Back to Journals » Journal of Inflammation Research » Volume 14
Estrogen Protects Articular Cartilage by Downregulating ASIC1a in Rheumatoid Arthritis
Authors Hang X, Zhang Z, Niu R, Wang C, Yao J, Xu Y, Tao J, Li L, Chen F
Received 4 December 2020
Accepted for publication 25 February 2021
Published 12 March 2021 Volume 2021:14 Pages 843—858
DOI https://doi.org/10.2147/JIR.S295222
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Xiaoyu Hang,1,2 Zhenyu Zhang,1,2 Ruowen Niu,1,2 Chen Wang,1,2 Jing Yao,1,2 Yayun Xu,1,2 Jingjing Tao,1,2 Lanlan Li,1,2 Feihu Chen1,2
1Anhui Key Laboratory of Bioactivity of Natural Products, School of Pharmacy, Anhui Medical University, Hefei, 230032, People’s Republic of China; 2The Key Laboratory of Anti-Inflammatory and Immune Medicine, Ministry of Education, Anhui Medical University, Hefei, 230032, People’s Republic of China
Correspondence: Feihu Chen
School of Pharmacy, Anhui Medical University, No. 81 Meishan Road, Hefei, 230032, Anhui Province, People’s Republic of China
Tel/Fax +86 551651611166
Email [email protected]
Purpose: The severity of rheumatoid arthritis (RA) in women is generally lower than that in men. RA is mediated, at least in part, by the protective effects of estradiol. However, the mechanisms underlying the protective effect of estradiol on RA are still unclear. Recent studies have demonstrated that activation of acid-sensing ion channel 1a (ASIC1a) by tissue acidosis plays an important role in the injury of cartilage in RA. Here, we assessed the effects of estradiol on acid-mediated cartilage injury both in vitro and in vivo and explored the involvement of ASIC1a in RA and its underlying mechanism.
Methods: Cultured primary articular chondrocytes were subjected to acidosis-mediated injury in vitro. Beclin1, LC3, p62, GPER1, and ASIC1a expression was detected through Western blotting, quantitative real-time PCR, and immunofluorescence analysis. Adjuvant arthritis (AA) was induced in rats through intradermal immunization by injecting 0.25 mL heat-killed mycobacteria (10 mg/mL) suspended in complete Freund’s adjuvant into the left hind metatarsal footpad. The levels of estrogen and related inflammatory factors in the serum were measured using enzyme-linked immunosorbent assay. The expression of ASIC1a and autophagy-related proteins was detected through immunohistochemical analysis and Western blot.
Results: Treatment of primary articular chondrocytes with estradiol decreased the expression of ASIC1a and autophagy level. The symptoms of cartilage damage and levels of inflammatory cytokines in the serum were reduced after estradiol treatment in the rats with AA. In addition, estradiol treatment reduced ASIC1a expression via the PI3K-AKT-mTOR pathway, among which G-protein coupled estradiol receptor 1 (GPER1) plays a regulatory role. Finally, the level of autophagy in chondrocytes was decreased by the selective ASIC1a blocker psalmotoxin-1 (PCTX-1).
Conclusion: Estradiol can protect the cartilage of rats with AA against acidosis-mediated damage and autophagy by suppressing ASIC1a expression through GPER1.
Keywords: rheumatoid arthritis, estradiol, GPER1, ASIC1a, autophagy, PI3K-AKT-mTOR
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease involving chronic inflammation in multiple joints. The obvious histopathological changes in RA patients are persistent inflammation and cell proliferation in the synovial membrane of the joint, infiltration of various inflammatory cells, pannus formation, and destruction of cartilage and bone tissue, eventually leading to joint deformity and loss of function.1 Several clinical reports have highlighted that joint injury is sometimes independent of synovitis, and subsequently, chondrocytes have gradually become the focus of research.2 Chondrocytes, a cellular component of joint tissue, are involved in cartilage formation, metabolism, and repair. They contribute to RA pathology by changing their cellular fate, thus exacerbating cartilage destruction. Although current therapies have been unable to inhibit chondrocyte damage in RA,3 different approaches have identified potential mechanisms that can explain chondrocyte destruction, including the reduction of pro-inflammatory factors and oxygen consumption at sites of inflammation. The degree of joint synovial acidosis has recently attracted attention, as radiological studies have reported that the low pH associated with joint synovial acidosis leads to the destruction of joints in patients with RA.4 Interestingly, indirect evidence indicating the therapeutic potential of acid-sensing ion channel 1a (ASIC1a) against articular cartilage destruction in RA is based on the fact that articular chondrocyte damage is caused by the ASIC1a-mediated overload of intracellular calcium ([Ca2+]i).5 ASICs are voltage-independent cation channels that belong to the epithelial sodium channel protein family and are activated by a drop in extracellular pH.6 Currently, seven ASIC subunit proteins (ASIC1a, ASIC1b, ASIC1b2, ASIC2a, ASIC2b, ASIC3, and ASIC4) and four independent genes (ACCN1, ACCN2, ACCN3, and ACCN4) have been identified.7 In pathological conditions, such as when the extracellular pH drops to 4, ASICs are activated. They, in turn, trigger the influx of extracellular Na+ and Ca2+, thereby activating various signaling pathways and ultimately exacerbating the course of RA.8 Our group previously observed an increase in the protein and mRNA expression of ASIC1a, ASIC2a, and ASIC3 in the articular cartilage of rats with adjuvant arthritis (AA) compared with that in the articular cartilage of normal rats.9 Furthermore, it has been shown that ASIC1a triggers [Ca2+]i overload and promotes acid-induced autophagy of chondrocytes; however, these effects can be reversed by psalmotoxin-1 (PCTX-1), a selective ASIC1a blocker.10 In recent studies, we investigated the role of ASIC1a-mediated autophagy in chondrocytes and the regulation of RA-associated articular cartilage injury and found that the activation of estradiol and its receptor regulates ASIC1a-mediated autophagy in rats with AA.
Some clinical trials have highlighted the critical role of circulating estradiol in the pathology of RA. Epidemiological studies have shown that the incidence of RA in women increases after menopause, and the median age at presentation of secondary clinical symptoms of RA is about 45 years, which is consistent with the age at which menopause occurs.11 Additionally, the use of oral contraceptives or estradiol replacement therapy leads to increased levels of estradiol and decreased levels of pro-inflammatory factors in the chondrocytes of postmenopausal patients with RA compared with those in the chondrocytes of healthy individuals.12 The impact of fluctuating estradiol levels on chondrocytes renders them more responsive to estradiol treatments, and this may be used as a complementary approach to existing therapies for RA.13
Estrogen is a steroid hormone composed of estradiol, estrogen, and its metabolite estradiol. Of these, 17β-estradiol (E2) has the highest bioactivity.11,14 Currently, three main estrogen receptors (ERs) have been identified: ERα, ERβ, and G-protein coupled estradiol receptor (GPER1/GPR30). ERs are constitutively expressed in most adult mammalian tissues.15 GPER1, the principal G-protein coupled receptor, has high affinity and specificity for estradiol and is composed of 375 amino acid residues.1 Unlike nuclear receptors, it can indirectly regulate transcription through the second messenger system and rapidly trigger signal transduction, including the activation of ion channels, kinases, and signaling pathways.16,17 Free Ca2+ in the cytoplasm, which acts as the second messenger, also plays a key role in the signaling process. Previous studies have shown that the selectiveASIC1a blocker PCTX-1 can protect chondrocytes by strongly blocking acid-induced Ca2+ influx into chondrocytes.10 Studies have also reported that estradiol attenuates H2O2- and glutamate-induced cell death by inhibiting Ca2+ influx through the L-type voltage-gated Ca2+ channels.3,18 ASIC1a activation by extracellular acidification leads to [Ca2+]i overload caused by extracellular Ca2+ and triggers autophagy in chondrocytes. However, the mechanism by which estradiol and its receptor subtypes regulate ASIC1a-mediated [Ca2+]i overload is worth exploring.
Autophagy is the process of packaging cytoplasmic proteins or organelles into vesicles, fusing of the vesicles with lysosomes to form autophagosomes, and degrading the lysosomes to meet the metabolic needs of cells and renew certain organelles.16 Autophagy disorders have been reported to alter cell fate in the pathogenesis of osteoarthritis (OA).16 In fibroblast-like synoviocytes (FLSs) in RA, the mechanism of resistance to programmed cell death is diverse, including an increase in the levels of anti-apoptotic factors and an increase in autophagy, where increased autophagy of FLSs promotes RA-associated synovitis.17 It is worth noting that the dysregulation of osteoclast and osteoblast autophagy is the key to cell fate change in RA. In patients with RA, various homeostatic changes, including are duction in the number of osteoclasts, increase in the number of osteoblasts, and bone resorption, lead to the erosion and hypertrophy of the subchondral bone and synovial interface. In summary, autophagy is key to changes in cell fate and functions in a variety of cell types, including osteoclasts and osteoblasts.19 Therefore, regulating autophagy in a tissue-specific or cell-specific manner helps balance the fate of multiple cell types in patients with RA. However, the association of GPER1 with ASIC1a and the role of these proteins in the mechanism of acidosis-induced autophagy in chondrocytes remain largely unknown.
Here, we tested the hypothesis that GPER1 is an estradiol receptor that contributes to reducing ASIC1a-mediated cell autophagy in chondrocytes and the downregulation of ASIC1a protein. Our results showed that estradiol played a protective role by inhibiting the expression of ASIC1a protein in the articular cartilage of rats with AA. Furthermore, high levels of estradiol in the intra-articular cartilage ameliorated the severity of arthritis as well as autophagy in the chondrocytes in the inflamed joint microenvironment in rats with AA. Taken together, our data show that estradiol ameliorates ASIC1a-mediated autophagy through GPER1 in chondrocytes and protects against RA.
Materials and Methods
Reagents and Antibodies
Dulbecco’s modified Eagle medium (DMEM) (HyClone SH30022.01), phosphate-buffered saline (PBS) solution (HyClone SH30256.01), and other standard cell culture products were obtained from Hyclone (MA, USA). Fetal bovine serum was obtained from Gibco (Thermo Fisher Scientific, Waltham, MA, USA) (Gibco 10099-141). Mouse anti-β-actin antibody was purchased from ZSGB-BIO (Beijing, China) (TA09). Rabbit anti-ASIC1a antibody was purchased from Bioss (Beijing, China) (bs-2586R). Rabbit anti-Beclin1 (ab207612), rabbit anti-LC3 (ab192890), and rabbit anti-GPER1 (ab260033) antibodies were purchased from Abcam (Cambridge, UK). Rabbit anti-mTOR (2983T), anti-p-mTOR (5536T), anti-AKT (56855), anti-p-AKT (4060T), anti-S6K1 (9209S), and anti-pS6K1 (9204S) antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Estradiol was purchased from Sigma-Aldrich (St. Louis, MO, USA) (Sigma E8875). GPER1 agonist G1 (B5455) and its inhibitor G15 (B5469), the AKT inhibitor MK2206 (A3010), and the ERα inhibitor MPP (B6910) were purchased from APExBIO (Houston, TX, USA). Freund’s adjuvant was purchased from Chondrex (Redmond, WA, USA) (Chondrex7027). Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used to measure the concentrations of IL-1β (E-EL-R0012c), TNF-α (E-AB-60065), and estrogen (E-EL-R1006c) (Elabscience Biotechnology Co., Ltd, Wuhan, China) according to the manufacturer’s instructions. The assay sensitivities for proinflammatory factors IL-1β (E-EL-R0012c), TNF-α (E-AB-60065), and estrogen (E-EL-R1006c) were 18.75 pg/mL, 1 mg/mL, and 0.10 ng/mL, respectively, with an intra-assay variation of less than 10.0% and an inter-assay variation of <10.0%.
Animals
Sprague-Dawley (SD) rats, aged 7–8 weeks and weighing 180–220 g, were purchased from the Experimental Animal Center of Anhui Medical University, China. Some of them were ovariectomized, and some of them had adipose tissue around the ovaries removed as the sham group. Animals were randomly assigned to different treatment groups, and the person who performed the animal studies was blinded to the treatments. Rats were ovariectomized, maintained at 22 °C under a 12 h light/dark cycle, and were provided free standard pellet food and water. All experiments involving animals followed the Animal Management Regulations of the Ministry of Health (Document No. 55 of 2001) and were approved by the Animal Experiment Ethics Review Committee of Anhui Medical University (LLSC20180328).
Establishment of AA in Rats
AA was initiated in the rats by intradermal immunization into the left hind metatarsal footpad with 0.25 mL heat-killed mycobacteria (10 mg/mL) suspended in complete Freund’s adjuvant according to our preliminary experimental results. Animals were randomly assigned to different treatment operation: some of them ovariectomized, and some of them remove adipose tissue around ovaries as the sham group. The rats with ovariectomized were divided into the following groups (n = 7): the normal group, the OVX + estradiol- injected group (estradiol was injected at 0.1, 0.2, and 0.3 mg/kg)20 and the OVX + triamcinolone acetonide (TA)-treated group (positive control, 1 mg/kg/three days). Treated AA groups received injections of estradiol, positive drug (triamcinolone acetonide)(TA, 1 mg/kg, Selleck, USA, once every 3 days, total eight times), or solvent on the 14th day after immunization, once every three days and the foot swelling was measured by the foot swelling instrument.21 The severity of joint surface damage was categorized and scored as follows: (a). Intact surface or normal in appearance = 0 point, (b). Surface rough with minimal fibrillation or a slight yellowish discoloration =1 point, (c). Cartilage erosion extending into the superficial or middle layers = 2 points, (d). Cartilage erosion extending into the deep layer = 3 points, (e). Complete cartilage erosion with subchondral bone exposed = 4 points. The average scores were obtained by summing the cartilage scores of the lesions in femur condyle and tibia plateau cartilage in eight knees of each group.
Hematoxylin and Eosin Staining
The rat right hind ankle joint was soaked in EDTA decalcifying solution for two months, and the secondary ankle joints and spleens were harvested and fixed in 4% paraformaldehyde for 48 h, decalcified in 10% ethylenediaminetetraacetic acid, and embedded in paraffin. Serial sections (4 μm) were cut, stained with hematoxylin and eosin (Beyotime, Shanghai, China), and examined microscopically, as described previously.5
Determination of Cytokine and Estrogen Levels in Serum
Serum samples were extracted through abdominal aortic dissection of the animals. The extraction and measurement of estrogen, proinflammatory factor IL-1β and TNF-α were performed according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA).
Isolation of Primary Rat Articular Chondrocytes
Rat articular chondrocytes were extracted as previously described.10 The cartilage of the knee was broken down into small pieces and digested in PBS with 0.2% type II collagenase (Sigma-Aldrich, St. Louis, MO, USA). The cells were then centrifuged for 10 min and washed thrice with PBS. Freshly isolated chondrocytes were cultured in DMEM/F-12 supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 mol/mL streptomycin. The cultures were maintained under sterile conditions at 37°C and 5% CO2 in humidified incubators for the first three generations.
Western Blotting Analysis
Cultured chondrocytes and articular cartilage from rats were lysed using RIPA lysis buffer (Beyotime). The total protein concentration was estimated using a BCA protein detection kit (Boster, Wuhan, China). Lysates were mixed with loading buffer and subjected to SDS-PAGE (initially at 80 V for 30 min, then at 120 V for 60 min) to separate proteins. Segregated proteins were then electrically transferred to a PVDF membrane (Millipore, Billerica, MA, USA). After transfer, the PVDF membrane was incubated with 5% skim milk for 1 h at room temperature or overnight at 4°C. The blots were then incubated with the following primary antibodies: mouse anti-actin antibody (1:800), rabbit anti-ASIC1a antibody (1:1000), rabbit anti-Beclin1 antibody (1:1000), rabbit anti-LC3 antibody (1:1000), rabbit anti-mTOR antibody (1:1000), rabbit anti-P-mTOR antibody (1:1000), rabbit anti-AKT antibody (1:1000), rabbit anti-P-AKT antibody (1:1000), rabbit anti-S6K1 antibody (1:1000), and rabbit anti-p-S6K1 antibody (1:1000). After incubation with primary antibodies, the blots were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (1:500). The membrane was rinsed three times (15 min each) with TBST. Finally, the blots were imaged using ECL-Plus (Thermo Fisher Scientific, Waltham, MA, USA). The Image J software was used for densitometric analysis of the blots, and β-actin was used as an internal reference.
Immunofluorescence Staining
Cultured chondrocytes were inoculated into a six-well plate pre-laid with cover glass. The cells were fixed with 4% paraformaldehyde solution for 20 min, and then washed three times with PBS. The cells were then transfused with 0.3% Triton X-100 for 30 min and washed three times with PBS. Next, they were incubated with 1% bovine serum albumin solution for 2 h at 37°C, followed by overnight incubation with rabbit anti-rat antibodyASIC1a, GPER1, LC3, and Beclin1 (1:100) solution. The negative control group was incubated with PBS instead of the primary antibody. After incubation with primary antibodies, the cells were incubated with a FITC-labeled secondary antibody at room temperature for 1 h. Finally, glycerol was added to prevent fluorescence quenching, and ASIC1a, GPER1, LC3, and Beclin1 green fluorescently labeled cells were observed using an inverted microscope (Olympus, Tokyo, Japan).
Construction of Short Hairpin RNA Plasmids and Cell Transfection
Chondrocytes were transfected with 5 μg of ASIC1a-specific short hairpin RNA (shRNA) or control shRNA in serum-free Opti-MEM (Invitrogen, Carlsbad, CA, USA) using Lipofectamine™ reagent. An shRNA with a known sequence without homology interference was used as the negative control. Cells were grown for 24 h after transfection. Control shRNA and DNA oligonucleotide sequences encoding the ASIC1a-specific shRNA were designed by Genepharma (Shanghai, China).
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen), and cDNA was synthesized using the iScript Select cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer’s protocols. The diluted RNA stock solution analyzed via ultra-micro UV-Vis spectrophotometry to determine the RNA concentration and the value of A260/A280, which was repeated three times to obtain the average value. All A260/A280 values of the RNA samples in this experiment were in the range of 1.8–2.0. RT negative control performed to ensure no DNA contamination in the RNA extraction. Total RNA (500 ng) was reverse-transcribed using a first-strand cDNA kit (Fermentas; Thermo Fisher Scientific, Inc.) into cDNA, according to the manufacturer’s protocol and analyzed via qPCR using a SYBR-Green PCR Master Mix (Takara Biotechnology Co., Ltd., Dalian, China) on a Step One platform (Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR amplification cycles consisted of denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 5 s and annealing/extension at 61°C for 10 s, and a melting curve was prepared by detection from 65°C to 95°C. The gene accession numbers were: β-actin (11461), LC3 (32001), Atg5 (9474) and Beclin1 (8678). The primers used were synthesized by Invitrogen as follows:
β-actin forward: 5′-CCCATCTATGAGGGTTACGC-3′, β-actin reverse: 5′-TTTAATGTCACGCACGATTTC-3′; LC3 forward: 5′-GATGTCCGACTTATTCGAGAGC-3′, LC3 reverse: 5′-TTGAGCTGTAAGCGCCTTCTA-3′; Beclin1 forward: 5′-TTCAAGATCCTGGACCGAGTGAC-3′, Beclin1 reverse: 5′-AGACACCATCCTGGCGAGTTTC-3′; ATG5 forward: 5′-TGAAGGAAGTTGTCTGGATAGCTCA-3′, ATG5 reverse: 5′-AAGGTCTGGTCCTTCCGCAGTC-3′. β-actin was used as an endogenous control, and ΔΔCt values were calculated after β-actin normalization. Relative levels of target mRNAs were calculated as 2−ΔΔCt. All experiments were repeated three times.
Immunohistochemistry and Toluidine Blue Staining
Rat primary articular chondrocytes were fixed in ice-cold acetone for 15 min and permeabilized with 0.3% Triton X-100 in PBS for 15 min. Immunohistochemistry and toluidine blue staining were performed as previously described.14 Cells were washed and imaged using an inverted fluorescence microscope (Olympus).
Statistical Analysis
Data were tested for normality and uniformity of variance using the Shapiro–Wilk test and the Brown-Forsythe test, respectively. If the data were found to be normally distributed with uniform variance, then one-way analysis of variance (ANOVA) and unpaired Student’s t-test were used to compare the means of different groups. When the results were found to be significant using ANOVA, Tukey’s honestly significant difference post hoc analysis was used to perform multiple comparisons. If the data did not follow a normal distribution, Kruskal–Wallis (with pairwise Dunn’s method, if applicable), and the Mann–Whitney U-test was used to compare the means. A P-value <0.05 was considered significant. Results are expressed as mean ± SEM.
Results
Estradiol Reduces Cartilage Damage and Protects Articular Cartilage in Ovariectomized AA Rats in vivo
To eliminate the interference of circulating estradiol in the body, we ovariectomized female SD rats at 4 weeks of age. Freund’s adjuvant was used to induce AA in OVX rats in order to investigate the role of estradiol. AA rats developed severe arthritis on the 28th day after immunization. As shown in Figure 1A, toluidine blue staining showed that bone erosion in AA rats, compared with the normal group. HE staining showed that synovial hyperplasia as well as cartilage and bone erosion in the cartilage of AA rats (Figure 1B–C). The right paws of rats with AA tended to be redder and more swollen, and a higher degree of foot swelling, as measured by a foot swelling instrument, compared with normal rats (68.5 ±3.72% increase) (Figure 1D). A lower increase in weight were observed in the AA group compared with the normal group (Figure 1E). These results indicated that the AA rat models were successfully induced. Immunohistochemical analysis showed that the expression of ASIC1a was decreased significantly (38.68 ±4.68% reduction) after the treatment of estradiol in inflamed articular cartilage compared with AA rats (Figure 1F–G), and the results were consistent with Western blot (20.76 ±3.58% reduction) (Figure 1H). Estradiol (0.1, 0.2 and 0.3 mg/kg) was administered by intramuscular injection into AA rats. Compared with the dissolved model group, the paw swelling of rats was significantly alleviated in estradiol-treated groups in a dose-dependent manner (Figure 1I–J). The reduction of paw swelling by E2 (0.3 mg/kg) (71.33 ± 3.72% reduction) is clearly larger than that in E2 (0.1 mg/kg) (15.37 ± 5.67% reduction) compared with that in the solvent group. HE staining and toluidine blue staining showed that synovial infiltration and cartilage damage was alleviated reduced in 0.1, 0.2 and 0.3 mg/kg estradiol-treated groups compared with the dissolved group (Figure 1K–L). The reduction of cartilage damage by E2 (0.3 mg/kg) (69.5 ± 1.72% reduction) is clearly larger than that in E2 (0.1 mg/kg) (20.3 ± 4.57% reduction) compared with that in the solvent group (Figure 1M). These results suggested that estradiol reduces cartilage damage in ovariectomized AA rats in vivo.
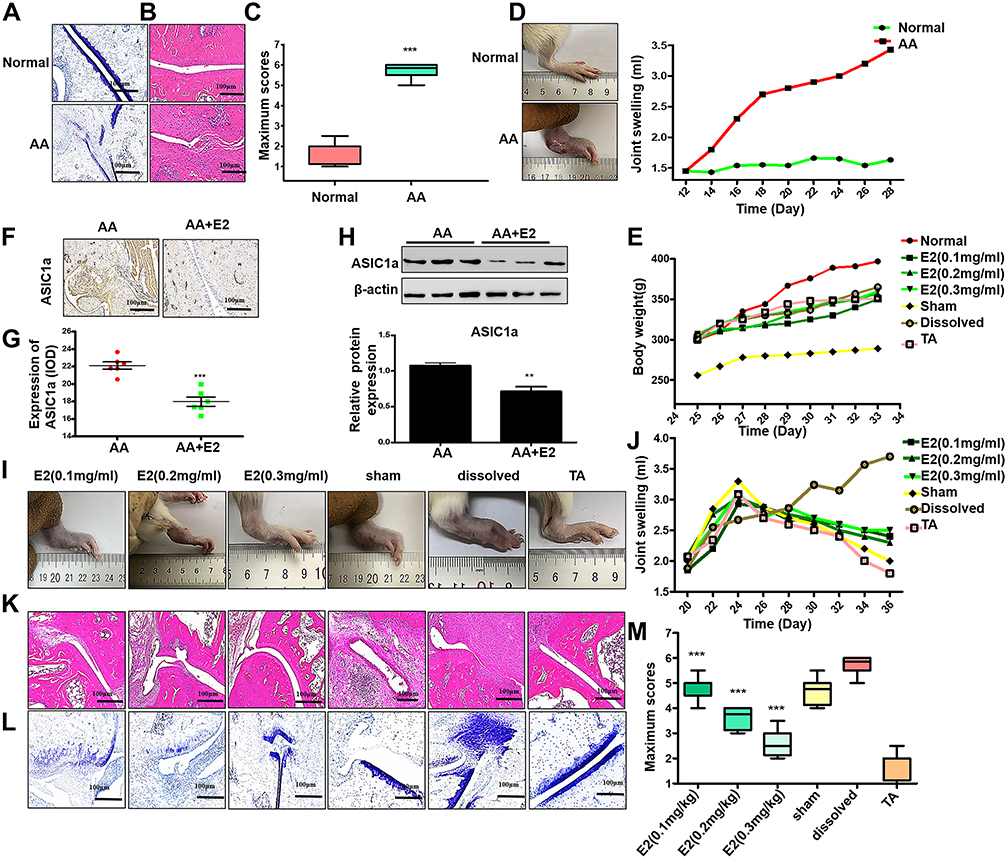 |
Figure 1 Estradiol reduces cartilage damage and protects articular cartilage in ovariectomized AA rats in vivo. (A) Normal and AA rat right hind ankle joint sections stained with toluidine blue staining. (B) Normal and AA rat right hind ankle joint sections stained with HE staining. (C) The scores of cartilage damage (n = 7), Semiquantitative analysis of ASIC1a protein expression in ankle joint of the E2 group and the AA group (n = 7) were calculated using the IPP 6.0 software. (D) Representative image of paw edema in rats and the joint swelling of the AA group and the normal group were measured by the joint swelling instrument (n = 7). (E) The body weight of all groups (n = 7). (F) Immunohistochemical of ASIC1a in ankle joint of the AA group and the E2 group (n = 7). (G) Semiquantitative analysis of ASIC1a protein expression in ankle joint of the E2 group and the AA group (n = 7) were calculated using the IPP 6.0 software (H) Western blot analysis of ASIC1a expressed in ankle joint of the E2 group and the AA group (n = 7). β-actin served as a loading normal, and the indicated proteins were quantified with Image J software. (I) Representative image of paw edema in rats with adjuvant-induced arthritis (n = 7). (J) The joint swelling of all groups was measured by the joint swelling instrument (n = 7). (K) HE staining in ankle joint of all groups (n = 7). (L) Toluidine blue staining in ankle joint of all groups (n = 7). (M) The scores of cartilage damage. Data are presented as the mean ± SEM of three independent experiments, (C) ***P < 0.001 compared with the Normal group. (G and H) **P < 0.01, ***P < 0.001 versus the AA group. (M) ***P < 0.001 compared with the solvent-treated group. |
Estradiol-Mediated Protection Against the Autophagy of Articular Cartilage in OVX Rats with AA in vivo
GPER1 and ERα were all detected immunohistochemically in cartilage tissue of rats with AA (Figure 2A). To assess the effect of estradiol modulation on the autophagy of articular cartilage, we conducted immunohistochemical staining for ASIC1a, LC3, p62 and Beclin1. The reduction of ASIC1a by E2 (0.3 mg/kg) (57.15 ± 8.56% reduction) is clearly larger than that in E2 (0.1 mg/kg) (28.18 ± 9.51% reduction) compared with that in the solvent group; the reduction of LC3 by E2 (0.3 mg/kg) (34.00 ± 10.56% reduction) is clearly larger than that in E2 (0.1 mg/kg) (18.87 ± 8.56% reduction), compared with that in the solvent group; the reduction of Beclin1 in the E2 (0.3 mg/kg) (29.88 ± 7.42% reduction) is clearly larger than the E2 (0.1 mg/kg) group (7.6 ± 7.11% reduction), compared with that in the solvent group; the increase of p62 were increased in the estradiol group E2 (0.3 mg/kg) (68.92 ± 8.90% increase) is larger than the E2 (0.1 mg/kg) group (11.29 ± 7.98% increase), compared with that in the solvent group (Figure 2B–C), and this result was consistent with that of the Western blot analysis (Figure 2D). Moreover, ELISA showed that IL-1β and TNF-α levels were significantly elevated in the serum of OVX rats with AA, and their levels were decreased after treatment with E2. Compared with that in the solvent group, the reduction of IL-1β by E2 (0.3 mg/kg) (55.54 ± 5.72% reduction) is clearly larger than that in E2 (0.1 mg/kg) (23.72 ± 5.43% reduction); the reduction of TNF-α by E2 (0.3 mg/kg) (58.74 ± 5.87% reduction) is clearly larger than that in E2 (0.1 mg/kg) (34.32 ± 3.56% reduction) (Figure 2E).
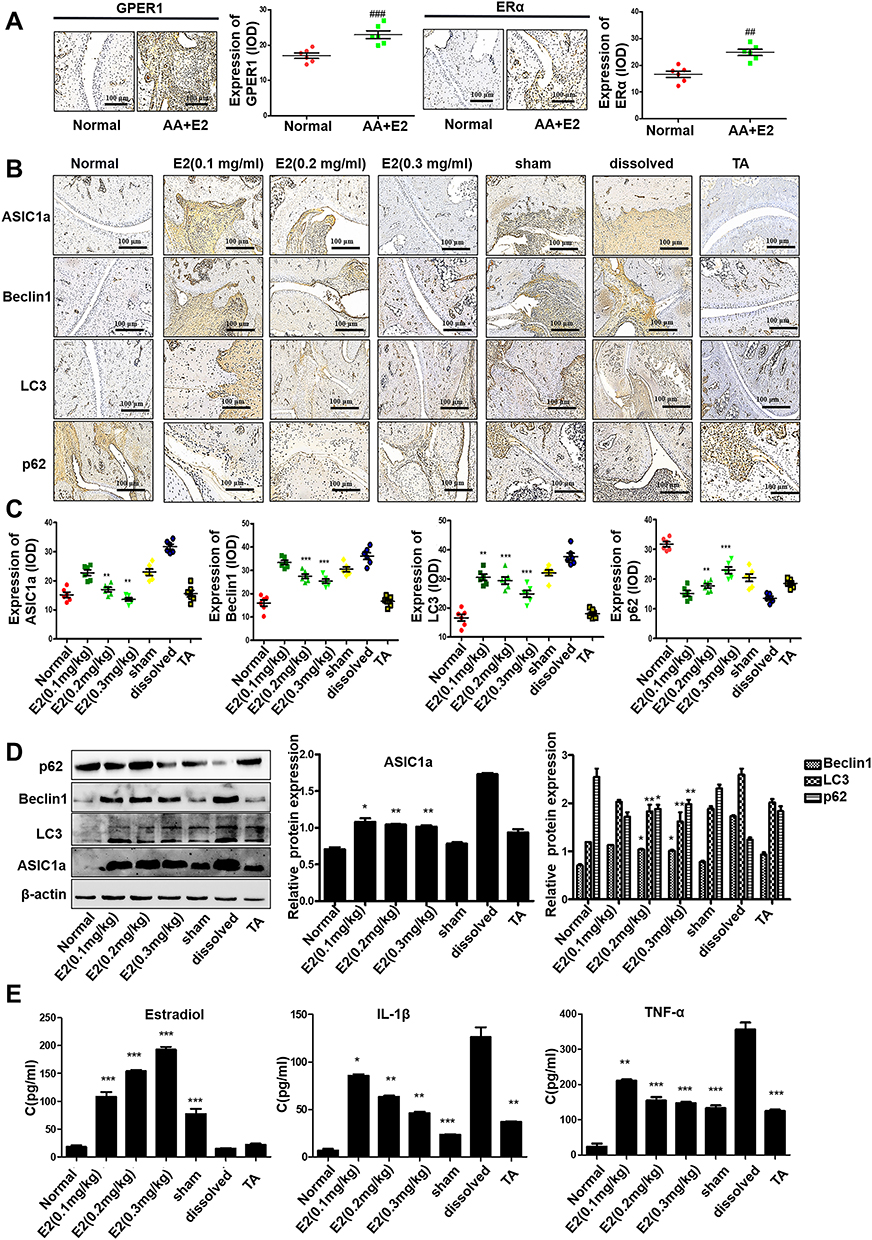 |
Figure 2 Estradiol-mediated protection against the autophagy of articular cartilage in OVX rats with AA in vivo. (A) The immunohistochemistry results of GPER1 and ERα of the Normal group and the AA+E2 group. Semiquantitative analysis of GPER1 and ERα expression in ankle joint of each group (n = 7) were calculated using the IPP 6.0 software. (B) Immunohistochemistry analysis of Beclin1, LC3, p62 and ASIC1a in articular cartilage tissue of all groups (n = 7). (C) Semi-quantitative analysis of ASIC1a, Beclin1, LC3, p62 expression in rat cartilage by integral optical density (IOD). (D) The protein expression levels of Beclin1, LC3, ASIC1a and p62 in articular cartilage tissue of all groups were measured by Western blot. β-actin served as a loading normal, and the indicated proteins were quantified with Image J software. (E) Estradiol, IL-1β and TNF-α levels in the serum of all groups were measured by ELISA assay (n = 7). Data are presented as the mean ± SEM of three independent experiments. (A) ##P < 0.01, ###P < 0.001 compared with the Normal group. (C–E) *P < 0.05, **P < 0.01, ***P < 0.001 compared with the solvent-treated group. |
Estradiol Contributes to Autophagy in Chondrocytes by Mediating the Downregulation of ASIC1a
The protective effects of estradiol on cartilage motivated us to evaluate the effect of estradiol on extracellular acidification of rat articular chondrocytes. As shown in Figure 3A and B, estradiol treatment led to a downregulation of ASIC1a protein in a time- and dose-dependent manner; Specifically, the ASIC1a expressions in chondrocytes after estradiol treatment for 24 h (45.67 ± 3.07% reduction) and 48 h (46.23 ± 1.11% reduction) were significantly decreased compared with 0 h; and after estradiol treatment at 500 nmol/mL (63.52 ± 3.21% reduction) and 1000 nmol/mL (52.96 ± 1.02% reduction) were significantly decreased compared with 0 nmol/mL. The downregulation of autophagy decreased by E2: the expression of LC3 reduced by estradiol in a time- and dose-dependent manner. Specifically, the LC3 expressions in chondrocytes after estradiol treatment for 24 h (43.85 ± 1.07% reduction) and 48 h (65.56 ± 6.66% reduction) were significantly decreased compared with 0 h; and after estradiol treatment at 500 nmol/mL (44.65 ± 2.22% reduction) and 1000 nmol/mL (52.34 ± 3.56% reduction) were significantly decreased compared with 0 nmol/mL; the expression of Beclin1 reduced by estradiol in a time- and dose-dependent manner. Specifically, the Beclin1 expressions in chondrocytes after estradiol treatment for 24 h (67.09 ± 3.22% reduction) and 48 h (60.98 ± 5.68% reduction) were significantly decreased compared with 0 h; and after estradiol treatment at 500nmol/mL (35.42 ± 3.33% reduction) and 1000nmol/mL (30.22 ± 1.65% reduction) were significantly decreased compared with 0 nmol/mL; the expression of p62 increased by estradiol in a time- and dose-dependent manner. Specifically, the p62 expressions in chondrocytes after estradiol treatment for 24 h (44.35 ± 3.02% increase) and 48 h (45.20 ± 2.01% increase) were significantly increased compared with 0 h; and after estradiol treatment at 500nmol/mL (33.52 ± 2.10% increase) and 1000nmol/mL (40.10 ± 3.21% increase) were significantly increased compared with 0 nmol/mL (Figure 3C and D). We found that treatment with 500 nmol/mL estradiol for 24 h had the highest effect on the expression of ASIC1a protein and autophagy-related proteins. Additionally, immunofluorescence staining showed that exposure to estradiol inhibited the expression of ASIC1a, which was upregulated in an inflammatory environment (Figure 3E).
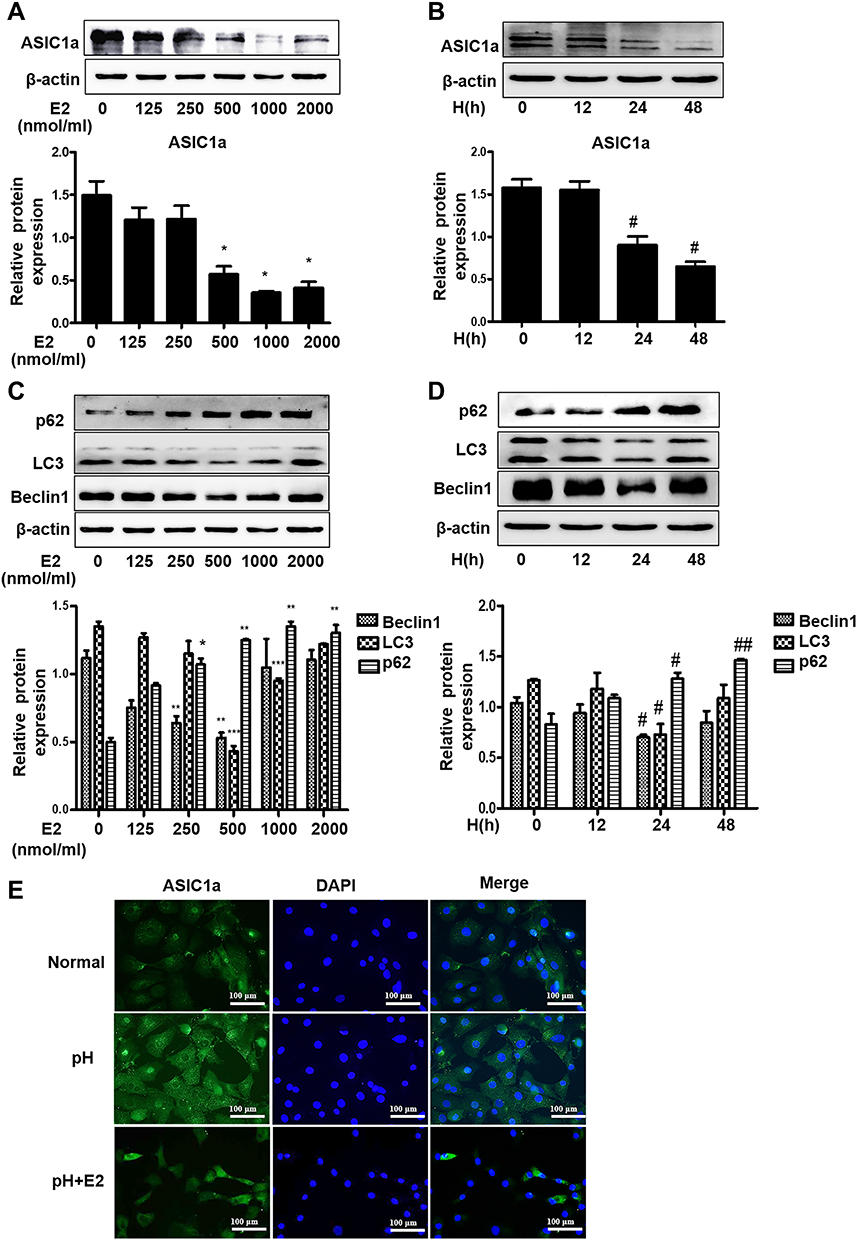 |
Figure 3 Estradiol contributes to autophagy in chondrocytes by mediating the downregulation of ASIC1a. (A and B) The expression of ASIC1a decreased in a time- and concentration-dependent manner following stimulation with 0–2000 nmol/mL estradiol for 0 h to 48 h in chondrocytes. (C and D) The expression of Beclin1, LC3 and p62 protein in chondrocytes in a time- and concentration-dependent manner following stimulation with 0–2000 nmol/mL estradiol for 0 h to 48 h. (E) Estradiol induced expression levels of ASIC1a in chondrocytes cultured in pH 6.0 medium for 24 h with or without E2 (500 nmol/mL) were analyzed by immunofluorescence. Representative views from each group are presented (original magnification, ×400). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus 0 nmol/mL group; #P < 0.05, ##P < 0.01 versus 0 h group. |
Estradiol Modulates ASIC1a Expression Through Its Receptor GPER1
To determine whether GPER1 is an effective estradiol target in chondrocytes, we estimated the expression of some representative estradiol receptors in chondrocytes. ERα and GPER1 were detected in chondrocytes, and the decreased expression of ASIC1a, which is mediated by E2, was reversed by the GPER1-specific inhibitor G15; however, the ERα-specific inhibitor MPP could not completely reverse this decrease in ASIC1a expression (Figure 4A). The increase of ASIC1a by G15 (68.98 ± 5.54% increase) is clearly larger than that in MPP (9.87 ± 2.65% increase), compared with that in the pH + E2 group. To further confirm the effects of ERα and GPER1, we silenced ERα and GPER1. As shown in Figure 4B, ERα- and GPER1-specific siRNAs were successfully transfected. As expected, ERα-siRNA could not completely reverse this decline mediated by E2, compared with the GPER1-siRNA group reduction (Figure 4C). The increase of ASIC1a by GPER1-siRNA (43.32 ± 2.45% increase) is clearly larger than that in ERα-siRNA (4.32 ± 8.07% increase), compared with that in the pH + E2 group. To determine the effect of GPER1 activation on ASIC1a-induced autophagy in chondrocytes, we treated cells with different concentrations of G1 (GPER1 specific agonist) (0–1000 nmol/L). Compared with the 0 nmol/L, the maximum decline in autophagy and ASIC1a expression (49.56 ± 5.55% reduction) and the maximum activation of GPER1 (50.69 ±3.65% increase) were observed at a G1 concentration of 100 nmol/L (Figure 4D–E); therefore, this concentration was used for subsequent tests. Compared with those in 0 nmol/L group, the Beclin1 expressions in chondrocytes after G1 treatment at 10 nmol/L (45.37 ± 6.88% reduction) and 100nmol/L (60.87 ± 8.66% reduction) were significantly decreased; the LC3 expressions in chondrocytes after G1 treatment at 10 nmol/L (32.02 ± 5.37% reduction) and 100nmol/L (58.66 ± 4.68% reduction) were significantly decreased; the p62 expressions in chondrocytes after G1 treatment at 10 nmol/L (22.51 ± 3.74% increase) and 100nmol/L (63.74 ± 3.20% increase) were significantly increased (Figure 4F). The results also showed that the effects of G1, including the upregulation of ASIC1a protein, inhibition of autophagy stimulation (downregulation of Beclin1 and LC3 protein, upregulation of p62 protein), and activation of GPER1, were blocked by co-treatment with G15, a GPER1 antagonist (Figure 5A–C). Furthermore, results of qRT-PCR (Figure 5D–E) and immunofluorescence (Figure 5F–I) analyses were consistent with those of the Western blot analysis.
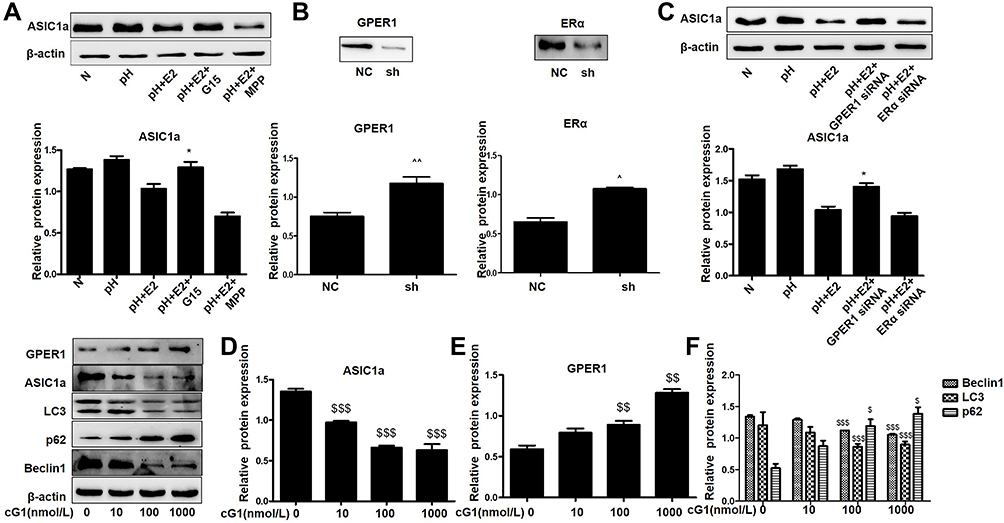 |
Figure 4 Estradiol modulates ASIC1a through its receptor GPER1.(A) Western blot analysis of the protein expression of ASIC1a with or without for estradiol (500 nmol/mL), MPP (20 mmol/L), and G15 (15 μmol/L). (B) Western blot analysis of ERα and GPER1 protein expressions in chondrocytes that without transfection or transfected with negative control (NC) or siRNA for ERα or GPER1. (C) Western blot analysis of the protein expression of ASIC1a with or without for estradiol (500 nmol/mL) and ERα-siRNA or GPER1-siRNA. (D–F) Chondrocytes in serum-free confluent monolayer cultures were stimulated by with increasing concentrations of G1 (0–1000 nmol/L) and levels of ASIC1a, GPER1, Beclin1, LC3 and p62 proteins were measured by Western blot. Data are presented as mean ± SEM, *P < 0.05, versus pH + E2 group, ^P < 0.05, ^^P < 0.01 versus the NC group, $P < 0.05, $$P < 0.01, $$$P < 0.001 versus 0 nmol/L. |
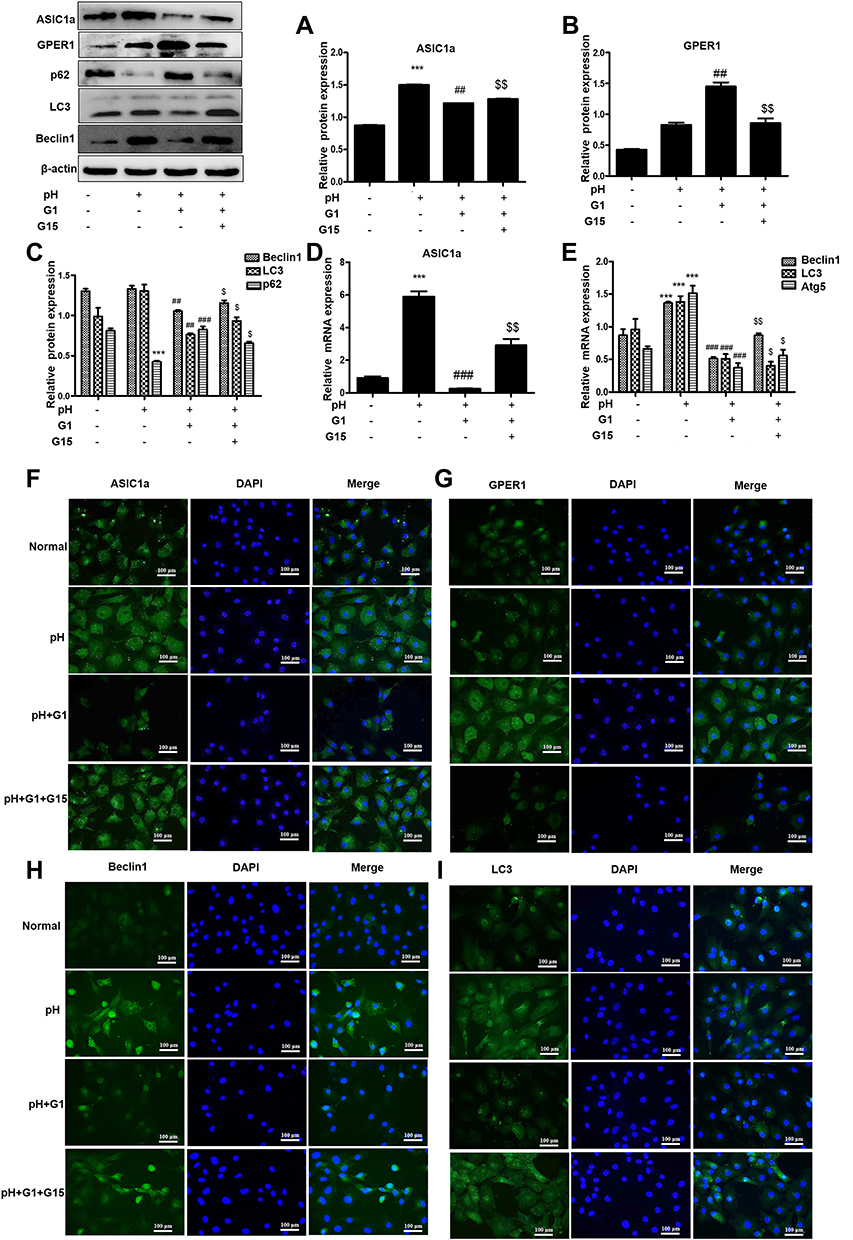 |
Figure 5 GPER1 inhibits autophagy by down-regulating the expression of ASIC1a. (A–C) Western blot analysis of ASIC1a, GPER1, Beclin1, LC3 and p62 protein in chondrocytes were pretreated with or without agonist for GPER1 (G1) followed by stimulation with G15. (D and E) The mRNA expression of ASIC1a, Beclin1, LC3 and Atg5 in chondrocytes were pretreated with or without G1 followed by stimulation with G15. (F–I) G1 and G15-induced expression levels of ASIC1a, GPER1, Beclin1 and LC3 were analyzed by immunofluorescence in chondrocytes. Representative views from each group are presented (original magnification, ×400). Data are presented as mean ± SEM. ***P < 0.001 versus normal group; ##P < 0.01, ###P < 0.001 versus pH group; $P < 0.05, $$P < 0.01 versus G1 group. |
GPER1 Inhibits the Expression of ASIC1a by Activating the PI3K-AKT-mTOR Signaling Pathway
To investigate the underlying mechanism of the GPER1-mediated decline in ASIC1a protein expression, we treated articular chondrocytes with G1 and G15 and determined the expression levels of phosphorylated AKT, mTOR, and S6K1 by Western blot analysis. G1 caused an obvious increase in the protein levels of phospho-AKT (56.43 ± 3.64% increase), phospho-mTOR (50.31 ± 6.77% increase), and phospho-S6K1 (57.89 ± 5.78% increase), with smaller changes in the levels of the respective total proteins. In contrast, treatment with G15 decreased the levels of the phosphorylated proteins (Figure 6A). To further explore the potential role of the GPER1-induced activation of the PI3K-AKT-mTOR pathway in chondrocytes, a selective inhibitor (MK2206) was used to block the signaling pathway. MK2206 was found to block the G1-induced phosphorylation of AKT (54.37 ±4.33% reduction), mTOR (65.43 ±5.34% reduction), and S6K1 (55.55 ±4.26% reduction) (Figure 6B). Moreover, qPCR analysis also showed that, compared with the G1+pH group, MK2206 treatment inhibited the G1-induced decrease in ASIC1a (69.66 ±3.75% reduction), Beclin1 (54.98 ±6.78% reduction), LC3 (66.89 ±3.43% reduction), and Atg5 (57.87 ±6.98% reduction) mRNA expression, which increased in response to acid stimulation in chondrocytes (Figure 6C–D).
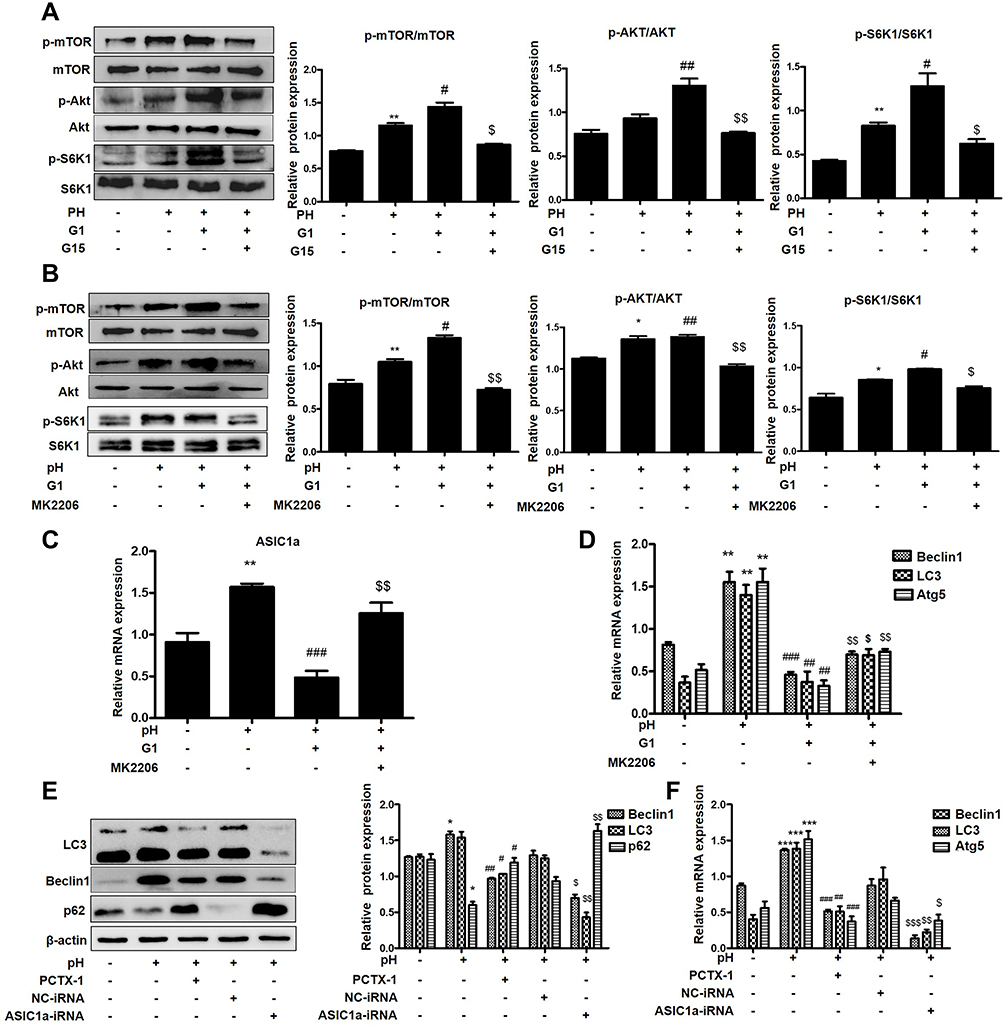 |
Figure 6 GPER1 inhibits the expression of ASIC1a by activating the PI3K-AKT-mTOR signaling in chondrocytes. (A) Cultured chondrocytes were pretreated with or without agonist for GPER1 (G1) followed by stimulation with G15 and levels of phosphorylated and total mTOR, AKT and S6K1 proteins were measured by Western blot. (B) Cultured chondrocytes were pretreated with or without inhibitors for AKT (MK2206, 1000 nmol/mL) followed by stimulation with G1 and levels of phosphorylated and total mTOR, AKT and S6K1 proteins were measured by Western blot. (C and D) qRT-PCR analysis of ASIC1a, Beclin1, LC3 and Atg5 mRNA expression levels in chondrocytes. (E) Western blot analysis of Beclin1, LC3, p62 and ASIC1a protein expressions in chondrocytes that without transfection or transfected with negative control (NC), ASIC1a-RNAi or PCTX-1 (100 nmol/L). (F) qRT-PCR analysis of Beclin1, LC3 and Atg5 protein expressions in chondrocytes that without transfection or transfected with negative control (NC), ASIC1a-RNAi or PCTX-1 (100 nmol/L). Data are presented as mean ± SEM. (A–D) *P < 0.05, **P < 0.01 versus normal group; #P < 0.05, ##P < 0.01, ###P < 0.001 versus pH group; $P < 0.05, $$P < 0.01 versus G1 group. (E and F) *P < 0.05, ***P < 0.001 versus normal group; #P < 0.05, ##P < 0.01, ###P < 0.001 versus pH group; $P < 0.05, $$P < 0.01, $$$P < 0.001 versus NC group. |
To further confirm that ASIC1a mediates autophagy in chondrocytes, we silenced and blocked ASIC1a expression. Western blotting analysis showed that the protein expression of Beclin1 (63.45 ± 5.43% reduction) and LC3 (47.86 ± 5.54% reduction) was increased and that of p62 (55.56 ± 5.78% increase) was decreased significantly in the pH 6.0 group compared to that in the normal group, but was reversed in response to ASIC1a-siRNA and PCTX-1 treatments (Figure 6E). This result was consistent with the results of qRT-PCR analysis (Figure 6F). In summary, our results indicate that the PI3K-AKT-mTOR signaling pathway is a major effector of ASIC1a-mediated autophagy in chondrocytes.
Discussion
In this study, we explored the effect of estradiol on RA and determined its underlying mechanisms. We first showed that estradiol protected cartilage against injury in AA by reducing the expression of ASIC1a, autophagy, and the secretion of pro-inflammatory factors in OVX rats with AA. We then demonstrated that E2 helps ameliorate arthritis, but at the same time, blocking GPER with G15 should be used to treat arthritis. Furthermore, we showed that blocking chondrocyte autophagy with the inhibitor of GPER1, G15, or the inhibitor of the PI3K-AKT-mTOR pathway, MK2206, is a safe and effective strategy for treating patients with RA. Together, these results indicated that estrogen protects articular cartilage by downregulating ASIC1a through GPER1, and the potential mechanism may be related to the PI3K-AKT-mTOR pathway.
Rheumatoid arthritis is a systemic autoimmune disease involving chronic inflammation at multiple joints, and is a cause of long-term disability worldwide. Unfortunately, the effectiveness of current therapies is limited. For decades, even if the inflammation of the joint site was successfully controlled, the destruction of articular cartilage and the gradual loss of joint function continue after first-line drug therapy for RA. Therefore, it is of great clinical significance to actively search for new drug targets and treatment strategies. Local tissue acidification is a common feature of RA, which induces persistent inflammation and cell proliferation in the synovial membrane of the joint, infiltration of various inflammatory cells, pannus formation, and destruction of cartilage and bone tissue. ASICs are widely distributed in joint tissue, and previous studies by us and others have clearly demonstrated that activation of ASIC1a is largely responsible for acidosis-induced cartilage injury.10 In addition, the blockade of ASIC1a activity by PcTX1 can ameliorate arthritis symptoms.5 Thus, the ASIC1a channel may serve as a novel therapeutic target for RA.
Hormone replacement therapy (HRT) is widely used to treat diseases such as systemic lupus erythematosus (SLE) and osteoarthritis (OA). Previous studies have shown that estradiol has an effect on voltage-gated and ligand-gated ion channels such as BK channels, T-type, and L-type Ca2+ channels and glutamate receptor channels.12,22 Several studies23 have reported that estradiol can exert a rapid potentiating effect on the functional activity of acid-sensing24 ion channels (ASICs), which mediated the acidosis-induced events.25 17β-estradiol could acutely enhance ASIC3-like currents in rat dorsal root ganglia neurons. However, the role of β-estradiol in modulating the activity/function of ASIC1a in RA was unknown. Our present results showed that chronic estradiol exposure (24 h) could reduce the protein expression of ASIC1a. These findings probably since ASIC1a and ASIC3 channels have distinct responses to modulators and pharmacological agents.
Estrogen has a protective effect on bone. It has been reported that cortical bone vascular porosity was increased by estrogen deficiency, with ovariectomized. In addition, estrogen deficiency resulted in resorption of subchondral bone and degeneration of articular cartilage in an ovariectomized rat model of postmenopausal OA. Our present studies provided the first and strong evidence suggesting that estrogen is effective in protecting against acidosis-induced and ASIC1a-mediated cartilage injury. Consistent with a reduction of ASIC1a activity/expression by estradiol, relative protection against acidosis-induced damage by PcTX1 is higher in the absence of β-estradiol than that in the presence of β-estradiol.26 Previous studies by us have clearly demonstrated that26 17β-E2 induced ASIC1a protein degradation through the ERα receptor, to protect chondrocytes from acid-induced apoptosis. However, whether estradiol plays a protective role in the cartilage of AA rats in vivo was unknown. Our present results showed that estradiol not only alleviated cartilage destruction in AA rats, but also reduced serum levels of inflammatory cytokines (IL-1β and TNF-α). These results are consistent with a prospective case-control study of women recently diagnosed with RA, in which the expression of inflammatory cytokines such as IL-6 and TNF-α was significantly increased after menopause. Under estrogen deprivation, circulating levels of IL-6 are elevated.27–30 TNF-α is responsible for monocytes/macrophages’ high-level production of downstream mediators, the proinflammatory cytokines IL-6 and IL-1β, which in turn, promote Th17-IL-17 production.30–32 In murine models of RA, IL-17-producing cells were shown to be restricted to the inflamed synovium and IL-17 was shown to play a central role in bone loss and joint destruction.33–35 IL-17 activates inflammatory cell recruitment and acts at the top of RA-related inflammatory cytokine cascades.36,37 It stimulates joint FLS and macrophages to produce more proinflammatory mediators, such as IL-1β, IL-6, and TNF-α as well as various chemokines; Furthermore, IL-17 has the capacity to stimulate the production of TNF-α and IL-6 in the context of estrogen deficiency.38 Given that TNF-α is critical for IL-17-regulated bone loss, and that elevated levels of IL-6 are thought to favor Th17 cell differentiation, the elevated circulating levels of TNF-α, IL-6, and IL-17 may act concomitantly to mediate postmenopausal bone loss. Taken together, the data gathered from postmenopausal women and OVX mouse studies support the concept of a key role played by IL-17 as an important mediator of the pathogenesis of estrogen deficiency-induced bone loss. This forms a positive feedback loop that amplifies the differentiation of Th17 cells,39,40 all of which, in turn, govern bone erosion by up-regulating osteoclastogenesis.32 The above results show that the mechanism of action of estrogen on RA is complex, the specific potential mechanism still need us to further explored.
Long-term use of HRT has been associated with increased risk of breast cancer, thrombosis, and possibly also stroke. Accordingly, there is great need for new activators of estrogen receptors selectively reproducing only the beneficial effects of estrogen. The biological effects of estradiol are mainly mediated through its interaction with the nuclear ERs, ERα, and ERβ and the membrane receptors which represented by GPER1. The distribution of ERs varies in different tissues. And we have previously shown that ERα and GPER1 are abundant in articular cartilage afflicted with RA while ERβ expression is limited.41 The majority of studies have focused on the nuclear activity of ligand-bound ERs, with an emphasis on specific gene regulation. However, new tools to study membrane-initiated ER signaling may help to understand more rapid responses to estradiol in innate immune cells.42,43 In this study, we found that the decrease in ASIC1a protein expression after E2 treatment was not fully reversed by administration of the ERα-specific inhibitor MPP or ERα-siRNA, but was reversed by pretreatment with the GPER1-specific inhibitor G15 or GPER1-siRNA. This GPER1-dependent mechanism is probably since changes in extracellular pH is rapidly sensed by GPER1, and consequently, the activity of ASIC1a which regulated by H+ in extracellular is inhibited, thus protecting chondrocytes.24 On the other hand, estrogens could have different effects on different receptors, depending on serum concentration expression of oestrogen receptors (ERs) or intracellular metabolism or the affinity to the target gene or protein. However, to confirm this hypothesis, future and larger studies are needed. Genome-wide analyses of epigenetic marks or ER binding sites in response to estradiol/ER signaling in innate immune cells would provide a more comprehensive picture of regulatory mechanisms. This type of work would reveal whether ERs act to mediate epigenetic changes at multiple genes linked in a pathway, or alternately, if ERs regulate specific pathways by acting to promote or inhibit a master regulator. Ultimately, investigation of ER-mediated regulatory mechanisms with new available molecular tools will increase our understanding of how estrogens and sex differences lead to profound effects on immunity and autoimmunity.
In recent years, epidemiological studies have shown that the incidence of RA in women increases significantly after menopause, and the peak incidences consistent with time of menopause. An interesting study analyzed the incidence of RA in users of anti-estrogen agents in a large US national database for breast cancer. The use of anti-estrogen agents, both selective estrogen receptors modulators and aromatase inhibitors, was associated with RA, with a dose- and duration-dependent effect, indicating that estrogen has a protective effect.42 The removal of estrogen by ovariectomy resulted in an aggravation of the cartilage damage can be reversed by exogenously administered estrogen in OA rats.20,44 Our present results also showed that the OVX rats are more likely to get RA compared with the sham group. In addition, we observed a clear difference in the expression level of ASIC1a, TNF-α and IL-1β in articular cartilage of OVX rats and sham rats. Consistent with this difference, ASIC1a inhibitor PCTX1 provided more protection against cartilage injury in sham mice than in OVX mice. These differences were, however, attenuated by the treatment of estradiol, further supporting a role for estradiol in regulating the level of ASIC1a expression and protecting articular cartilage from damage. In line with our current findings, a recent study has shown that the expression levels of ASIC1 and ASIC2 were increased in bone tissue and bone marrow cells from ovariectomized female mice compared with sham female mice.8
This is the first study to indicate that the cartilage protection of estradiol is, at least partially, mediated by down-regulating ASIC1a expression and function. Our findings highlight a novel mechanism underlying the protective effect of estradiol and disclose potential future therapeutic strategies for RA treatment without compromising systemic homeostasis in normal cells and offer a safer and novel additional approach for combination therapy, independent of systemic immunosuppression, against RA.
Conclusion
Estradiol can protect the cartilage of rats with AA against acidosis-mediated damage and autophagy by suppressing ASIC1a expression through GPER1.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81873986).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barton M, Filardo EJ, Lolait SJ, et al. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J Steroid Biochem Mol Biol. 2018;176:4–15. doi:10.1016/j.jsbmb.2017.03.021
2. Bouta E, Bell RD, Rahimi H, et al. Targeting lymphatic function as a novel therapeutic intervention for rheumatoid arthritis. Nat Rev Rheumatol. 2018;14(2):94–106. doi:10.1038/nrrheum.2017.205
3. Dai D, Bradford A, Prossnitz E. Endometrial cancer: molecular and cellular basis of tumor development, novel biomarkers and therapeutic agents, and innovative research approaches. Obstet Gynecol Int. 2014;2014:710405. doi:10.1155/2014/710405
4. Luo Y, Sinkeviciute D, He Y, et al. The minor collagens in articular cartilage. Protein Cell. 2017;8(8):560–572. doi:10.1007/s13238-017-0377-7
5. Yuan F, Chen F-H, Lu W-G, et al. Inhibition of acid-sensing ion channels in articular chondrocytes by amiloride attenuates articular cartilage destruction in rats with adjuvant arthritis. Inflamm Res. 2010;59(11):939–947. doi:10.1007/s00011-010-0206-4
6. Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–177. doi:10.1038/386173a0
7. Carattino M, Montalbetti N. Acid-sensing ion channels in sensory signaling. Am J Physiol Renal Physiol. 2020;318(3):F531–F543. doi:10.1152/ajprenal.00546.2019
8. Tan Z, Lu Y, Whiteis CA, et al. Acid-sensing ion channels contribute to transduction of extracellular acidosis in rat carotid body glomus cells. Circ Res. 2007;101(10):1009–1019. doi:10.1161/circresaha.107.154377
9. Jahr H, van Driel M, van Osch G, Weinans H, van Leeuwen J. Identification of acid-sensing ion channels in bone. Biochem Biophys Res Commun. 2005;337(1):349–354. doi:10.1016/j.bbrc.2005.09.054
10. Zhou R, Dai -B-B, Xie -Y-Y, et al. Interleukin-1β and tumor necrosis factor-α augment acidosis-induced rat articular chondrocyte apoptosis via nuclear factor-kappaB-dependent upregulation of ASIC1a channel. Biochim Biophys Acta Mol Basis Dis. 2018;1864(1):162–177. doi:10.1016/j.bbadis.2017.10.004
11. Islander U, Jochems C, Lagerquist M, Forsblad-d’Elia H, Carlsten H. Estrogens in rheumatoid arthritis; the immune system and bone. Mol Cell Endocrinol. 2011;335:14–29. doi:10.1016/j.mce.2010.05.018
12. Sribnick E, Del Re A, Ray S, Woodward J, Banik N. Estrogen attenuates glutamate-induced cell death by inhibiting Ca2+ influx through L-type voltage-gated Ca2+ channels. Brain Res. 2009;1276:159–170. doi:10.1016/j.brainres.2009.04.022
13. MacDonald A, Murphy E, Capell H, Bankowska U, Ralston S. Effects of hormone replacement therapy in rheumatoid arthritis: a double blind placebo-controlled study. Ann Rheum Dis. 1994;53(1):54–57. doi:10.1136/ard.53.1.54
14. Lingueglia E. Acid-sensing ion channels in sensory perception. J Biol Chem. 2007;282(24):17325–17329. doi:10.1074/jbc.R700011200
15. Kotula-Balak M, Pawlicki P, Milon A, et al. The role of G-protein-coupled membrane estrogen receptor in mouse Leydig cell function-in vivo and in vitro evaluation. Cell Tissue Res. 2018;374(2):389–412. doi:10.1007/s00441-018-2861-7
16. Rockel J, Kapoor M. Autophagy: controlling cell fate in rheumatic diseases. Nat Rev Rheumatol. 2017;13(3):193. doi:10.1038/nrrheum.2017.17
17. Karami J, Masoumi M, Khorramdelazad H, et al. Role of autophagy in the pathogenesis of rheumatoid arthritis: latest evidence and therapeutic approaches. Life Sci. 2020;254:117734. doi:10.1016/j.lfs.2020.117734
18. Wang X, Dykens JA, Perez E, et al. Neuroprotective effects of 17β-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH Cells. Mol Pharmacol. 2006;70(1):395–404. doi:10.1124/mol.106.022384
19. Wang S, Deng Z, Ma Y, et al. The role of autophagy and mitophagy in bone metabolic disorders. Int J Biol Sci. 2020;16(14):2675–2691. doi:10.7150/ijbs.46627
20. Xu X, Li X, Liang Y, et al. Estrogen modulates cartilage and subchondral bone remodeling in an ovariectomized rat model of postmenopausal osteoarthritis. Med Sci Monit. 2019;25:3146–3153. doi:10.12659/MSM.916254
21. Zhang Y, Qian X, Yang X, et al. ASIC1a induces synovial inflammation via the Ca(2+)/NFATc3/RANTES pathway. Theranostics. 2020;10(1):247–264. doi:10.7150/thno.37200
22. Song M, Li C, Liu X, Xiao J, Zhao H. Effect of 17β-oestradiol on T-type calcium channels in the lateral habenula. J Neuroendocrinol. 2018;30(9):e12629. doi:10.1111/jne.12629
23. Popolo A, Piccinelli AL, Morello S, et al. Cytotoxic activity of nemorosone in human MCF-7 breast cancer cells. Can J Physiol Pharmacol. 2011;89(1):50–57. doi:10.1139/y10-100
24. Yagi J, Wenk H, Naves L, McCleskey E. Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ Res. 2006;99(5):501–509. doi:10.1161/01.RES.0000238388.79295.4c
25. Qu Z, Liu -T-T, Ren C, et al. 17β-estradiol enhances ASIC activity in primary sensory neurons to produce sex difference in acidosis-induced nociception. Endocrinology. 2015;156(12):4660–4671. doi:10.1210/en.2015-1557
26. Song S, Tao -J-J, Li S-F, et al. 17β-estradiol attenuates rat articular chondrocyte injury by targeting ASIC1a-mediated apoptosis. Mol Cell Endocrinol. 2020;505:110742. doi:10.1016/j.mce.2020.110742
27. Axmann R, Böhm C, Krönke G, et al. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009;60(9):2747–2756. doi:10.1002/art.24781
28. Pfeilschifter J, Koditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause. Endocr Rev. 2002;23:90–119. doi:10.1210/edrv.23.1.0456
29. Ding C, Parameswaran V, Udayan R, Burgess J, Jones G. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: a longitudinal study. J Clin Endocrinol Metab. 2008;93(5):1952–1958. doi:10.1210/jc.2007-2325
30. Zhang Y, Ren G, Guo M, et al. Synergistic effects of interleukin-1beta and interleukin-17A antibodies on collagen-induced arthritis mouse model. Int Immunopharmacol. 2013;15(2):199–205. doi:10.1016/j.intimp.2012.12.010
31. Nishimoto N, Yoshizaki K, Miyasaka N, et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50(6):1761–1769. doi:10.1002/art.20303
32. Sadik CD, Kim ND, Alekseeva E, Luster AD. IL-17RA signaling amplifies antibody-induced arthritis. PLoS One. 2011;6(10):e26342. doi:10.1371/journal.pone.0026342
33. Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi:10.1084/jem.20061775
34. Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41(2):84–91. doi:10.1016/j.cyto.2007.09.014
35. Mangnus L, van Steenbergen HW, Lindqvist E, et al. Studies on ageing and the severity of radiographic joint damage in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):222. doi:10.1186/s13075-015-0740-0
36. Daoussis D, Andonopoulos AP, Liossis SN. Wnt pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin Arthritis Rheum. 2010;39(5):369–383. doi:10.1016/j.semarthrit.2008.10.008
37. Paulissen SM, van Hamburg JP, Davelaar N, et al. Synovial fibroblasts directly induce Th17 pathogenicity via the cyclooxygenase/prostaglandin E2 pathway, independent of IL-23. J Immunol. 2013;191(12):1364–1372. doi:10.4049/jimmunol.1300274
38. Zhao R. Immune regulation of bone loss by Th17 cells in oestrogen-deficient osteoporosis. Eur J Clin Invest. 2013;43(11):1195–1202. doi:10.1111/eci.12158
39. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–898. doi:10.1056/NEJMra0707449
40. Witowski J, Ksiazek K, Jorres A. Interleukin-17: a mediator of inflammatory responses. Cell Mol Life Sci. 2004;61(5):567–579. doi:10.1007/s00018-003-3228-z
41. Feng Y, Madungwe N, da Cruz Junho C, Bopassa J. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol. 2017;174(23):4329–4344. doi:10.1111/bph.14033
42. Lu Y, Jiang Q, Yu L, et al. 17β-estradiol rapidly attenuates P2X3 receptor-mediated peripheral pain signal transduction via ERα and GPR30. Endocrinology. 2013;154(7):2421–2433. doi:10.1210/en.2012-2119
43. Small K, Nag S, Mokha S. Activation of membrane estrogen receptors attenuates opioid receptor-like1 receptor-mediated antinociception via an ERK-dependent non-genomic mechanism. Neuroscience. 2013;255:177–190. doi:10.1016/j.neuroscience.2013.10.034
44. Zimmerman M, Budish R, Kashyap S, Lindsey S. GPER-novel membrane oestrogen receptor. Clin Sci (Lond). 2016;130(12):1005–1016. doi:10.1042/cs20160114
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.