Back to Journals » OncoTargets and Therapy » Volume 11
Estrogen affects cell growth and IGF-1 receptor expression in renal cell carcinoma
Authors Sun L, Gao Z, Luo L, Tan H, Zhang G ![]()
Received 24 April 2018
Accepted for publication 29 June 2018
Published 17 September 2018 Volume 2018:11 Pages 5873—5878
DOI https://doi.org/10.2147/OTT.S172149
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Federico Perche
Lijiang Sun,* Zhemin Gao,* Lei Luo, Hailin Tan, Guiming Zhang
Department of Urology, The Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China
*These authors contributed equally to this work
Purpose: Both obesity and gender are important etiological factors in renal cell carcinoma (RCC) development, suggesting a pivotal role of sex hormone signaling pathway and insulin-like growth factor (IGF) family in RCC carcinogenesis. Here, we aimed to investigate the effect of estrogen on RCC growth and the possible interaction between estrogen/estrogen receptor (ER) signaling pathway and the IGF axis.
Methods: ER-α and ER-β were detected in four human RCC cell lines. Cells were treated with 17β-estradiol (E2), and cell proliferation was determined using the cell counting kit-8 assay. Using siRNA, ER-β was downregulated in RCC cells and the effect of E2 on cell growth and IGF-1 receptor (IGF-1R) expression was examined.
Results: E2 inhibited 786-O cell but not A498 cell growth significantly. After the downregulation of ER-β, E2 showed no obvious inhibitory role in 786-O cells. E2 stimulation increased the expression of IGF-1R in 786-O cells. Downregulation of ER-β, as well as fulvestrant, attenuated the stimulatory effect of E2 on IGF-1R expression.
Conclusion: Our results revealed that estrogen induced RCC growth inhibition via an ER-β-dependent pathway. Estrogen also upregulated the expression of IGF-1R, suggesting a link between estrogen/ER and IGF axis.
Keywords: renal cell carcinoma, estrogen, insulin-like growth factor-1 receptor
Introduction
Renal cell carcinoma (RCC) is a common malignancy derived from kidney parenchyma, which accounts for 2%–3% of all adult cancers.1 Nearly one-third of RCC patients develops recurrence and/or metastasis after complete surgical removal of the primary tumors.2 RCC is characterized by a very low sensitivity to chemotherapy and radiotherapy. Recent development of targeted therapy has yielded improved overall survival in patients with late-stage RCC, however, with limited long-term prognosis. Therefore, a deeper insight into the mechanisms underlying RCC initiation and progression is required.
RCC shows obvious gender differences in several aspects. First, the incidence of RCC in males is about twice higher than that in females.3 Second, the RCC incidence increases in females after hysterectomy.4 Third, chromophobe RCC is more likely to be diagnosed in females while papillary RCC is less likely to be in females, compared with clear cell RCC as the major histological subtype of RCC.5 Fourth, a recent study reported that premenopausal female RCC patients showed lower cancer-specific mortality compared with their male counterparts, while the gender disparities were diminished and even reversed in postmenopausal women.6 These differences supposed that estrogen might play an inhibitory effect on RCC development. Estrogen functions through estrogen receptors (ERs), including ER-α and ER-β. Yu et al4 found that ER-β might act as a tumor suppressor in RCC: upregulation of ER-β could reduce RCC cell growth rate and inhibit cell migration in vitro.
Obesity has been demonstrated to be a definite etiology of RCC. Obesity is associated not only with increased long-term RCC risk but also with adverse oncological outcome.7,8 However, recent studies have found a phenomenon in terms of “the obesity paradox”. Zhang et al9 performed a meta-analysis and concluded that obesity is related to favorable cancer-specific postoperative survival in RCC patients. The obesity paradox suggests a complicated role and mechanisms by which obesity is involved in RCC development. Obesity can trigger a cascade of secondary metabolic disorders, such as dyslipidemia and diabetes. These pathologies, either alone or in combination, can promote RCC carcinogenesis through a series of complicated signaling pathways, among which the insulin-like growth factor (IGF) family functions as an important molecular mediator.10 IGF-1 and IGF-1 receptor (IGF-1R) are crucial members of the IGF family and have been investigated in many studies to understand their role in RCC. IGF-1, which is bound to IGF-1R, can stimulate tumor angiogenesis, promote cell mitosis and migration, and inhibit cell apoptosis.11,12 Previous studies have revealed the crosstalk between IGF-1R and ER signaling in breast cancer.13 Kanagaraj et al14 reported that estrogen could exert its anticancer function through affecting IGF system in prostate cancer. In this study, we aimed to find the evidence for an interaction between estrogen/ER pathway and the IGF family, providing a possible explanation and deeper understanding of RCC evolution.
Materials and methods
Cell lines and reagents
Four human RCC cell lines (786-O, ACHN, A498, and Caki-1) were obtained from the Institute of Cell Research of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and used for <3 months after resuscitation. 786-O cells were cultured in Roswell Park Memorial Institute 1640 medium, ACHN and A498 cells were cultured in minimum Eagle’s medium, and Caki-1 cells were cultured in McCoy’s 5A medium. All cells were maintained in the media (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) in CO2 incubator at 37°C. Before use, cells were cultured in serum-free media for 24 hours, aiming to excluding the effect of exogenous estrogen.
The following reagents and antibodies were used: 17β-estradiol (E2) and fulvestrant (Sigma-Aldrich Co., St Louis, MO, USA), cell counting kit (CCK)-8 kit (Dojindo, Shanghai, People’s Republic of China), Lipofectamine 2000 (Thermo Fisher Scientific), rabbit polyclonal antibodies against ER-α, ER-β, and IGF-1R (Abcam, Cambridge, MA, USA), mouse monoclonal antibody against β-actin and horseradish peroxidase (HRP)-conjugated secondary antibodies (goat antimouse IgG HRP and goat antirabbit IgG HRP) (Santa Cruz Biotechnology Inc., Dallas, TX, USA), and protease inhibitors (Hoffman-La Roche Ltd., Basel, Switzerland). E2 was dissolved in absolute ethanol and stored at −5°C in the dark. Before use, E2 was prepared with a working concentration of 10 nM and fulvestrant was prepared with a working concentration of 1 μM.
siRNA transfection
Three pairs of the ER-β-targeted siRNA were chemically synthesized in Genepharma (Shanghai, People’s Republic of China). The sequence of siRNA that was chosen for further experiments because of its high efficiency in reducing ER-β expression was as follows: 5′-CACUUCUGCGCUGUCUGCAGCGAUU-3′. siRNA transfection was performed using Lipofectamine 2000 according to the manufacturer’s instruction. Briefly, cells were seeded in six-well plate and cultured overnight. Then, the transfection complex containing Lipofectamine 2000 and diluted siRNA was added to the plate, followed by continued incubation for 48 hours. Then, the transfected cells were collected for further experiments.
Cell proliferation assay
Cell proliferation was determined using the CCK-8 assay. Cells were seeded in 96-well plates. Twenty-four hours later, the media were replaced by those mixed with CCK-8 (10:1). After incubation for 2 hours, absorbance was measured by a microplate reader at 450 nm.
Western blotting analysis
Western blotting analysis was performed as described previously.15 Briefly, cells were harvested using the CelLytic extraction kit supplemented with protease inhibitors. Protein concentration was measured using the BCA Protein Assay reagent kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. After separated by SDS-PAGE in 12% (w/v) polyacrylamide gels, proteins were transferred to polyvinylidene fluoride membranes and blocked in 5% nonfat milk. Then, the primary antibodies and secondary antibodies were used to probe the target proteins. Immunoreactivity was then visualized by the ECL Plus Western Blotting System (Thermo Fisher Scientific).
Statistical analyses
All experiments were performed in triplicate. Data were expressed as mean ± SD. Comparison between groups was performed using Student’s t-test. A two-sided P-value of <0.05 was considered statistically significant. Statistical analyses were performed with STATA Version 12.0.
Results
The expression levels of ER-α and ER-β in four human RCC cell lines were detected by Western blotting. As shown in Figure 1, all cell lines had no ER-α protein expression and positive ER-β expression. Among the four cell lines, 786-O cells and A498 cells indicated the strongest and weakest ER-β expressions, respectively (Figure 1A and B). Therefore, we chose 786-O cells and A498 cells for further experiments.
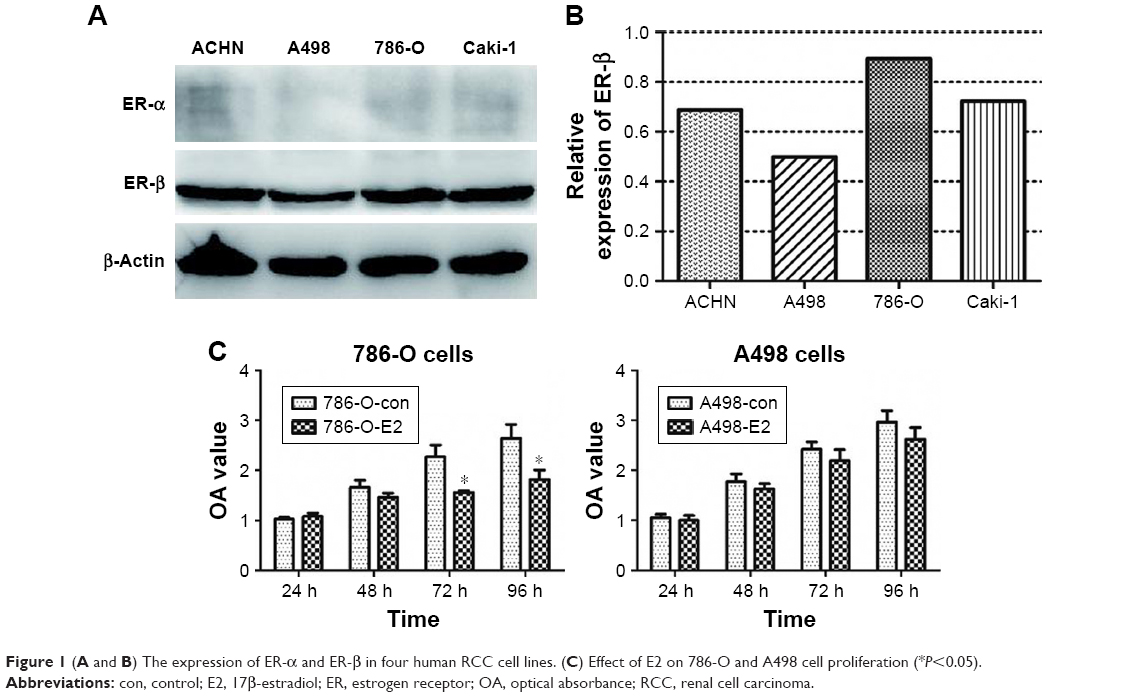 | Figure 1 (A and B) The expression of ER-α and ER-β in four human RCC cell lines. (C) Effect of E2 on 786-O and A498 cell proliferation (*P<0.05). |
The effect of E2 on 786-O and A498 cell viabilities was tested using the CCK-8 assays. The results showed that E2 inhibited 786-O cell growth significantly 72 hours after E2 administration, while there was no obvious difference between the E2 group and the control group in A498 cells (Figure 1C). Then, we downregulated the expression of ER-β in 786-O cells by siRNA, and the effect of downregulation is shown in Figure 2. Because of its highest efficiency in decreasing ER-β expression (reduced by 46%), siER-β3 was selected for further experiments.
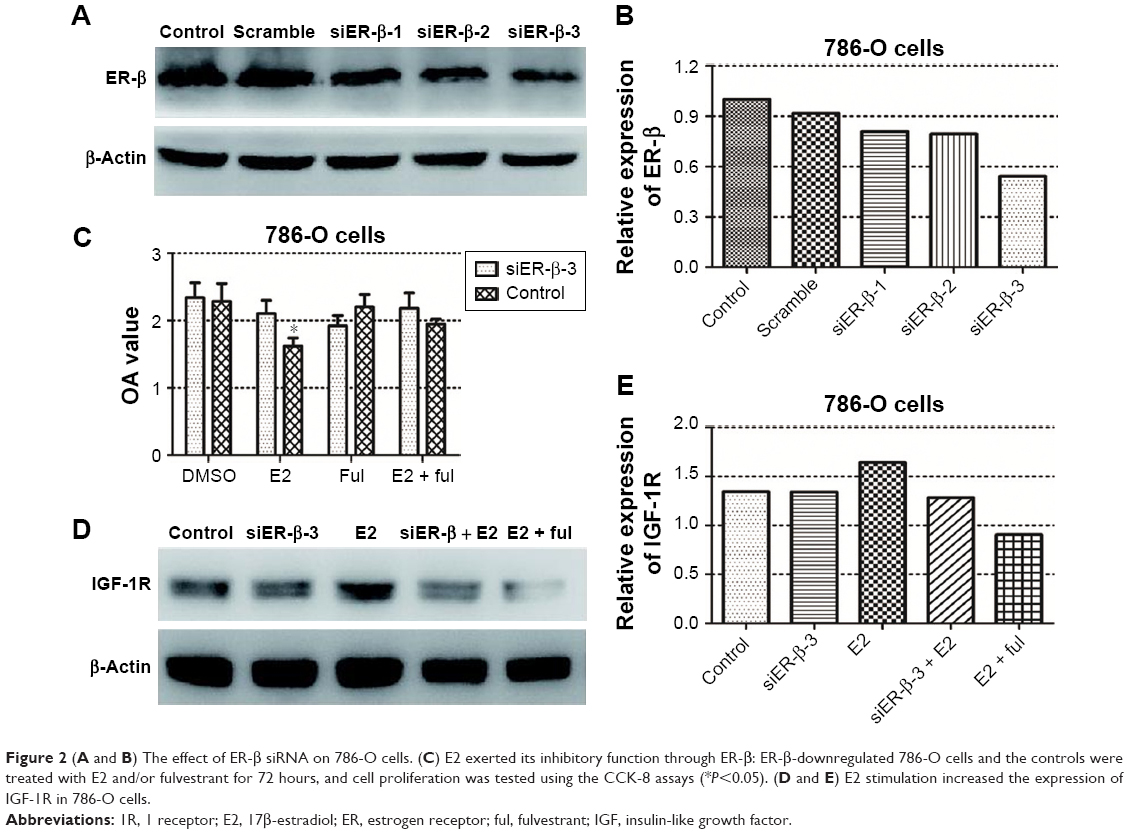 | Figure 2 (A and B) The effect of ER-β siRNA on 786-O cells. (C) E2 exerted its inhibitory function through ER-β: ER-β-downregulated 786-O cells and the controls were treated with E2 and/or fulvestrant for 72 hours, and cell proliferation was tested using the CCK-8 assays (*P<0.05). (D and E) E2 stimulation increased the expression of IGF-1R in 786-O cells. |
Next, we investigated whether E2 exerted its inhibitory function through ER-β. As shown in Figure 2C, E2 reduced 786-O cell growth rate significantly. After the downregulation of ER-β, the inhibitory effect of E2 could not be taken. In addition, this inhibitory effect could be reversed by estrogen inhibitor (fulvestrant). All these results suggest that E2 inhibits RCC cell viability through ER-β.
Then, we examined the relationship between estrogen/ER pathway and the IGF-1R. E2 stimulation increased the expression of IGF-1R by 23% in 786-O cells. Downregulation of ER-β, as well as fulvestrant, attenuated the stimulatory effect of E2 on IGF-1R expression (Figure 2D, E2), suggesting a crosstalk between estrogen/ER-β and IGF axis.
Discussion
The present study confirms the antiproliferative effect of estrogen in 786-O cells. This inhibitory effect was not observed in A498 cells, which showed relatively lower expression of ER-β. In ER-β-downregulated 786-O cells, the antiproliferative effect of estrogen was not found, either. All these results revealed an important role of ER-β in RCC viability. We also proved an association between estrogen/ER-β and IGF axis, suggesting a complicated mechanism linking estrogen, obesity, and RCC carcinogenesis.
The role of estrogen in cancer development remains controversial. Recent studies have demonstrated that estrogen promotes the malignant progression not only in target organs, such as breast cancer, but also in other nontarget organs, such as lung cancer.16,17 However, other studies reported that estrogen can inhibit cell growth, induce cell apoptosis in prostate cancer, and exert a protective function in laryngeal cancer and colorectal cancer.14,18,19 A possible explanation is the ununiform expression of ERs in different organs. The expression subtypes and levels of ER are primary elements that determine organ-specific estrogen responsiveness. After binding to ER-α, estrogen initiates a series of complicated biological processes involving the transcription of growth-related factors, which enhance gene expression and mitosis, resulting in tumor development. As far as ER-β is concerned, previous studies have reported that ER-β indicated antiproliferative and apoptosis-inducing potential.20,21 Yu et al4 investigated the expression of ER-α and ER-β in kidney and RCC tissues and observed the negative expression of ER-α in normal kidney and RCC tissues, as well as relatively lower expression of ER-β in RCC tissues.4 Using immunohistochemical staining, our previous study also found positive expression of ER-β in 22.58% of clear cell RCC specimens (data not shown). In the present study, we observed negative expression of ER-α and positive expression of ER-β in four RCC cell lines. Therefore, we deduced that estrogen might take its effect on RCC through ER-β rather than through ER-α. Further experiments confirmed our speculation. Exogenous estrogen administration could inhibit RCC cell growth in 786-O cells and not in A498 cells. In addition, in ER-β-downregulated 786-O cells, we did not observe the inhibitory effect of estrogen. Taken together, we speculated that estrogen inhibits RCC cell proliferation through ER-β.
Previous studies have deeply investigated the role of IGF family in cancer development, including RCC. Compared with normal kidney tissue, RCC tissue indicated high expression levels of IGF-1R.22 IGF-1R activation is considered as prerequisite for malignant transformation.23 Transgenic overexpression of IGF-1R can induce mammary epithelial hyperplasia and tumor formation in vivo.24 Cardillo et al25 reported that targeting both IGF-1R and mTOR synergistically inhibited the growth of RCC in vitro. Thus, IGF-1R serves as an oncogenic mediator in RCC. According to previous studies, there exists an interaction between estrogen/ER signaling and IGF axis. Estrogen may regulate the expression of key molecules in the IGF axis.17 In a hamster model, estrogen stimulation could induce the elevated expression of IGF-1R.26 Administration of estrogen with IGF-1 could promote lung adenocarcinoma development in vivo.17 In the present study, we validated that estrogen stimulation increased the expression of IGF-1R in 786-O cells. Downregulation of ER-β, as well as estrogen inhibitor, attenuated the stimulatory effect of estrogen on IGF-1R expression. These results seemed to be self-contradictive: estrogen can inhibit RCC cell growth and promote IGF-1R expression. IGF-1R is a positive stimulatory factor for RCC development. There are several possible explanations: first, whether a series of signaling reactions can be initiated by IGF-1R without the stimulation of IGF-1 is unknown; second, the promotion effect of IGF-1R might be weakened by the inhibitory effect of estrogen; and third, our results were based on the findings in only one cell line and whether there are different phenomena in other cell lines or in vivo remains unclear. Thus, further experiments about the complicated crosstalk between estrogen/ER-α/ER-β and the IGF family, as well as their role in RCC carcinogenesis, are warranted.
Conclusion
Our results demonstrated that estrogen induced RCC growth inhibition via an ER-β-dependent pathway. Estrogen also upregulated the expression of IGF-1R, suggesting a link between estrogen/ER and IGF axis. These data revealed the role of ER-β as a tumor suppressor and facilitated the potential estrogen-based therapeutic strategies in RCC.
Acknowledgments
This study was partly funded by the National Natural Science Foundation of China (grant numbers NSFC 81502195 and NSFC 81672512) and the Medicine and Health Science Technology Development Project of Shandong Province (number 2016WS0258).
Disclosure
The authors report no conflicts of interest in this work.
References
International Agency for Research on Cancer; World Health Organization. Cancer Today. GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Available from: http://globocan.iarc.fr/Default.aspx. Accessed August 22, 2018. | ||
Rydzanicz M, Wrzesiński T, Bluyssen HA, Wesoły J. Genomics and epigenomics of clear cell renal cell carcinoma: recent developments and potential applications. Cancer Lett. 2013;341(2):111–126. | ||
Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212–236. | ||
Yu CP, Ho JY, Huang YT, et al. Estrogen inhibits renal cell carcinoma cell progression through estrogen receptor-β activation. PLoS One. 2013;8(2):e56667. | ||
Lipworth L, Morgans AK, Edwards TL, et al. Renal cell cancer histological subtype distribution differs by race and sex. BJU Int. 2016;117(2):260–265. | ||
Qu Y, Chen H, Gu W, et al. Age-dependent association between sex and renal cell carcinoma mortality: a population-based analysis. Sci Rep. 2015;5:9160. | ||
Zhai T, Zhang B, Qu Z, Chen C. Elevated visceral obesity quantified by CT is associated with adverse postoperative outcome of laparoscopic radical nephrectomy for renal clear cell carcinoma patients. Int Urol Nephrol. 2018;50(5):845–850. | ||
Leiba A, Kark JD, Afek A, et al. Adolescent obesity and paternal country of origin predict renal cell carcinoma: a cohort study of 1.1 million 16 to 19-year-old males. J Urol. 2013;189(1):25–29. | ||
Zhang J, Chen Q, Li ZM, Xu XD, Song AF, Wang LS. Association of body mass index with mortality and postoperative survival in renal cell cancer patients, a meta-analysis. Oncotarget. 2018;9(17):13959–13970. | ||
Zhang GM, Zhu Y, Ye DW. Metabolic syndrome and renal cell carcinoma. World J Surg Oncol. 2014;12:236. | ||
Ibrahim YH, Yee D. Insulin-like growth factor-I and cancer risk. Growth Horm IGF Res. 2004;14(4):261–269. | ||
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56(4):549–580. | ||
Fagan DH, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(4):423–429. | ||
Kanagaraj P, Vijayababu MR, Ilangovan R, et al. Effect of 17beta-estradiol on apoptosis, IGF system components and gelatinases A and B in prostate cancer cells (PC-3). Clin Chim Acta. 2007;377(1–2):70–78. | ||
Zhang GM, Luo L, Ding XM, et al. MicroRNA-126 inhibits tumor cell invasion and metastasis by downregulating ROCK1 in renal cell carcinoma. Mol Med Rep. 2016;13(6):5029–5036. | ||
Chlebowski RT, Anderson GL. Changing concepts: menopausal hormone therapy and breast cancer. J Natl Cancer Inst. 2012;104(7):517–527. | ||
Tang H, Liao Y, Xu L, et al. Estrogen and insulin-like growth factor 1 synergistically promote the development of lung adenocarcinoma in mice. Int J Cancer. 2013;133(10):2473–2482. | ||
Schwartz N, Verma A, Muktipaty C, Bivens C, Schwartz Z, Boyan BD. Estradiol receptor profile and estrogen responsiveness in laryngeal cancer and clinical outcomes. Steroids. Epub December 20, 2017. | ||
Ji J, Sundquist J, Sundquist K. Use of hormone replacement therapy improves the prognosis in patients with colorectal cancer: a population-based study in Sweden. Int J Cancer. 2018;142(10):2003–2010. | ||
Treeck O, Lattrich C, Springwald A, Ortmann O. Estrogen receptor beta exerts growth-inhibitory effects on human mammary epithelial cells. Breast Cancer Res Treat. 2010;120(3):557–565. | ||
Lindberg K, Helguero LA, Omoto Y, Gustafsson JÅ, Haldosén LA. Estrogen receptor β represses Akt signaling in breast cancer cells via downregulation of HER2/HER3 and upregulation of PTEN: implications for tamoxifen sensitivity. Breast Cancer Res. 2011;13(2):R43. | ||
Braczkowski R, Białożyt M, Plato M, Mazurek U, Braczkowska B. Expression of insulin-like growth factor family genes in clear cell renal cell carcinoma. Contemp Oncol. 2016;20(2):130–136. | ||
Werner H. Tumor suppressors govern insulin-like growth factor signaling pathways: implications in metabolism and cancer. Oncogene. 2012;31(22):2703–2714. | ||
Jones RA, Campbell CI, Gunther EJ, et al. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26(11):1636–1644. | ||
Cardillo TM, Trisal P, Arrojo R, Goldenberg DM, Chang CH. Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro. BMC Cancer. 2013;13:170. | ||
Narayan S, Roy D. Insulin-like growth factor 1 receptors are increased in estrogen-induced kidney tumors. Cancer Res. 1993;53(10 suppl):2256–2259. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.