks. There was a reported case of serious cardiac fractional shortening in one patient, but this was attributed to a DMD-related and not a drug-related complication. Safety issues external to the expected DMD phenotype were not found with respect to lung, kidney, liver, or bone marrow function. Inflammatory infiltrates, as revealed by IHC on muscle biopsies with a set of T-cell-specific antibodies, were found at generally decreased frequencies among treated patients. This is consistent with the nonimmunogenic nature of PMOs described earlier.54,55 Anti-dystrophin antibody production was not induced by eteplirsen treatment.
A similar safety profile for eteplirsen was observed in NCT01396239/NCT01540409 at 48 weeks posttreatment with 30 mg/kg/week or 50 mg/kg/week of eteplirsen. At this time posttreatment, the drug was well tolerated, neither hepatic nor renal function were compromised, serum chemistry and properties appeared to be within expectations given the progression of DMD, and no T-cell-based immune response was stimulated. Three years into the trial, eteplirsen was still well tolerated. Note, though, that there were some generally observed adverse effects in patients during the course of the entire study that were deemed related to eteplirsen treatment. These included, among others, vomiting, headaches, balance disorder, and proteinuria. These events were manifest in about half of patients treated with eteplirsen (independently of dose) for 168 weeks. Treatment schedules remained uninterrupted and as planned amid these events.
Patient-focused perspectives
The approval and use of eteplirsen are seen as a welcome hope, widely supported and celebrated by DMD patients and a number of advocacy groups.59,60 However, there is the issue of efficacy that has to be resolved by Sarepta. At present, eteplirsen is far from curative: trial results have shown eteplirsen to have a marginal effect on improving DMD clinical manifestations and all while the drug is administered with the standard of care for DMD, eg, corticosteroid treatment. Sarepta has also announced that eteplirsen will cost ~US$300,000 a year on average, with the price varying depending on patient weight.61 The price is thought to be reasonable for a rare disorder, but whether patients should spend so much for a drug with disputed efficacy remains contested. Eteplirsen is indeed a landmark achievement for the DMD community; however, stronger evidence of efficacy is undoubtedly required for cementing its place as a viable DMD therapeutic.
On the other hand, while much investment has been made in eteplirsen, it is still only applicable to a highly specific subset of DMD patients, ie, the ~14% of patients with mutations amenable to exon 51 skipping.32 Therapeutics aimed at skipping other exons or that use a cocktail of exon-skipping AOs must be developed to cover other patients.62–64 Currently, Sarepta has two other exon-skipping AOs for DMD in Phase III clinical trials. These are SRP-4045 and SRP-4053, PMOs that target exons 45 and 53, respectively (NCT02500381). There is also another PMO-based drug, NS-065/NCNP-01, developed by Nippon Shinyaku and the National Center of Neurology and Psychiatry (Tokyo, Japan), that acts by exon 53 skipping. A Phase II trial (NCT02740972) to test this drug is currently recruiting. Trial results for the aforementioned drugs are not yet available in the literature. In relation to this, it has been shown that besides AOs, small chemical compounds that affect the phosphorylation of SR proteins can also be used to promote DMD exon skipping in DMD patients.65 Also, besides exon skipping, another promising strategy is nonsense suppression, wherein premature stop codons in the DMD gene are bypassed or ignored by translational machinery, leading to successful dystrophin production.5 One drug working under this principle is PTC Therapeutics’ (South Plainfield, NJ, USA) ataluren or Translarna.66 Ataluren has performed satisfactorily in clinical trials to obtain conditional approval from the European Medicines Agency in 2014 (~2 years ahead of eteplirsen, making it the first drug ever approved for DMD) for DMD treatment in the EU.5,67 Like eteplirsen, a confirmatory clinical trial is required for its final approval in the EU.
On a different note, the highly specific nature of exon-skipping therapy presents additional concerns for patients. Under the current regulatory framework, AOs targeting different sequences or with different chemistry are seen as different therapeutics. Each chemistry-unique, sequence-specific AO will thus have to be individually checked for safety and efficacy in the form of lengthy preclinical and clinical trials.68 The same situation applies for therapies aiming to use exon-skipping AOs in combination. All in all, this equates to high costs accrued over long periods of time spent for each therapeutic, for a few DMD cases: an assemblage of factors bound to discourage potential drug manufacturers. There thus exists a strong need to rethink the regulatory procedure for AO-based therapeutics, in the interests of time – recall the rapid progression of DMD – and of providing treatment accessibility to a huge number of DMD patients.68,69
Another related issue is AO patenting by pharmaceutical companies, academic institutes, and others. Numerous patents claiming exclusive use of AOs covering entire DMD-exon sequences have been granted to such entities70–72 and are seen to hamper the development of new DMD therapies using antisense technology. Revisions to patent policy, eg, redefining what constitutes as patentable,73 and the adaptation of less stringent licensing policies by patent holders, eg, by using patent pools or making licenses more affordable,74 could prove vital to streamlining the DMD therapy-development process and speeding up the creation of therapeutics for patients. As this issue of gene-based patenting is not specific to DMD,75,76 such changes would also be beneficial for the development of therapies and molecular diagnostic tools74,75,77 for other genetic disorders.
All that aside, as a therapeutic technique, AO-based exon skipping of at most two exons is only applicable to about 83% of all patients with amenable deletions, duplications, and small mutations.78 Patients with mutations in key DMD protein domain-coding regions are not amenable to treatment by exon skipping. Alternative options to AO-based therapy must thus be explored for the treatment of these patients.
Conclusion
Multidisciplinary management of symptoms is currently the standard of treatment for DMD, with interventions primarily focused on delaying disease progression.5 None of these directly addresses the molecular etiology of DMD. The envisioned place of eteplirsen in therapy would thus be to serve as a curative treatment option for patients, as it acts directly on the DMD gene itself to restore dystrophin production. While accelerated approval of the therapeutic has paved the way for early patient access to eteplirsen, it was faced with heated controversy over the observed efficacy of the drug in clinical trials.23,31 A confirmatory Phase III trial is ongoing and recruiting participants to resolve this issue; results from this trial are also vital for eteplirsen to obtain final approval from the FDA.
Eteplirsen is beneficial for patients with amenable DMD mutations, comprising ~14% of the entire DMD-patient population.32 This leaves a majority of DMD patients without a treatment option that directly addresses the molecular cause of the disorder. In the meantime, while eteplirsen development is under way, it would be most beneficial if research on improving present antisense-based therapeutic strategies were continued (not only for exon 51 skipping but also for the skipping of other exons) or if other avenues for treatment of the disorder were explored. For instance, recent advances in enhancing PMO uptake and efficacy through its conjugation with cell-penetrating peptides (called peptide-conjugated PMOs) are showing promising results in animal models79,80 and have great potential to enter clinical trials soon. Concurrent with these scientific advancements, however, must be a reevaluation of the current regulatory process to accommodate the personal nature of antisense therapies better. Above anything else, collaborative efforts among the scientific, regulatory, and patient communities must be sustained to help keep the DMD-therapy scene moving forward.
Acknowledgments
This work was supported by Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, Canada Foundation for Innovation (CFI), Alberta Advanced Education and Technology, and the Women and Children’s Health Research Institute (WCHRI).
Disclosure
The authors report no conflicts of interest in this work.
References
Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. | ||
Emery AE. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul Disord. 1991;1(1):19–29. | ||
Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–313. | ||
Manzur A, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93(11):986–990. | ||
Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 2016;12(3):1795–1807. | ||
Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219–228. | ||
Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122(4):809–823. | ||
Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66(6):1121–1131. | ||
Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772(2):108–117. | ||
Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90(8):3710–3714. | ||
Nichols B, Takeda S, Yokota T. Nonmechanical roles of dystrophin and associated proteins in exercise, neuromuscular junctions, and brains. Brain Sci. 2015;5(3):275–298. | ||
van Deutekom JC, van Ommen GJ. Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet. 2003;4(10):774–783. | ||
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–517. | ||
Roberts RG, Coffey AJ, Bobrow M, Bentley DR. Exon structure of the human dystrophin gene. Genomics. 1993;16(2):536–538. | ||
Buzin CH, Feng J, Yan J, et al. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005;25(2):177–188. | ||
Takeshima Y, Yagi M, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55(6):379–388. | ||
White S, Kalf M, Liu Q, et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71(2):365–374. | ||
Goto M, Komaki H, Takeshita E, et al. Long-term outcomes of steroid therapy for Duchenne muscular dystrophy in Japan. Brain Dev. 2016;38(9):785–791. | ||
Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16(4):249–255. | ||
Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. 2015;87:104–107. | ||
Guncay A, Yokota T. Antisense oligonucleotide drugs for Duchenne muscular dystrophy: how far have we come and what does the future hold? Future Med Chem. 2015;7(13):1631–1635. | ||
US Food and Drug Administration. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy [press release]. Silver Spring (MD): FDA; 2016 [September 19]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm. Accessed November 18, 2016. | ||
US Food and Drug Administration. Application number 206488Orig1s000: summary review. 2016. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_summaryreview_redacted.pdf. Accessed November 5, 2016. | ||
Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. | ||
Lee JJA, Yokota T. Antisense therapy in neurology. J Pers Med. 2013;3(3):144–176. | ||
Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A. 1993;90(18):8673–8677. | ||
Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–271. | ||
Arechavala-Gomeza V, Graham IR, Popplewell LJ, et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum Gene Ther. 2007;18(9):798–810. | ||
Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7(3):187–195. | ||
Leiden University Medical Center. DMD (dystrophin). 2004. Available from: http://www.dmd.nl/DMD_home.html. Accessed January 22, 2017. | ||
Kesselheim AS, Avorn J. Approving a problematic muscular dystrophy drug: implications for FDA policy. JAMA. 2016;316(22):2357–2358. | ||
Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. | ||
Aoki Y, Nakamura A, Yokota T, et al. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol Ther. 2010;18(11):1995–2005. | ||
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605. | ||
Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–647. | ||
Sarepta Therapeutics. Exondys 51 [prescribing information]. 2016. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/206488lbl.pdf. Accessed November 6, 2016. | ||
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. | ||
Burki U, Keane J, Blain A, et al. Development and application of an ultrasensitive hybridization-based ELISA method for the determination of peptide-conjugated phosphorodiamidate morpholino oligonucleotides. Nucleic Acid Ther. 2015;25(5):275–284. | ||
Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–928. | ||
Sarepta Therapeutics. Confirmatory study of eteplirsen in DMD patients (PROMOVI). Available from: https://clinicaltrials.gov/ct2/show/study/NCT02255552. NLM identifier: NCT02255552. Accessed November 5, 2016. | ||
Han G, Gu B, Cao L, et al. Hexose enhances oligonucleotide delivery and exon skipping in dystrophin-deficient mdx mice. Nat Commun. 2016;7:10981. | ||
Echigoya Y, Mouly V, Garcia L, Yokota T, Duddy W. In silico screening based on predictive algorithms as a design tool for exon skipping oligonucleotides in Duchenne muscular dystrophy. PLoS One. 2015;10(3):e0120058. | ||
Mamchaoui K, Trollet C, Bigot A, et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle. 2011;1:34. | ||
Yokota T, Duddy W, Partridge T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007;26(3):179–184. | ||
Yokota T, Takeda S, Lu QL, Partridge TA, Nakamura A, Hoffman EP. A renaissance for antisense oligonucleotide drugs in neurology: exon skipping breaks new ground. Arch Neurol. 2009;66(1):32–38. | ||
Ricotti V, Muntoni F, Voit T. Challenges of clinical trial design for DMD. Neuromuscul Disord. 2015;25(12):932–935. | ||
Straub V, Balabanov P, Bushby K, et al. Stakeholder cooperation to overcome challenges in orphan medicine development: the example of Duchenne muscular dystrophy. Lancet Neurol. 2016;15(8):882–890. | ||
Mayhew A, Mazzone ES, Eagle M, et al. Development of the performance of the upper limb module for Duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55(11):1038–1045. | ||
Bushby K, Connor E. Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig (Lond). 2011;1(9):1217–1235. | ||
Alfano L, Lowes L, Berry K, et al. Pilot study evaluating motivation on the performance of timed walking in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2014;24(9–10):860. | ||
Parent Project Muscular Dystrophy. Guidance for industry – Duchenne muscular dystrophy: developing drugs for treatment over the spectrum of disease. 2014. Available from: http://www.parentprojectmd.org/site/DocServer/Guidance_Document_Submission_Duchenne_Muscular_Dystrop.pdf?docID=15283. Accessed February 13, 2017. | ||
Ricotti V, Evans MR, Sinclair CD, et al. Upper limb evaluation in Duchenne muscular dystrophy: fat-water quantification by MRI, muscle force and function define endpoints for clinical trials. PLoS One. 2016;11(9):e0162542. | ||
Forbes SC, Willcocks RJ, Triplett WT, et al. Magnetic resonance imaging and spectroscopy assessment of lower extremity skeletal muscles in boys with Duchenne muscular dystrophy: a multicenter cross sectional study. PLoS One. 2014;9(9):e106435. | ||
Moulton JD. Guide for morpholino users: toward therapeutics. J Drug Discov Dev Deliv. 2016;3(2):1023. | ||
Murphy K. Janeway’s Immunobiology. 8th ed. New York: Garland Science; 2012. | ||
Sazani P, Weller DL, Shrewsbury SB. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int J Toxicol. 2010;29(2):143–156. | ||
Sazani P, Van Ness KP, Weller DL, Poage DW, Palyada K, Shrewsbury SB. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of Duchenne muscular dystrophy. Int J Toxicol. 2011;30(3):313–321. | ||
Sazani P, Van Ness KP, Weller DL, Poage D, Nelson K, Shrewsbury AS. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int J Toxicol. 2011;30(3):322–333. | ||
Parent Project Muscular Dystrophy. PPMD applauds FDA for landmark approval of first-ever disease-modifying drug to treat Duchenne muscular dystrophy. 2016. Available from: http://www.prnewswire.com/news-releases/ppmd-applauds-fda-for-landmark-approval-of-first-ever-disease-modifying-drug-to-treat-duchenne-muscular-dystrophy-300330263.html?tc=eml_cleartime. Accessed November 18, 2016. | ||
Global Genes. Duchenne community rejoices over FDA approval of eteplirsen! 2016. Available from: https://globalgenes.org/raredaily/duchenne-community-rejoices-over-fda-approval-of-eteplirsen. Accessed November 18, 2016. | ||
Black A, Radke J. Sarepta’s Duchenne drug to cost $300k annually. 2016. Available from: http://www.raredr.com/news/duchenne-drug-to-cost-300k. Accessed January 29, 2017. | ||
Echigoya Y, Yokota T. Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic Acid Ther. 2014;24(1):57–68. | ||
Aoki Y, Yokota T, Wood MJ. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed Res Int. 2013;2013:402369. | ||
Yokota T, Duddy W, Echigoya Y, Kolski H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: too many mutations, too few patients? Expert Opin Biol Ther. 2012;12(9):1141–1152. | ||
Nishida A, Kataoka N, Takeshima Y, et al. Chemical treatment enhances skipping of a mutated exon in the dystrophin gene. Nat Commun. 2011;2:308. | ||
Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487. | ||
PTC Therapeutics. PTC Therapeutics receives conditional approval in the European Union for Translarna for the treatment of nonsense mutation Duchenne muscular dystrophy. 2014. Available from: http://ir.ptcbio.com/ReleaseDetail.cfm?ReleaseID=863914. Accessed November 18, 2016. | ||
Aartsma-Rus A, Ferlini A, Goemans N, et al. Translational and regulatory challenges for exon skipping therapies. Hum Gene Ther. 2014;25(10):885–892. | ||
Douglas AG, Wood MJ. Splicing therapy for neuromuscular disease. Mol Cell Neurosci. 2013;56:169–185. | ||
Sazani P, Kole R, inventors; Sarepta Therapeutics, assignee. Multiple exon skipping compositions for DMD. United States patent US9234198B1. 2016 Jan 12. | ||
Wilton SD, Fletcher S, McClorey G, inventors; University of Western Australia, assignee. Antisense oligonucleotides for inducing exon skipping and methods of use thereof. United States patent US9035040B2. 2015 May 19. | ||
Platenburg G, De Kimpe J, van Deutekom JC, van Ommen GJ, Aartsma-Rus A, inventors; Biomarin Technologies, assignee. Methods for efficient exon (44) skipping in duchenne muscular dystrophy and associated means. United States patent US9139828B2. 2015 Sep 22. | ||
Cook-Deegan R. Law and science collide over human gene patents. Science. 2012;338(6108):745–747. | ||
Andrews LB. Genes and patent policy: rethinking intellectual property rights. Nat Rev Genet. 2002;3(10):803–808. | ||
Caulfield T, Cook-Deegan RM, Kieff FS, Walsh JP. Evidence and anecdotes: an analysis of human gene patenting controversies. Nat Biotechnol. 2006;24(9):1091–1094. | ||
Cook-Deegan R, Heaney C. Patents in genomics and human genetics. Annu Rev Genomics Hum Genet. 2010;11:383–425. | ||
Huys I, Matthijs G, Van Overwalle G. The fate and future of patents on human genes and genetic diagnostic methods. Nat Rev Genet. 2012;13(6):441–448. | ||
Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–299. | ||
Yin H, Moulton HM, Seow Y, et al. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17(24):3909–3918. | ||
Yin H, Moulton HM, Betts C, et al. Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Mol Ther. 2010;18(10):1822–1829. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.