")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Etanercept Protected Against Cigarette Smoke Extract-Induced Inflammation and Apoptosis of Human Pulmonary Artery Endothelial Cells via Regulating TNFR1
Authors Xue H, Xie B, Xu N, Li H, Chen Q, Xie W, Wang H
Received 3 December 2020
Accepted for publication 21 March 2021
Published 11 May 2021 Volume 2021:16 Pages 1329—1345
DOI https://doi.org/10.2147/COPD.S295580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Hong Xue,1,2 Baosong Xie,1,2 Nengluan Xu,1,2 Hongru Li,1,2 Qianshun Chen,2,3 Weiping Xie,4 Hong Wang4
1Department of Respiratory and Critical Care Medicine, Fujian Provincial Hospital, Fuzhou, Fujian, People’s Republic of China; 2Provincial School of Clinical Medicine, Fujian Medical University, Fuzhou, Fujian, People’s Republic of China; 3Department of Thoracic Surgery, Fujian Provincial Hospital, Fuzhou, Fujian, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital with Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
Correspondence: Baosong Xie; Hong Xue
Department of Respiratory and Critical Care Medicine, Fujian Provincial Hospital, No. 134, East Street, Fuzhou, Fujian, 350000, People’s Republic of China
Tel +86 591-88217531
Email [email protected]; [email protected]
Purpose: Etanercept (ETN), a tumor necrosis factor-α (TNF-α) inhibitor, has been applied in the treatment of many diseases. However, whether it has effects on chronic obstructive pulmonary disease (COPD) and its interaction with tumor necrosis factor receptor 1 (TNFR1) remained unknown.
Methods: Histopathological analysis of lung tissues from non-smokers and smokers with or without COPD was conducted using hematoxylin–eosin (H&E) staining, Van Gieson (VG) staining, and terminal transferase-mediated biotin dUTP nick end labeling (TUNEL). TNF-α content was measured using Immunohistochemistry. Correlation analysis among apoptosis rate, smoke index, the FEV1/FVC ratio, and TNF-α-positive cells was performed. After ETN treatment and transfection of overexpressed or silenced TNFR1, levels of inflammatory cytokines, apoptosis and related genes expressions in cigarette smoke extract (CSE)-treated human pulmonary artery endothelial cells (HPAECs) were detected using enzyme-linked immunosorbent assay (ELISA), Hoechst 33342 staining, flow cytometry, quantitative real-time PCR (qRT-PCR) and Western blot.
Results: Pulmonary arterial remodeling and increased apoptotic and TNF-α+ HPAECs were found in lung tissue of smokers with or without COPD, with higher degrees in smokers with COPD. The numbers of apoptotic and TNF-α+ HPAECs were positively correlated with smoke index, while the FEV1/FVC ratio was negatively correlated with apoptotic HPAECs. In HPAECs, ETN downregulated the expressions of proteins related to CSE-induced apoptosis and the TNF receptor family, decreased CSE-induced cell apoptosis and inflammatory cytokine levels, and inhibited TNFR1 expression and p65 phosphorylation. Overexpressed TNFR1 reversed the effects of ETN on CSE-treated HPAECs, whereas silencing TNFR1 did the opposite.
Conclusion: ETN protected HPAECs against CSE-induced inflammation and apoptosis via downregulating TNFR1, thus providing a potential therapy for smoking-induced COPD.
Keywords: chronic obstructive pulmonary disease, etanercept, apoptosis, cigarette smoke extract, human pulmonary artery endothelial cells, tumor necrosis factor receptor 1
Introduction
Chronic obstructive pulmonary disease (COPD) refers to airflow obstruction, which is not fully reversible and usually progressive, and it is a prevalent disease that causes significant morbidity and mortality.1 As estimated by the World Health Organization (WHO), COPD will become the third most common cause of death and disability around the world by the year of 2030.2 The onset of COPD has been found to be induced by various factors, among which cigarette smoke (CS) has been recognized as the most common one.3 Despite that great effort has been exerted to search for therapeutic medication for COPD, at present, there is no available efficient medicine for COPD intervention.
One characteristic of COPD was lung inflammation, which persists after smoking cessation, and in COPD patients with an elevated level of tumor necrosis factor-α (TNF-α), one of the major inflammatory cytokines, active smoking was found to be related with a higher degree of systemic inflammation.4,5 Also, it was addressed that an increased systemic TNF-α level was implicated in cachexia and skeletal muscle weakness in COPD patients.6 Therefore, it could be reasonably assumed that blocking TNF-α might have ameliorative effects on patients with COPD.
Etanercept (ETN) has been found to be an anti-TNF-α agent, which elicited its pharmacological effects via binding to and neutralizing the endogenous TNF-α molecule.7 Prior studies suggested that ETN could reduce disease activity and limit joint damage progression in rheumatoid arthritis (RA), and demonstrated a favorable profile for immunogenicity, drug survival and infections at the same time.8 Also, ETN treatment was found to exert an evident ameliorating effect on leg and back pain.9 Additionally, based on a rat model, ETN could attenuate cigarette smoke extract (CSE) exposure-induced pulmonary arterial remodeling and activities of matrix metallopeptidase-2 (MMP-2) and MMP-9 via suppressing the activation of the TNF-α/nuclear factor-κB (NF-κB) pathway.10 However, whether ETN had the same effect in human pulmonary artery endothelial cells (HPAECs) and its interaction with tumor necrosis factor receptor 1 (TNFR1) remained to be further explored. In our study, we analyzed the pathological changes in the lungs of non-smokers and smokers with or without COPD and constructed a cigarette smoke extract (CSE)-induced COPD model in vitro in order to uncover the protective effect of ETN against CSE-induced inflammation and apoptosis in HPAECs and its interaction with TNFR1, hoping to find out a potential prevention method for smoking-induced COPD.
Methods
Ethics Statement
The current study was carefully reviewed and approved by the Ethics Committee of Fujian Provincial Hospital (approval number: NK20190816-06). All enrolled patients have signed and provided written informed consent and agreed that their tissues would be used for clinical research. The study was conducted in accordance with the Declaration of Helsinki.
Clinical Samples
In our present study, clinical lung tissue samples were collected from 32 male patients who underwent lobotomy because of lung carcinoma in Fujian Provincial Hospital. Only male patients were selected for treatment in this study due to the fairly small proportion of females among smokers and COPD patients, the unavailability of lung tissues of female COPD patients, and given that gender is not considered as a confounding factor for COPD diagnosis and treatment. Based on the current ethical considerations, it is generally impossible to obtain lung tissue from patients without tumors for research, and therefore, only tumor patients were selected for the current study. In order to minimize errors, we selected lung tissue far away from the tumor to reduce the impact of the tumor on our research indicators, Patients included in this study comprised non-smokers (Non-smokers, n=8) who neither smoked nor had been exposed to second-hand smoke with normal lung ventilation function), and smokers (n=24). Among the smokers, those with normal lung ventilation function were assigned to smokers without COPD group (smokers without COPD, n=13), and the others, who were diagnosed with stable COPD according to the Global initiative for COPD (GOLD-2017) guidelines (patients with an FEV1/FVC (forced expiratory volume in the first second/forced vital capacity) ratio of less than 70% were diagnosed with COPD),11 were assigned to Smokers with COPD group (Smokers with COPD, n=11). None of the patients diagnosed with COPD experienced acute exacerbation (defined as worsened respiratory symptoms that require medical treatment) within 4 weeks before our study. All enrolled patients received no intravenous injection or oral administration of medication including antibiotics, glucocorticoids, and theophylline within 4 weeks prior to our study, except lobotomy and bronchodilator used for pulmonary function test. None of these patients were atopic or had a history of allergic diseases such as asthma or rhinitis. Surgical patients involved basically had no underlying diseases or serious cardiopulmonary diseases. The clinical characteristics of patients enrolled in our present study are provided in Table 1. Smoke index = number of cigarettes smoked per day × number of years of smoking.
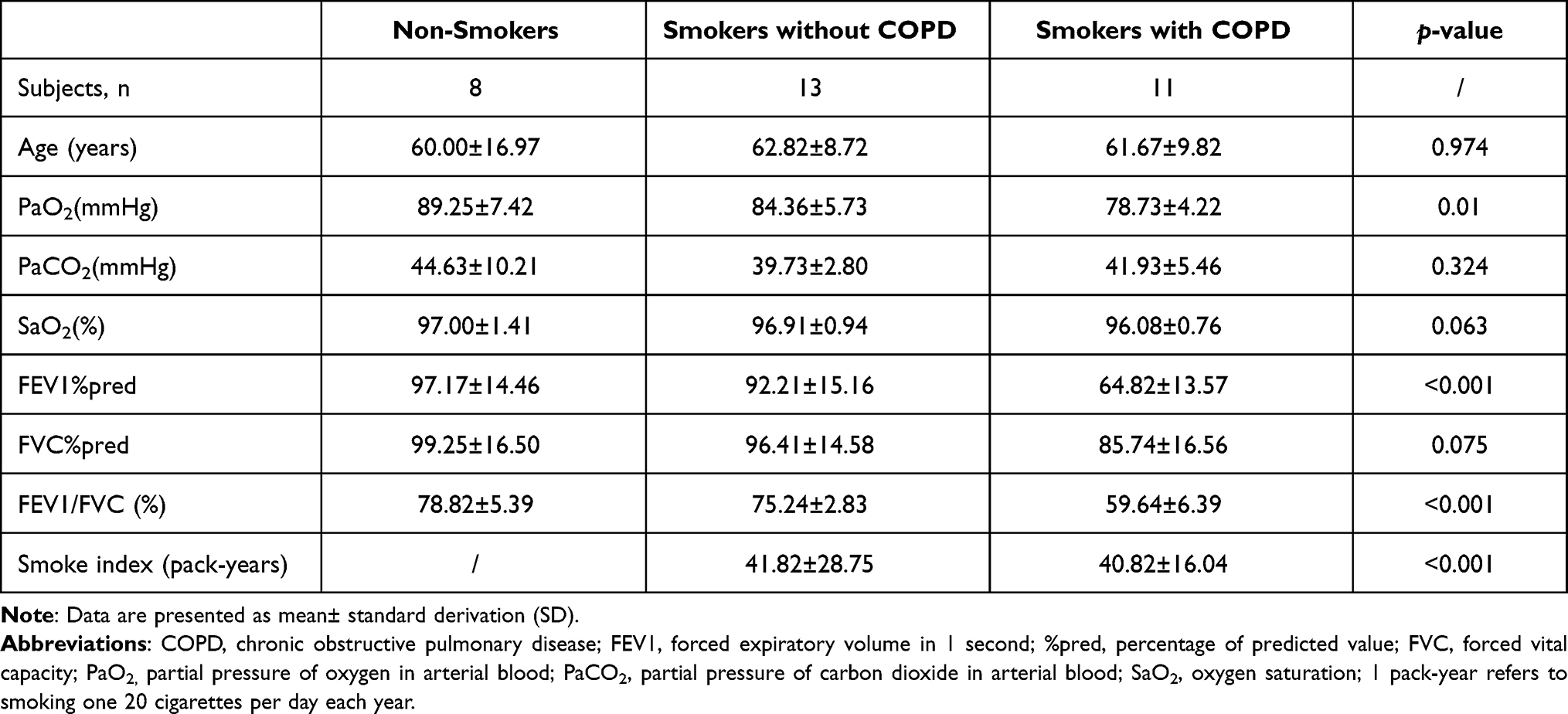 |
Table 1 Demographic and Lung Function Data |
For histological examination, all clinical samples were obtained after surgical operation, and then stored at −80 °C after being washed with phosphate-buffered saline (PBS).
Hematoxylin–Eosin (H&E) Staining
Lung tissue samples from non-smokers and smokers with or without COPD were firstly collected, and then immersed in 4% paraformaldehyde solution. After 48 hours, samples were embedded in dissolved paraffin wax and cut into 5-μm-thick sections, and subsequently stained with hematoxylin–eosin (H&E) staining reagent (G1120, Solarbio, Beijing, China) at room temperature for 10 minutes. Images of tissue samples were captured using an Eclipse 50i light microscope (Nikon, Tokyo, Japan) at 100 × magnification.
At least 10 muscular arteries per section were examined in our study in a blinded fashion with a computerized Image Processing and Analysis system Quantimet 500 (Leica, Cambridge, UK). The percentage of medial wall thickness (MWT%) of fully muscularized arteries with an external diameter ranging from 50 to 100 μm was evaluated via calculating MWT% along the shortest curvature using the following formula: MWT% = (medical wall thickness × 2/external diameter) × 100 in line with prior publication.10
Van Gieson (VG) Staining
VG staining was performed as previously described.12 Lung tissue samples were firstly rinsed with normal saline, and then soaked in 4% paraformaldehyde solution for 48 hours, followed by dehydration, transparentization, and paraffin embedment. Afterwards, each sample was cut into 4-μm-thick sections and stained using a Verhoeff Van Gieson staining kit (25089–1, Polysciences, Inc., Warrington, PA). Images of all tissue samples were captured using an Eclipse 50i light microscope (Nikon, Tokyo, Japan).
Terminal Transferase-Mediated Biotin dUTP Nick End Labeling (TUNEL) Assay
The experiment was conducted using an in situ Cell Death Detection kit (11684817910, Roche, Basel, Switzerland) in accordance with the previous description.13 Samples were deparaffinized and treated with 20 μg/mL DNase-free proteinase K (AM2548, Thermo Fisher Scientific, Waltham, MA) at room temperature for 20 minutes, and then washed with PBS three times for 5 minutes. After being fixed at room temperature for 15 minutes, samples were washed with PBS for three times for another 5 minutes. Next, each sample was added with 50 μL of labeling liquid, which was prepared by 2 μL of terminal deoxynucleotidyl transferase (TdT) enzyme and biotin and 48 μL of dUTP, and incubated for an hour at 37 °C. Afterwards, the color was developed using a 3,3ʹ-diaminobenzidine (DAB) kit (DA1015; Solarbio, China), and the samples were subsequently washed with PBS. Then, the samples were sealed after transparentization with xylene for 5 minutes. Finally, cells were observed under a stereomicroscope (SZX10; Olympus, Tokyo, Japan), and the apoptosis rate was calculated.
Immunohistochemical Analysis
The immunohistochemical analysis was conducted on the parenchyma of lung tissue collected during surgery. Samples collected were first fixed in 4% paraformaldehyde overnight, then sliced into 5 μm thick sections, and subsequently embedded in paraffin for staining. The paraffin sections were then stained using anti-human (TNF-α) antibody (MABF351; 1:100, Sigma-Aldrich, St Louis, MO) at 4 °C overnight, and then stained using horseradish peroxidase (HRP)-conjugated goat anti-human IgG H&L secondary antibody (goat, ab6858, 1:1000, Abcam, Cambridge, UK) and a diaminobenzidine visualization (DAB) kit (P0202, Beyotime, Shanghai, China) at room temperature for 15 minutes. Then, the nuclei were counterstained using hematoxylin. Finally, positive cells from five randomly chosen fields were counted and observed under a stereomicroscope (SZX10; Olympus, Japan).
Cell Culture and Treatment
HPAECs (catalog no. PCS-100-022) used for our study were obtained from American Type Culture Collection (ATCC; Manassas, VA) and cultured in endothelial cell medium (ECM, 210–500, Sigma-Aldrich) with 5% fetal bovine serum (FBS; F2442, Sigma-Aldrich), 1% endothelial cell growth supplement (211-GS, Sigma-Aldrich) and 1% penicillin/streptomycin (P4333, Sigma-Aldrich) at 37°C with 5% CO2. Cells without any treatment was applied as control group.
CSE was prepared in our study as previously described.14 Research cigarettes were obtained from the Chongqing tobacco industry Co. (Chongqing, China). Components of tobacco included tar (11 mg/cigarette), carbon monoxide (17 mg/cigarette) and nicotine (1.1 mg/cigarette). In detail, cigarettes without a filter were installed on pumping apparatus and completely combusted in 2 minutes, and then mainstream smoke was bubbled through a glass vessel that contained 10 mL of non-serum ECM. PH value of the resulting liquid mixture was adjusted to 7.4 and filtered through a 0.22-μm Millipore filter (GSWP04700, Millipore, Billerica, MA) for removal of large particles and bacteria. Concentration of the resulting solution was designated as 100% and CSE solutions employed in our study were diluted to 5% and used within 30 minutes after preparation.
In our present study, ETN (catalog no. Y0001969) was obtained from Sigma-Aldrich. HPAECs were pretreated with different concentrations of ETN (1, 5 and 10 mg/L) for 30 minutes, and then were exposed to CSE for 12 hours. Cells were subsequently collected for further studies.
Hoechst 33342 Staining
Hoechst 33342 staining was performed as previously described.15 In detail, 1 ×105 HPAECs were seeded on a 12-well plate at 37 °C with 5% CO2. Twenty-four hours after culture, HPAECs were treated with 10 mg/L ETN for 30 min and exposed to CSE for 12 hours. After washing twice with PBS, the cells were stained using a Hoechst 33342 staining kit (C0030; Solarbio, China) at room temperature for 10 minutes. Finally, HPAECs were collected, washed with PBS and observed under a fluorescence microscope (BX51; Olympus, Japan).
Transfection
Short hairpin RNA for TNFR1 (sh-TNFR1, catalog no. SHRNA) and its negative control (sh-NC, catalog no. SHC016) were synthesized and obtained from Sigma-Aldrich. The TNFR1 overexpression plasmid was successfully established using pcDNA3.1 plasmid (V79020; Thermo Fisher Scientific), and pcDNA3.1 plasmid was applied as negative control (NC). Then, for transfection, HPAECs were seeded in a 96-well plate at 2 ×104 cells/well, and when growing to reach 90% confluence, the cells were transfected with 50 nmol sh-TNFR1 and its negative control as well as the TNFR1 overexpression plasmid using Lipofectamine 2000 transfection reagent (11668–019, Invitrogen, Carlsbad, CA) at 37 °C for 48 hours. Sequences for transfection are listed in Table 2.
 |
Table 2 Sequence for Transfection |
Enzyme-Linked Immunosorbent Assay (ELISA)
Levels of inflammatory cytokines TNF-α (catalog no. BMS223-4), Interleukin-6 (IL-6, catalog no. EH2IL6) and Interleukin-8 (IL-8, catalog no. KAC1301) in HPAEC supernatant were quantified with their corresponding ELISA kits (Invitrogen) following the manufacturers’ manuals. OD values were measured using a Crocodile 4-in-one assay miniWorkstation (11050011, Titertek-Berthold, Pforzheim, Germany).
Flow Cytometry
For apoptosis detection, 1×105 HPAECs were treated with Annexin V (5 μL) and propidium iodide (PI) (5 μL) together for 15 minutes at room temperature in the dark. After cells were collected and washed with cold PBS twice, cell apoptosis was detected using an Annexin V-FITC/PI cell apoptosis kit (C1062L, Beyotime, China). Cell apoptosis was detected with Guava easyCyte Benchtop Flow Cytometer (BR168323; Luminex, Austin, TX) and data were analyzed using Kaluza C Analysis Software (version 2.1, Beckman Coulter, Indianapolis, IN).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA from HPAECs was extracted with Trizol (15596–026, Invitrogen) and preserved at −80 °C. Concentration of total RNA was quantified with a NanoDrop Lite spectrophotometer (ND-LITE, Thermo Fisher Scientific). One μg of total RNA was used to synthesize cDNA with a RevertAid H Minus First-strand cDNA Synthesis Kit (K1632, Thermo Fisher Scientific). QRT-PCR was performed with an Easy-A One-Tube RT-PCR kit (600182, Agilent Technologies, Santa Clara, CA) in real-time PCR Detection system (LineGene 9600 Plus; Biosan; Riga, Latvia) under the conditions as follows: at 95 °C for 5 minutes, followed by 35 cycles of at 95 °C for 1 minute, at 73 °C for 1 minute and at 72 °C for 5 minutes. Primer sequences are listed in Table 3. β-Actin was used as the internal control. Relative gene expressions were later quantified using the 2−ΔΔCT method.16
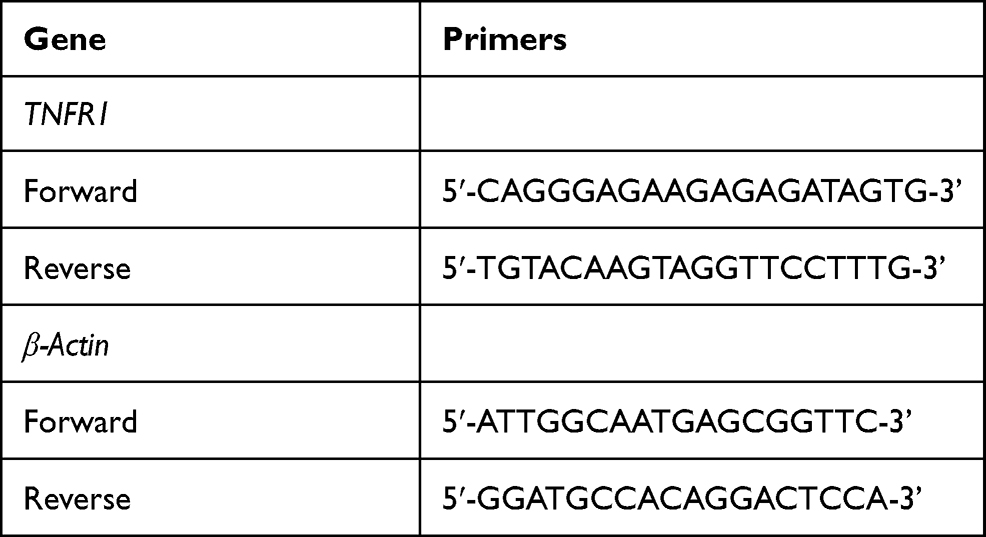 |
Table 3 Primers for qRT-PCR |
Western Blot
In our study, Western blot was used to quantify protein expressions in accordance with previous description.17 After HPAECs were collected, protein was firstly lysed and extracted using RIPA Lysis Buffer (R0278, Sigma-Aldrich), and its concentration was then measured with a bicinchoninic acid (BCA) protein kit (BCA1, Sigma-Aldrich). Sample lysates of protein (30 μg) were first electrophoresed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; P0012A; Beyotime, China), and then transferred into Immun-Blot® polyvinylidene fluoride (PVDF) membrane (P2938, Sigma-Aldrich). After being blocked with fat-free milk (5%) for 2 hours, the membrane was incubated with primary antibodies: anti-cleaved caspase-3 antibody (rabbit, 1:1000, ab2302, Abcam, UK), anti-caspase-3 antibody (rabbit, 1:5000, ab32351, Abcam, UK), anti-caspase-8 antibody (rabbit, 1:1000, ab25901, Abcam, UK), anti-cleaved caspase-8 antibody (mouse, 1:1000, #9748, Cell Signialing Technology (CST), Danvers, MA), anti-cleaved caspase-9 antibody (rabbit, 1:1000, #20750, CST), anti-caspase-9 antibody (rabbit, 1:1000, #9502, CST), anti-Bax (Bcl-2-associated X protein) antibody (rabbit, 1:1000, #5023, CST), anti-Bcl-2 (B-cell lymphoma 2) antibody (mouse, 1:1000, #15071, CST), anti-Fas antibody (mouse, 1:1000, #8023, CST), anti-FasL (Fas ligand) antibody (rabbit, 1:1000, #68405, CST), anti-TNFR1 antibody (rabbit, 1:1000, #3736, CST), anti-p-p65 (phosphorylated-p65) antibody (rabbit, 1:1000, ab86299, Abcam, UK), anti-p65 antibody (rabbit, 1:100, ab16502, Abcam, UK), and anti-β-actin antibody (mouse, 1:2000, ab8226, Abcam, UK) at 4°C overnight, with β-actin as the internal control. After that, the membrane was incubated with secondary HRP-conjugated antibodies: goat anti-rabbit IgG H&L (HRP) (1:5000, A00098, GenScript, Nanjing, China) and goat anti-mouse IgG H&L (HRP) (1:5000, A00160, GenScript, China) at room temperature for an hour and washed with tris-buffer saline tween (TBST) three times. Protein band was collected and analyzed using an enhanced chemiluminescence (ECL) kit (P0013S, Beyotime, China). Gray values of the strips were gathered and calculated using ImageJ 5.0 (National Institutes of Health, Bethesda, MD) in the iBright CL750 Imaging System (A44116, Thermo Fisher Scientific).
Statistical Analysis
All experiments were performed independently in triplicate. Data were expressed as mean ± standard error (SE). Statistical analysis was performed with SPSS 20.0 software (SPSS, Chicago, IL). Statistical significance was assessed by one-way ANOVA followed by Tukey’s post hoc test, and correlation analysis was performed with Pearson’s correlation test. Statistical significance was defined at P-value<0.05.
Results
Pulmonary Arterial Remodeling
The microstructures of pulmonary muscular arteries from non-smokers and smokers with or without COPD were also observed. As depicted in Figure 1A–C, the results from H&E and VG staining showed that smokers with or without COPD displayed thicker intimae and larger lumens than non-smokers (Figure 1A–C). These results suggested that pulmonary arterial remodeling occurred in patients with or without COPD, with little higher severity in patients with COPD.
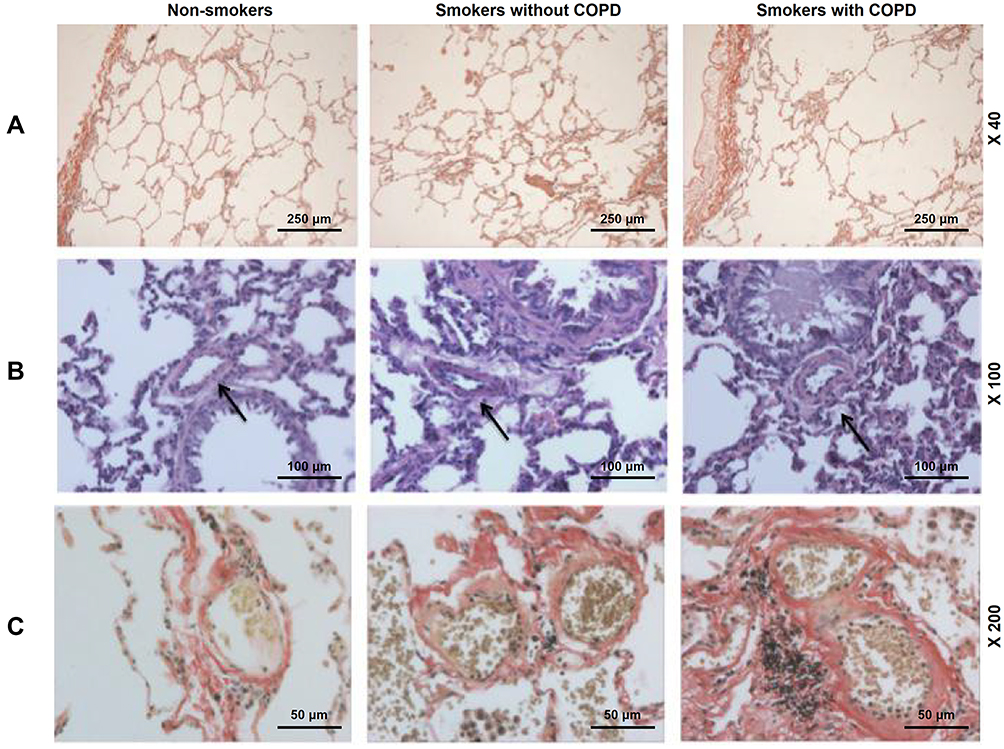 |
Figure 1 Pulmonary arterial remodeling was evidenced in smokers, with a higher severity in smokers with COPD. (A) Histology of lung tissues from non-smokers and smokers with or without COPD were observed via Van Gieson (VG) staining, at 40× magnification. (B) Histology of lung tissues from non-smokers and smokers with or without COPD were observed via Hematoxylin–eosin (H&E) staining, at 100× magnification. (C) Histology of lung tissues of the alveolus from non-smokers and smokers with or without COPD were observed via VG staining, at 200× magnification. Arrows pointed to thicker intimae and larger lumens. Abbreviation: COPD, chronic obstructive pulmonary disease. |
Apoptotic and TNF-α+ HPAECs
In Figure 2A and D, results from TUNEL staining indicated that the apoptosis rate of lung tissues were evidently lower in non-smokers than in smokers with or without COPD (Figure 2A and D, P<0.05). However, there was no significant difference in Smokers with or without COPD, yet a higher apoptosis rate was found in Smokers with COPD.
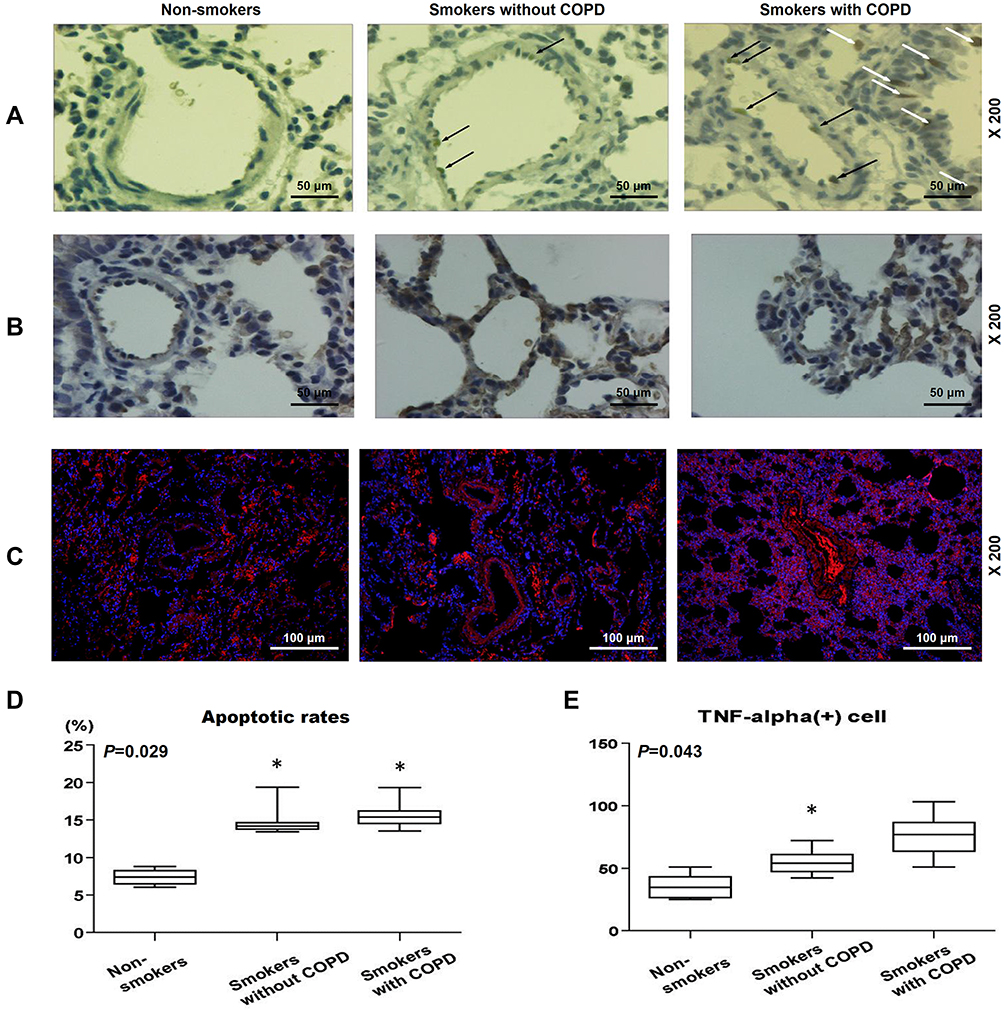 |
Figure 2 Pulmonary vascular endothelial apoptosis and higher TNF expression were observed in smokers, with a higher severity in smokers with COPD. (A) Representative TUNEL staining images of lung tissues from non-smokers and smokers with or without COPD at 200× magnification. (B) Immunohistochemistry analysis of TNF-α in lung tissue from non-smokers and smokers with or without COPD. (C and E) Immunofluorescence analysis of TNF-α from non-smokers and smokers with or without COPD. (D) Apoptosis rate of lung tissues from non-smokers and smokers with or without COPD was quantified. Black and white arrows were all positive cells. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs Non-smokers. Abbreviations: TUNEL, terminal transferase-mediated biotin dUTP nick end labeling; TNF-α, tumor necrosis factor-α; HPAECs, human pulmonary artery endothelial cells. |
In Figure 2B, C and E, it was found that TNF-α+ cells were increased in patients with or without COPD as compared to non-smokers (Figure 2B, C and E, P<0.05). However, in smokers with or without COPD, no significant difference was found in the number of TNF-α+ cells.
Apoptotic and TNF-α+ HPAECs Correlated with Smoke Index
In Figure 3A–C and Table 4, correlation analysis suggested that the number of apoptotic HPAECs was positively correlated with smoke index (Figure 3A, r=0.700, P<0.01). Meantime, we also found a positive correlation between the number of TNF-α+ HPAECs and smoke index (Figure 3C, r=0.417, P=0.027). Furthermore, the FEV1/FVC ratio and the number of apoptotic HPAECs were found to be negatively correlated (Figure 3B, r=−0.547, P=0.026). The blood vessel wall medial thickness (WMT%) of alveoli was correlated with smoke index in smokers without COPD, but was not correlated with smoke index in smokers with COPD (Table 4).
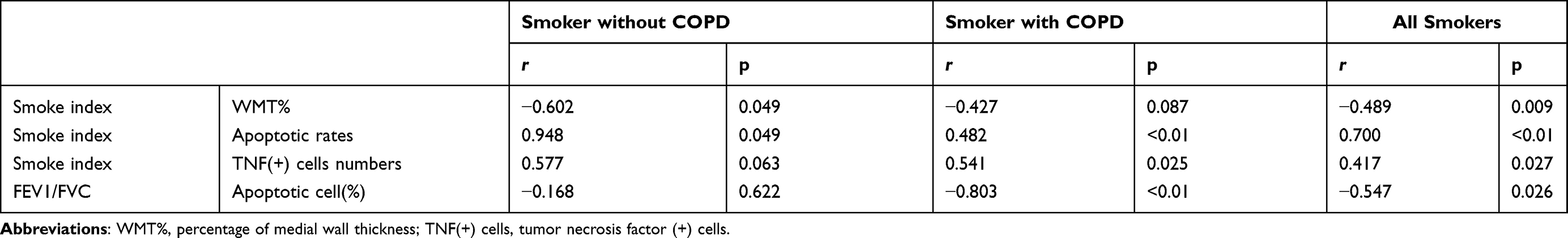 |
Table 4 Correlation Analysis Between the Percentage of Medial Wall Thickness, Lung Function, Apoptosis Index, TNF(+) Cell and Smoke Index |
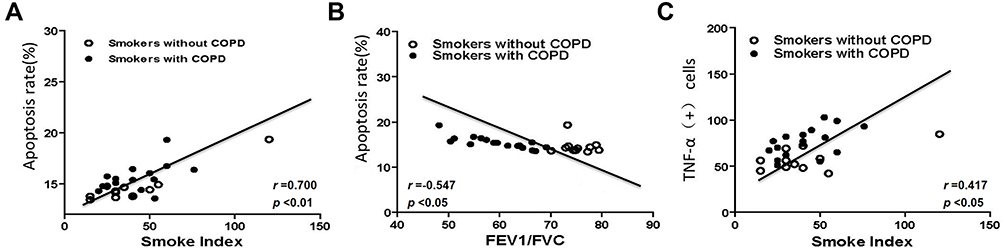 |
Figure 3 Correlation analysis of lung function, apoptosis rates, TNF-α+ cells and smoke index. (A) A positive correlation between smoke index and apoptosis rates in smokers with or without COPD. (B) A negative correlation between apoptosis rates and FEV1/FVC in smokers with or without COPD. (C) A positive correlation between smoke index and TNF-α+ cells in smokers with or without COPD. Abbreviations: FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity. |
Etanercept Alleviated CSE-Induced HPAEC Apoptosis
With the aim to discover the effects of ETN on CSE-induced HPAEC apoptosis, we firstly treated HPAECs with different concentrations of ETN (1, 5, 10 mg/L) and then exposed HPAECs to CSE. HPAEC apoptosis was then detected by Hoechst 33342 staining and flow cytometry. As shown in Figure 4A–D, the number of apoptotic HPAECs, induced by CSE, was reduced after ETN treatment (Figure 4A–D, P<0.05), which suggested that ETN could alleviate the effects of CSE on HPAEC apoptosis.
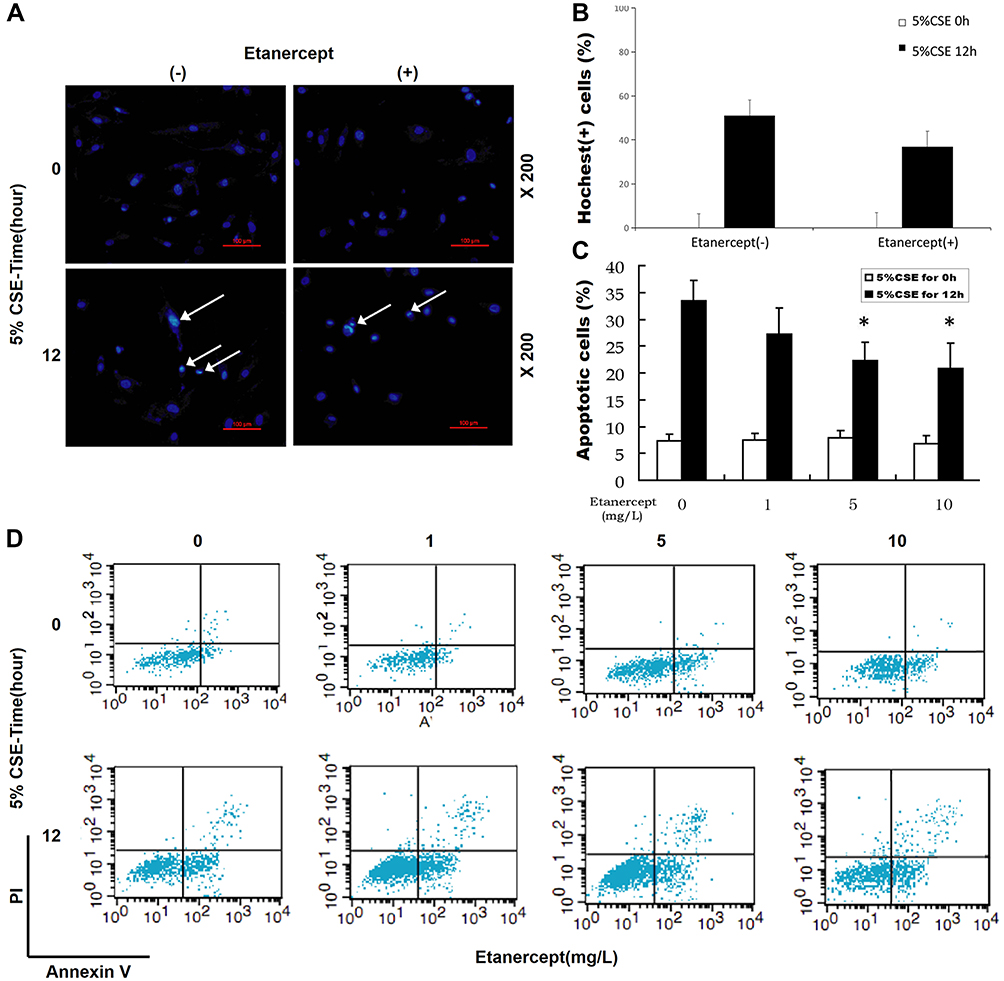 |
Figure 4 Etanercept reversed the effects of CSE on promoting HPAEC apoptosis. (Aand B) Hoechst 33342 staining was used for detecting HPAEC apoptosis after CSE exposure and ETN treatment. White arrows were apoptotic cells. A set of plots of a representative experiment was presented in panel. (C and D) HPAEC apoptosis was detected by flow cytometry after CSE exposure and etanercept treatment. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs etanercept 0 mg/L, with 5% CSE for 12 h. |
Etanercept Regulated Apoptotic-Proteins and TNF Receptor Family in CSE-HPAECs
Protein expressions of cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, Bax, Bcl-2, Fas and FasL in HPAECs were determined following exposure to CSE and treatment with ETN. Figures 5 and 6 demonstrated that after exposure to CSE, the protein expressions of Cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, Bax, and Fas were increased in HPAECs, yet that of Bcl-2 was decreased as compared to the control group, accompanied by an increase in the cleaved caspase-3/procaspase-3, cleaved caspase-8/procaspase-8, and cleaved caspase-9/procaspase-9 ratios (Figures 5 and 6, P<0.05). However, ETN treatment led to an opposite effect of CSE exposure on these proteins (Figures 5 and 6, P<0.05). Meanwhile, no significant difference in FasL protein expression was found before and after CSE exposure and ETN treatment.
 |
Figure 5 Etanercept attenuated the apoptosis of CSE-induced HPAECs via regulating levels of cell apoptosis-related caspases. (A) Protein expressions of cleaved caspase-3, cleaved caspase-8, and cleaved caspase-9 after CSE exposure and ETN treatment were measured by Western blot. β-Actin was used as the internal control. (B–D) Ratios of cleaved-caspase-3/procaspase-3 (B), cleaved-caspase-8/procaspase-8 (C), and cleaved-caspase-9/procaspase-9 (D) were determined. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs control; #P<0.05, vs CSE. Abbreviations: ETN, etanercept; CSE, cigarette smoke extract. |
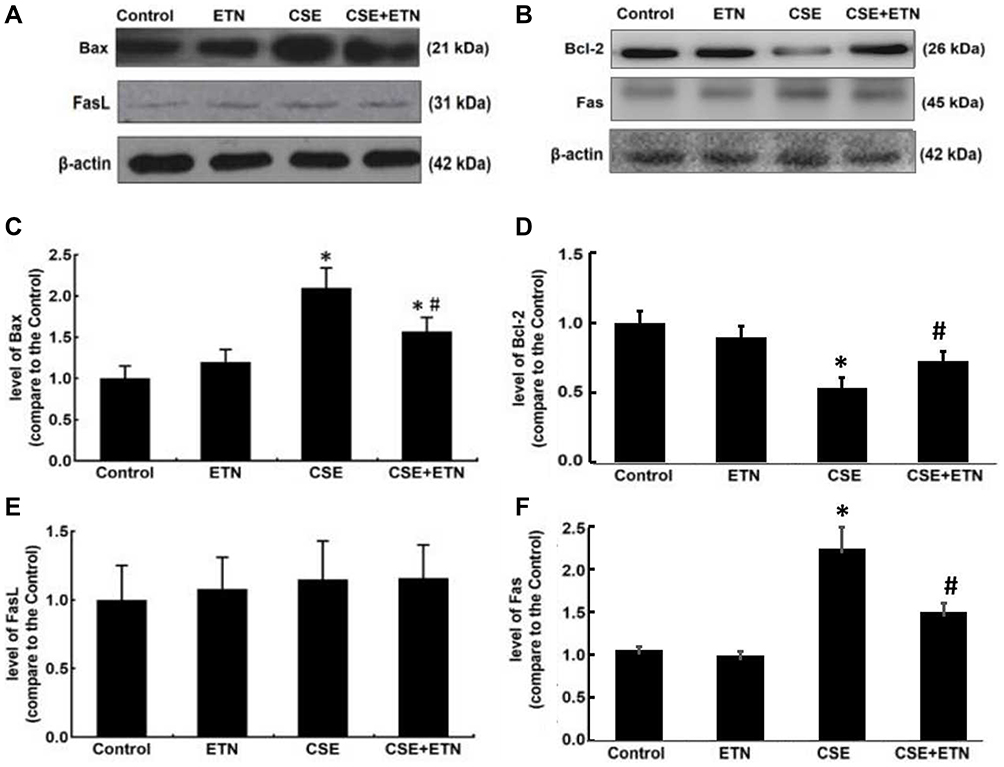 |
Figure 6 Etanercept attenuated the apoptosis of CSE-induced HPAECs via regulating levels of cell apoptosis-related proteins. (A–F) Protein levels of Bax (C), Bcl-2 (D), FasL (E) and Fas (F) after CSE exposure and ETN treatment were measured by Western blot. β-Actin was used as the internal control. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs control; #P<0.05, vs CSE. Abbreviations: Bax, Bcl-2 associated X protein; Bcl-2, B-cell lymphoma-2; FasL, Fas ligand. |
Etanercept Suppressed Inflammatory Cytokines in CSE-HPAECs
Levels of inflammatory cytokines (TNF-α; Interleukin-6, IL-6; IL-8) were measured after CSE exposure and ETN treatment. In Figure 7A–C, we found an increase in these levels after CSE exposure (Figure 7A–C, P<0.05), whereas ETN pretreatment caused no effect on these levels, as compared to the control group. Also, ETN treatment downregulated these levels in CSE-exposed HPAECs (Figure 7A–C, P<0.05), which suggested that ETN could downregulate inflammatory cytokine levels in CSE-treated HPAECs.
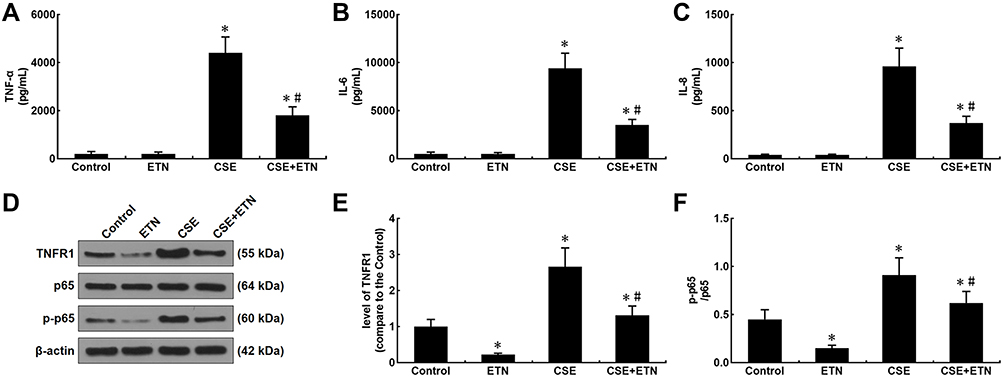 |
Figure 7 Etanercept reversed the effects of CSE on levels of inflammatory cytokines, TNFR1, p-p65/ p65 in HPAECs. (A and C) Levels of inflammatory cytokines TNF-α (A), IL-6 (B) and IL-8 (C) in HPAECs after CSE exposure and ETN treatment were quantified using enzyme-linked immunosorbent assay (ELISA). (D–F) Protein/β-actin expressions of TNFR1, p65 and p-p65 in HPAECs after CSE exposure and ETN treatment were determined using Western blot. β-Actin was used as the internal control. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs control; #P<0.05, vs CSE. Abbreviations: TNFR1, tumor necrosis factor receptor 1; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-8, interleukin-8; p-p65, phosphorylated-p65. |
Etanercept Ameliorated TNFR1 and p65 in CSE-HPAECs
Our data suggested that etanercept had the anti-apoptosis and anti-inflammatory properties in CSE-treated HPAECs, and therefore, we further investigated its possible molecular mechanisms. As shown in Figure 7D–F, TNFR1 expression and the p-p65/p65 ratio were increased after CSE exposure as compared to the control group, whereas ETN pre-treatment resulted in an opposite effect (Figure 7D–F, P<0.05). Meanwhile, in CSE-exposed HPAECs, ETN treatment could downregulate TNFR1 and the p-p65/p65 ratio (Figure 7D–F, P<0.05), showing that ETN could ameliorate the effects of CSE on TNFR1 expression and p65 phosphorylation in HPAECs.
TNFR1 and Etanercept on the Apoptosis of CSE-Treated HPAECs
To further confirm the role of TNFR1 implicated in the protective effects of ETN, TNFR1 expression in CSE-treated HPAECs was measured after transfection of shTNFR1 and the TNFR1 overexpression plasmid and ETN treatment. As shown in Figure 8A, in CSE-treated HPAECs, TNFR1 was downregulated after shTNFR1 transfection as compared to the CSE+sh-NC group (Figure 8A, P<0.05). Also, we found a decrease in TNFR1 expression in CSE-treated HPAECs after ETN treatment as compared to the CSE group (Figure 8A, P<0.05). However, TNFR1 overexpression was found to reverse the effects of ETN treatment on TNFR1 expression in CSE-treated HPAECs (Figure 8A, P<0.05). In Figure 8B and C, we found silencing TNFR1 caused a decreased apoptosis rate of CSE-treated HPAECs as compared to the CSE+sh-NC group, and ETN treatment also down-regulated the apoptosis rate of CSE-treated HPAECs as compared to the CSE group (Figure 8B and C, P<0.05). However, overexpressed TNFR1 reversed the down-regulatory effect of ETN treatment on the apoptosis of CSE-treated HPAECs (Figure 8B and C, P<0.05).
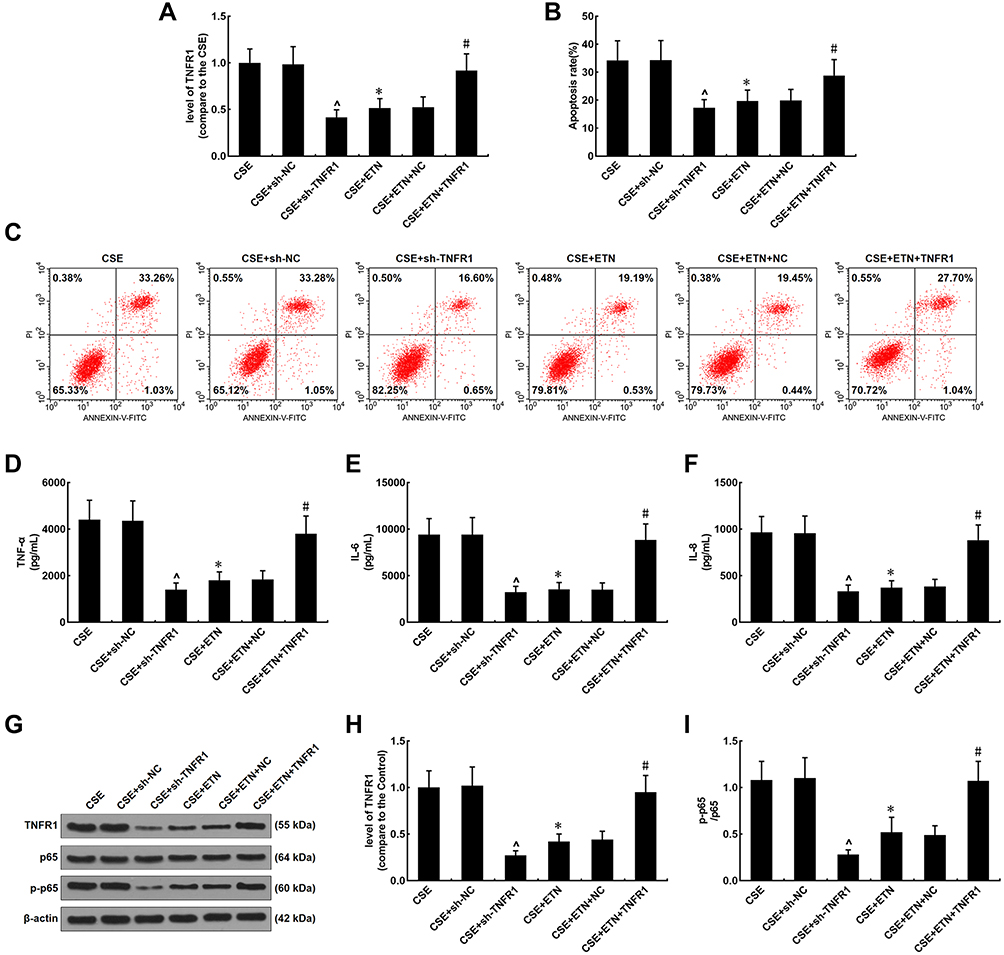 |
Figure 8 Etanercept alleviated the effects of CSE on TNFR1 expression, suppressed apoptosis and inflammatory cytokine levels in HPAECs, which was reversed by overexpressed TNFR1. (A) Expressions of TNFR1 after CSE exposure, transfection of shTNFR1 and the TNFR1 overexpression plasmid, and ETN treatment were measured using quantitative-real-time polymerase chain reaction (qRT-PCR). β-Actin was used as the internal control. (B and C) Apoptosis rates of HPAECs after CSE exposure, transfection of shTNFR1 and the TNFR1 overexpression plasmid, and ETN treatment were determined by flow cytometry. (D–F) Levels of inflammatory cytokines TNF-α (D), IL-6 (E) and IL-8 (F) in HPAECs after CSE exposure, transfection of shTNFR1 and the TNFR1 overexpression plasmid, and ETN treatment were quantified using ELISA. (G and H) Protein levels of TNFR1, p65 and p-p65 in HPAECs after CSE exposure, transfection of shTNFR1 and the TNFR1 overexpression plasmid, and ETN treatment were determined using Western blot. β-Actin was used as the internal control. (I) Ratios of p-p65/p65 were measured. All experiments were performed in triplicate and experimental data were expressed as mean ± standard errors (SE). *P<0.05, vs CSE; ^P<0.05, vs CSE+sh-NC; #P<0.05, vs CSE+ETN+NC. Abbreviations: sh-TNFR1, short hairpin RNA for TNFR1; sh-NC, short hairpin RNA for negative control. |
TNFR1 and Etanercept on Inflammatory Cytokines in CSE-Treated HPAECs
As depicted in Figure 8D–F, the levels of inflammatory cytokines TNF-α, IL-6 and IL-8 were evidently decreased after silencing TNFR1 as compared to the CSE+sh-NC group (Figure 8D–F, P<0.05), and they were also downregulated after ETN treatment when compared to the CSE group (Figure 8D–F, P<0.05). On the contrary, overexpressed TNFR1 reversed the effects of ETN on the levels of these inflammatory cytokines in CSE-treated HPAECs (Figure 8D–F, P<0.05).
TNFR1 and Etanercept on TNFR1 and NF-κB Pathway in CSE-Treated HPAECs
As shown in Figure 8G and H, after silencing TNFR1, both the protein expressions of TNFR1 and p-p65 were reduced as compared to the CSE+sh-NC group (Figure 8G and H), and compared with the CSE group, ETN treatment also led to downregulation of the protein expressions of TNFR1 and p-p65 (Figure 8G and H, P<0.05), while overexpressed TNFR1 was found to reverse the effects of ETN on the protein expressions of TNFR1 and p-p65 in CSE-treated HPAECs (Figure 8G and H, P<0.05).
Then, the p-p65/p65 ratio was determined as well. According to Figure 8I, silencing TNFR1 lowered the p-p65/p65 ratio as compared to the CSE+sh-NC group (Figure 8I, P<0.05), and ETN treatment also led to a reduced p-p65/p65 ratio when compared to the CSE group (Figure 8I, P<0.05). However, the down-regulated p-p65/p65 ratio induced by ETN treatment in CSE-treated cells was reversed by TNFR1 overexpression (Figure 8I, P<0.05).
Discussion
Etanercept consisted of an extracellular ligand-binding portion of the human TNFR linked to the Fc portion of human immunoglobulin G1 (IgG1), and could competitively suppress the binding of TNF to TNFR.18 Prior study suggested that etanercept could alleviate the short-term effects of CSE-induced pulmonary arterial remodeling in a rat model and proposed its interactions with the suppression on the TNF-α/NF-κB signaling pathway and MMP-2 and MMP-9.10 However, whether ETN had the same effects in HPAECs and its interaction with TNFR1 were largely unknown, which were the main focuses in our study.
One of the current diagnostic criteria for COPD is irreversible airflow limitation, and pulmonary vascular injury is not included in the clinical diagnostic criteria for COPD. However, pulmonary vascular injury/remodeling is the pathological basis of pulmonary hypertension and pulmonary heart disease in COPD. Therefore, improving pulmonary vascular damage is of great significance to improving the prognosis of COPD patients. In our study, there was no significant difference in pulmonary vascular inflammation between smoking COPD and smoking non-COPD, indicating that smoking is the cause of early pulmonary vascular damage, not airflow limitation. Hence, the exposure factor for the later experiment is smoke extract. Because the alveoli of the lungs and the vascular network of the alveoli have been destroyed in patients with COPD, being the pathological characteristics of COPD, changes in the structure of the alveoli lead to structural changes in the blood vessels. So there is no significant correlation between smoking index and blood vessel wall thickening in smokers with COPD, but significant correlation in smokers without COPD.
In the airway and lung, CSE upregulated TNF level, which was associated with COPD pathogenesis.19 TNFR1 is one of the receptors that mediate TNF signaling, and prior studies suggested that in human bronchial epithelial (HBE) cells, TNFR1 knockdown could suppress the activation of CSE-induced NF-κB and the high levels of inflammatory cytokines.20 Meanwhile, previous study uncovered that etanercept could attenuate pulmonary apoptosis and lung injury during ischemic acute kidney injury via inhibiting TNFR1.21 In our study, we found that the ameliorative effects of etanercept on inflammation and apoptosis in HPAECs were reversed by overexpressed TNFR1, indicating that etanercept could alleviate CSE-induced inflammation and apoptosis in HPAECs via downregulating TNFR1.
COPD was characterized by chronic bronchitis and/or emphysema which led to lung function impairment.22 And pulmonary arterial remodeling, an alteration of the structures of both small and big airways, has been demonstrated in patients with severe COPD.23,24 It was also demonstrated that CSE exposure could directly led to endothelium dysfunction, which was associated with vascular remodeling and vasoconstriction in smokers.25 In our present study, pulmonary arterial remodeling was evidenced in smokers, with a higher severity in smokers with COPD, which suggested that CSE exposure might be a factor for the pulmonary vasculature alteration and COPD.
In a previous study, CSE-induced airway and lung inflammation was recognized as a predominant feature of COPD.26,27 Several cytokines have also been recognized to play pivotal roles in various pathological processes in COPD.27 For example, TNF-α, which was overexpressed in COPD, had effects on innate host response to microbes and could recruit inflammatory cells to infection sites.28 IL-6 was promptly and transiently produced in response to infections, and contributed to host defense by stimulating acute phase responses, hematopoiesis, and immune reactions.29 IL-6 was also found overexpressed in COPD.30 IL-8 had a property to drive target cells to inflammatory sites in the inflammatory process, and it was found to exhibit a higher level in individuals with COPD than in smokers without airflow obstruction.31 Prior study suggested that etanercept could suppress the levels of inflammatory cytokines (TNF-α, IL-6, and interferon-γ, IFN-γ) in lethally influenza-infected mice.32 In our current study, we found that the proportion of TNF-α+ cells was increased in lung tissues in smokers, with a higher proportion in smokers with COPD, and it had a positive correlation with smoke index; meantime, CSE exposure caused an increase in the levels of inflammatory cytokines TNF-α, IL-6 and IL-8 in HPAECs, whereas etanercept led to downregulation of these levels, which indicated that CSE exposure-induced inflammation was also associated with the aggravation of lung pathology in smokers with COPD, and etanercept could alleviate COPD by suppressing CSE-induced inflammatory cytokines. Limited to the current research conditions, only a small sample research has been carried out at present, and it is hoped that further research with larger samples can be conducted in the future. As anti-TNF therapy have not been succeeded in COPD patients (infliximab, golimumab and etanercept), more experiments needed to be done to make it to be clinical application.
After stimulation of inflammation, NF-κB was activated by some post-translational modifications, and then translocated into the nucleus to induce pro-inflammatory gene transcription.33 It was uncovered in prior study that the sequestration of NF-κB was regulated by p65, a kind of heterodimer,10 and etanercept could attenuate the activation of CSE-induced NF-κB pathway through evidently reducing p65 translocation.10 In our present study, CSE exposure was found to increase p-p65 expression in HAPECs, while etanercept led to decreased p-p65 expression, which uncovered that etanercept could reverse the effects of CSE exposure on the activation of the NF-κB pathway.
Cell death process dysregulation was also involved in CSE-induced COPD, and in patients with COPD, increased apoptotic alveolar, bronchiolar and endothelial cells in lung tissues were observed.34 Several proteins were also found associated with apoptosis, such as caspases. Caspase-3 was a protein implicated in apoptosis and its suppression led to inhibited apoptosis.35 Caspase-8 played a role in receptor-mediated extrinsic pathway, while caspase-9 was involved in the mitochondrial intrinsic pathway, and both caspase-8 and caspase-9 were implicated in apoptosis following caspase-3 activation.36 Meanwhile, some proteins from the Bcl-2 family were also involved in apoptosis, such as Bcl-2 and Bax. Bax prevented the suppressive function of Bcl-2 via forming a heterodimer with Bcl-2, whereas Bcl-2 had an inhibitory effect on apoptosis.37,38 In addition, apoptosis mediated by Fas-FasL has also been recognized as an important mechanism for immune homeostasis maintenance.39 As pointed out in a previous study, in rats with streptozotocin (STZ)-induced diabetes, Fas and caspase-8 expressions were upregulated and the apoptosis rate of ganglion cells was enhanced, whereas etanercept decreased both the expressions and the apoptosis rate.40 In our present study, we found an increased apoptosis rate of HPAECs following CSE exposure, accompanied by increases in the protein expressions of cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, Bax and Fas and a decrease in Bcl-2 expression, whereas no evident change was observed in FasL expression. We additionally found that apoptosis rate was positively correlated with smoke index yet negatively correlated with the FEV1/FVC ratio. However, we found that etanercept led to an alleviating effect on the apoptosis of CSE-induced HPAECs, and reversed the effects of CSE on the expressions of apoptosis-related proteins, which unveiled that etanercept could ameliorate the apoptosis of CSE-induced HPAECs. As COPD is more of muco-obstructive disease and Bik is the known proapoptotic protein via BCL-pathway in airway epithelium because Bik-deficient mice fail to resolve allergen-induced mucous expression,41,42 Bik might function on etanercept and CSE treated HPAECs.
Conclusion
Taken together, our study was the first to put forward that CSE exposure could led to pulmonary arterial remodeling in smokers with COPD, and inflammation and apoptosis in HPAECs. We also found that etanercept could alleviate the effects of CSE on promoting inflammation and apoptosis in HPAECs, which could be possibly achieved by downregulating TNFR1. We hoped that our results could help bring further insights into smoking-induced COPD pathology and the interaction between etanercept and TNFR1 in smoking-induced COPD, which may be conducive to finding a possible therapeutic method for smoking-induced COPD prevention and treatment.
Funding
This work was supported by the Health Talent Training Project for Middle-young Age of Fujian Province under Grant [number 2019-ZQN-5]; Initial Funding of Fujian Medical University under Grant [number 2018QH1122]; and Natural Foundation of Fujian Science and Technology Ministry under Grant [number 2017J01175]; and Medical Innovation Project of Fujian Health and Family Planning Commission under Grant [number 2017-CX-5]; and Natural Science Foundation of Fujian Province under Grant [number 2019J01178].
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Yousuf A, Brightling CE. Biologic drugs: a new target therapy in COPD? COPD. 2018;15(2):99–107. doi:10.1080/15412555.2018.1437897
2. Hillas G, Perlikos F, Tsiligianni I, Tzanakis N. Managing comorbidities in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:95–109. doi:10.2147/COPD.S54473
3. Zong D, Liu X, Li J, Ouyang R, Chen P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenetics Chromatin. 2019;12(1):65. doi:10.1186/s13072-019-0311-8
4. King PT. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin Transl Med. 2015;4(1):68. doi:10.1186/s40169-015-0068-z
5. Tanni SE, Pelegrino NR, Angeleli AY, Correa C, Godoy I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. J Inflamm. 2010;7:29. doi:10.1186/1476-9255-7-29
6. Liu XJ, Bao HR, Zeng XL, Wei JM. Effects of resveratrol and genistein on nuclear factor-kappa B, tumor necrosis factor-alpha and matrix metalloproteinase9 in patients with chronic obstructive pulmonary disease. Mol Med Rep. 2016;13(5):4266–4272. doi:10.3892/mmr.2016.5057
7. Deng Y, Hu L, Qiang W, Cheng Z, Wang L, Wang X. TNF-alpha level affects etanercept clearance: TNF-alpha concentration as a new correction factor of allometric scaling to predict individual etanercept clearances in patients with ankylosing spondylitis. Clin Exp Pharmacol Physiol. 2018;45(7):643–651. doi:10.1111/1440-1681.12924
8. Zhao S, Mysler E, Moots RJ. Etanercept for the treatment of rheumatoid arthritis. Immunotherapy. 2018;10(6):433–445. doi:10.2217/imt-2017-0155
9. Jing S, Yang C, Zhang X, Wen S, Li Y. Efficacy and safety of etanercept in the treatment of sciatica: a systematic review and meta-analysis. J Clin Neurosci. 2017;44:69–74. doi:10.1016/j.jocn.2017.06.054
10. Xue H, Sun K, Xie W, et al. Etanercept attenuates short-term cigarette-smoke-exposure-induced pulmonary arterial remodelling in rats by suppressing the activation of TNF-a/NF-kB signal and the activities of MMP-2 and MMP-9. Pulm Pharmacol Ther. 2012;25(3):208–215. doi:10.1016/j.pupt.2012.02.006
11. Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Global initiative for chronic obstructive lung disease: the changes made. Cureus. 2019;11(6):e4985.
12. Gao L, Li SD, Zhang Y, Liu Y, Yang MH. Early intervention of tongxinluo () on right ventricular function assessed by echocardiography in rats with pulmonary arterial hypertension induced by monocrotaline. Chin J Integr Med. 2020;26(12):913–920. doi:10.1007/s11655-020-3229-x
13. Ding HB, Liu KX, Huang JF, Wu DW, Chen JY, Chen QS. Protective effect of exogenous hydrogen sulfide on pulmonary artery endothelial cells by suppressing endoplasmic reticulum stress in a rat model of chronic obstructive pulmonary disease. Biomed Pharmacother. 2018;105:734–741. doi:10.1016/j.biopha.2018.05.131
14. Chen H, Liao K, Cui-Zhao L, et al. Cigarette smoke extract induces apoptosis of rat alveolar type II cells via the PLTP/TGF-beta1/Smad2 pathway. Int Immunopharmacol. 2015;28(1):707–714. doi:10.1016/j.intimp.2015.07.029
15. Chen C, Han Z, Yang M, Jiang Z, Ou X. Induction of apoptosis by Xiakemycin A in human hepatoma HepG2 cells. Medicine. 2020;99(17):e19848. doi:10.1097/MD.0000000000019848
16. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
17. Andrade P, Hoogland G, Del Rosario JS, Steinbusch HW, Visser-Vandewalle V, Daemen MA. Tumor necrosis factor-alpha inhibitors alleviation of experimentally induced neuropathic pain is associated with modulation of TNF receptor expression. J Neurosci Res. 2014;92(11):1490–1498. doi:10.1002/jnr.23432
18. Langley RG, Kasichayanula S, Trivedi M, et al. Pharmacokinetics, immunogenicity, and efficacy of etanercept in pediatric patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2018;58(3):340–346. doi:10.1002/jcph.1029
19. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
20. Xu H, Sun Q, Lu L, et al. MicroRNA-218 acts by repressing TNFR1-mediated activation of NF-kappaB, which is involved in MUC5AC hyper-production and inflammation in smoking-induced bronchiolitis of COPD. Toxicol Lett. 2017;280:171–180. doi:10.1016/j.toxlet.2017.08.079
21. White LE, Santora RJ, Cui Y, Moore FA, Hassoun HT. TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury. Am J Physiol Lung Cell Mol Physiol. 2012;303(5):L449–L459. doi:10.1152/ajplung.00301.2011
22. Jones B, Donovan C, Liu G, et al. Animal models of COPD: what do they tell us? Respirology. 2017;22(1):21–32. doi:10.1111/resp.12908
23. Jindal SK. Remodeling in asthma and COPD-recent concepts. Lung India. 2016;33(1):1–2. doi:10.4103/0970-2113.173074
24. Wrobel JP, McLean CA, Thompson BR, et al. Pulmonary arterial remodeling in chronic obstructive pulmonary disease is lobe dependent. Pulm Circ. 2013;3(3):665–674. doi:10.1086/674339
25. Lu Q, Gottlieb E, Rounds S. Effects of cigarette smoke on pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2018;314(5):L743–L756. doi:10.1152/ajplung.00373.2017
26. Hikichi M, Mizumura K, Maruoka S, Gon Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis. 2019;11(Suppl 17):S2129–S2140. doi:10.21037/jtd.2019.10.43
27. Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412. doi:10.2147/COPD.S42544
28. Zheng J, Shi Y, Xiong L, et al. The expression of IL-6, TNF-alpha, and MCP-1 in respiratory viral infection in acute exacerbations of chronic obstructive pulmonary disease. J Immunol Res. 2017;2017:8539294. doi:10.1155/2017/8539294
29. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10):a016295. doi:10.1101/cshperspect.a016295
30. Wei J, Xiong XF, Lin YH, Zheng BX, Cheng DY. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PeerJ. 2015;3:e1199. doi:10.7717/peerj.1199
31. Huang AX, Lu LW, Liu WJ, Huang M. Plasma inflammatory cytokine IL-4, IL-8, IL-10, and TNF-alpha levels correlate with pulmonary function in patients with asthma-chronic obstructive pulmonary disease (COPD) overlap syndrome. Med Sci Monit. 2016;22:2800–2808. doi:10.12659/MSM.896458
32. Shi X, Zhou W, Huang H, et al. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit Care. 2013;17(6):R301. doi:10.1186/cc13171
33. Luo X, Zhang H, Wei X, et al. Aloin suppresses lipopolysaccharide-induced inflammatory response and apoptosis by inhibiting the activation of NF-kappaB. Molecules. 2018;23(3):517. doi:10.3390/molecules23030517
34. Sun X, Feng X, Zheng D, et al. Ergosterol attenuates cigarette smoke extract-induced COPD by modulating inflammation, oxidative stress and apoptosis in vitro and in vivo. Clin Sci. 2019;133(13):1523–1536. doi:10.1042/CS20190331
35. Xiong Y, Jin E, Yin Q, Che C, He S. Boron attenuates heat stress-induced apoptosis by inhibiting endoplasmic reticulum stress in mouse granulosa cells. Biol Trace Elem Res. 2021;199(2):611–621. doi:10.1007/s12011-020-02180-1
36. Gogebakan B, Bayraktar R, Ulasli M, Oztuzcu S, Tasdemir D, Bayram H. The role of bronchial epithelial cell apoptosis in the pathogenesis of COPD. Mol Biol Rep. 2014;41(8):5321–5327. doi:10.1007/s11033-014-3403-3
37. Tirapelli D, Lustosa IL, Menezes SB, et al. High expression of XIAP and Bcl-2 may inhibit programmed cell death in glioblastomas. Arq Neuropsiquiatr. 2017;75(12):875–880. doi:10.1590/0004-282x20170156
38. Warren CFA, Wong-Brown MW, Bowden NA. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019;10(3):177. doi:10.1038/s41419-019-1407-6
39. Soysa NS, Alles N. Positive and negative regulators of osteoclast apoptosis. Bone Rep. 2019;11:100225. doi:10.1016/j.bonr.2019.100225
40. Ye Q, Lin YN, Xie MS, et al. Effects of etanercept on the apoptosis of ganglion cells and expression of Fas, TNF-alpha, caspase-8 in the retina of diabetic rats. Int J Ophthalmol. 2019;12(7):1083–1088. doi:10.18240/ijo.2019.07.05
41. Hussain SS, George S, Singh S, et al. A Small Molecule BH3-mimetic Suppresses Cigarette Smoke-Induced Mucous Expression in Airway Epithelial Cells. Sci Rep. 2018;8(1):13796. doi:10.1038/s41598-018-32114-w
42. Comer DM, Kidney JC, Ennis M, et al. Airway epithelial cell apoptosis and inflammation in COPD, smokers and nonsmokers. Eur Respir J. 2013;41(5):1058–1067. doi:10.1183/09031936.00063112
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.