")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Estimating the Prevalence of AATD Patients in the UK to Identify Underdiagnosis and Determine the Eligibility for Potential Augmentation Therapy
Authors Newnham M , Quinn M, Turner AM
Received 10 January 2023
Accepted for publication 13 May 2023
Published 13 June 2023 Volume 2023:18 Pages 1197—1205
DOI https://doi.org/10.2147/COPD.S395663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Michael Newnham,1,2 Mark Quinn,1 Alice M Turner1,2
1Institute of Applied Health Research, University of Birmingham, Birmingham, UK; 2University Hospitals Birmingham, Birmingham, UK
Correspondence: Alice M Turner, Institute of Applied Health Research, University of Birmingham, Birmingham, UK, Email [email protected]
Purpose: Alpha 1 antitrypsin deficiency (AATD) is a genetic risk factor for chronic obstructive pulmonary disease (COPD). Whilst testing for the condition is relatively simple, there is a disconnect in published literature between genetic epidemiology and numbers of patients known to specialists. This makes planning services for patients difficult. We aimed to estimate the number of patients likely to have lung disease eligible for specific AATD therapy within the UK.
Patients and Methods: The THIN database was used to determine the prevalence of AATD and symptomatic COPD. This, and published rates of AATD were used to extrapolate THIN data to the population size of the UK to give an indicative population size for symptomatic AATD patients who have lung disease. The Birmingham AATD registry was used to describe age at diagnosis, rate of lung disease and symptomatic lung disease for patients with PiZZ (or equivalent) AATD, together with the time from symptom onset to diagnosis, in order to aid interpretation of the THIN data and improve modeling.
Results: THIN data showed COPD prevalence of 3%, and AATD prevalence of 0.005– 0.2%, depending on how stringently AATD diagnostic codes were applied. The majority of Birmingham AATD patients were diagnosed between the ages 46– 55, whilst patients recorded in THIN tended to be older. The rate of COPD was similar in the THIN and Birmingham patients diagnosed with AATD. Modelling to the size of the UK demonstrated a likely symptomatic AATD population of between 3016 and 9866 people.
Conclusion: AATD is likely to be under-diagnosed in the UK. Based on projected patient numbers an expansion to specialist services is desirable, in particular if specific therapy for AATD such as augmentation were to be introduced to the healthcare system.
Keywords: alpha 1 antitrypsin deficiency, chronic obstructive pulmonary disease, epidemiology
Introduction
AATD is an autosomal, co-dominant inherited genetic disorder that may result in lung or liver disease; AAT is encoded by the SERPINA1 gene and mutations in this gene cause deficiency. The normal allele is designated M and the normal genotype designated PiMM, with common deficiency genotypes being PiZZ and PiSZ. Symptoms of alpha-1 associated lung disease resemble those of classic chronic obstructive pulmonary disease (COPD) or emphysema. Therefore it remains undiagnosed in many patients and the underlying cause may not get treated; supporting this, a Europe wide survey of clinicians managing AATD patients found that most (77%) believed only 15% of AATD cases were diagnosed.1 Underdiagnosis was further demonstrated when comparing the clinicians’ estimates for confirmed cases of AATD diagnosis to published prevalence: on average the clinicians’ estimates were 90% lower than the figures ascertained from epidemiological studies.2 The diagnostic delay between symptom onset and diagnosis of AATD is generally reported as over 5 years and can result in worse clinical outcomes.3 To decrease the number of un- or misdiagnosed patients, guidelines recommend testing every COPD patient for AATD,4 although in practice low awareness and the cost of testing can result in low uptake.1 Nevertheless, even if prevalence is known, it will remain unclear how many individuals with severe AATD will develop severe lung disease during their life, which is the main indication for the only currently licensed specific treatment for PiZZ patients, namely intravenous AAT replacement, known as augmentation therapy.5 One approach to this would be newborn screenings with regular follow-ups to determine disease development. The onset of respiratory disease in smokers with AATD is characteristically between ages 40 and 50; in non-smokers, the onset can be delayed to the sixth decade.6 Therefore, this approach will take decades in AATD and carries the risk of bias due to lost follow-up visits from participants. Data from this approach in Sweden when participants were aged 34 showed little evidence of lung disease, and few differences according to genotype or smoking status,7 implying that follow up was required for even longer.
Another approach could be to use existing registries that are designed to prospectively collect data from a large sample of individuals followed in real life. The Birmingham AATD registry program was established in 1996 and includes many highly characterized patients with a range of AATD genotypes. The inclusion of both index (symptomatic) and non-index (typically diagnosed through family screening) patients, smokers and never smokers provides a unique opportunity to study progression. Use of registry data that describes time from symptoms to diagnosis, and age at onset of lung disease, together with population based data on AATD and COPD prevalence gives a route to estimate numbers of “healthy, not diagnosed”, “symptomatic lung disease, not diagnosed”, “diagnosed AATD with and without lung disease”. In this study we aimed to describe onset of lung disease in the Birmingham registry, as well as rate of symptomatic COPD and diagnosed AATD in a primary care database representative of the UK population (THIN). Using these figures and extrapolating to the population size of the country we estimate the numbers who might be present, and eligible for augmentation therapy, whether diagnosed or not, to inform health service design.
Materials and Methods
THIN Data
THIN (The Health Improvement Network) is a nationally representative electronic primary care records database that contains anonymized medical records of over 15 million patients from GP practices in the UK.8 Diagnoses of morbidities, symptoms observed, surgical procedures performed, and referrals made are systematically recorded in primary care using a hierarchical coding system called Read Codes; those pertaining to AATD, and COPD were selected (Tables S1 and S2). We adopted a case: control design within THIN where cases were defined by having COPD and controls no COPD, matching adult (age ≥18) patients by age, sex and ethnicity in a 1:2 ratio. Study flow is illustrated in Figure S1. THIN has multiple diagnostic codes relating to AATD, some of which relate to testing and others to genotype or clinical phenotypes. Using all Read Codes (Table S1) that mentioned “alpha-1-antitrypsin” gives a broad view of possible AATD patients (used to define the upper end of our estimated case numbers for modelling). A more stringent selection of codes (“Alpha-1-antitrypsin deficiency”) was used to define the lower bound of our modelled case number range.
Birmingham Registry Data
Processes for assessment of patients within the Birmingham registry have been described previously,9 but in brief comprise annual structured clinical history, lung function and liver assessment. For the following analyses the cohort (starting n=1410) has been filtered on (i) phenotype/genotype of AATD ZZ or Z null (n=998 individuals remaining) (ii) Liver disease and other (non-pulmonary) index cases removed (n=944 individuals remaining). Symptoms indicative of lung disease, time of onset of symptoms, COPD diagnosis and AATD diagnosis were noted, alongside demographic and pulmonary disease characteristics.
Modelling of Likely Patient Numbers
Data was taken from the 2011 UK census (https://www.ethnicity-facts-figures.service.gov.uk/uk-population-by-ethnicity/demographics/age-groups/latest, accessed 21st June 2021) to show the number of people in the age categories described within THIN and Birmingham AATD data. Data was then taken from Blanco et al10 for the likely prevalence of PiZZ AATD in England (1:4440), as this country comprises the largest proportion of THIN cohort patients. The proportion of the population covered by THIN was calculated by dividing the census population by the total patients registered within THIN and estimating AATD patients by dividing the census population by 4440. We used the stringent and permissive AATD codes to generate lower and upper bound of AATD case numbers. If we assume that all cases in THIN (or extrapolated from THIN to be diagnosed in the UK) are symptomatic index cases, and that asymptomatic or non-index cases occur in the proportions observed in Birmingham data we can then estimate the number of additional cases likely to be symptomatic even though they have not been diagnosed yet.
Study Ethics
Data from the original Birmingham registry was approved by the South Birmingham Research Ethics Committee (REC Ref: 3359a) and subsequently by the South Central Oxford Research Ethics Committee (REC Ref: 18/SC/0541, IRAS Project ID: 233675). The use of the THIN data for research was approved by the South-East Multicenter Research Ethics Committee in 2003, without the need for informed consent. As per the requirements for ethical approval, further registration and authorisation for this project were obtained from the Scientific Review Committee of the data provider. Data from the UK census was open access and anonymised and therefore separate ethical approval was not required.
Results
THIN Data
254911 COPD patients were identified, as well as 1754 AATD patients and 473,095 control patients (Figure S1), characteristics being summarised in Table 1. The majority of patients (63%, n=461,321) in the THIN cohort for this study were from England, with 19% (n=136,977) from Scotland, 13% (n=96,819) from Wales and 5% (n=34,643) from Northern Ireland. The vast majority of AATD patients were of Caucasian ethnicity and most had COPD. AATD patients comprised 0.2% of the whole and 0.5% of the COPD patients if using permissive AATD coding; when using more stringent coding these proportions were 0.005 and 0.2% respectively (n=534). Sensitivity analysis restricting results to Caucasian patients did not make a substantial difference to these proportions.
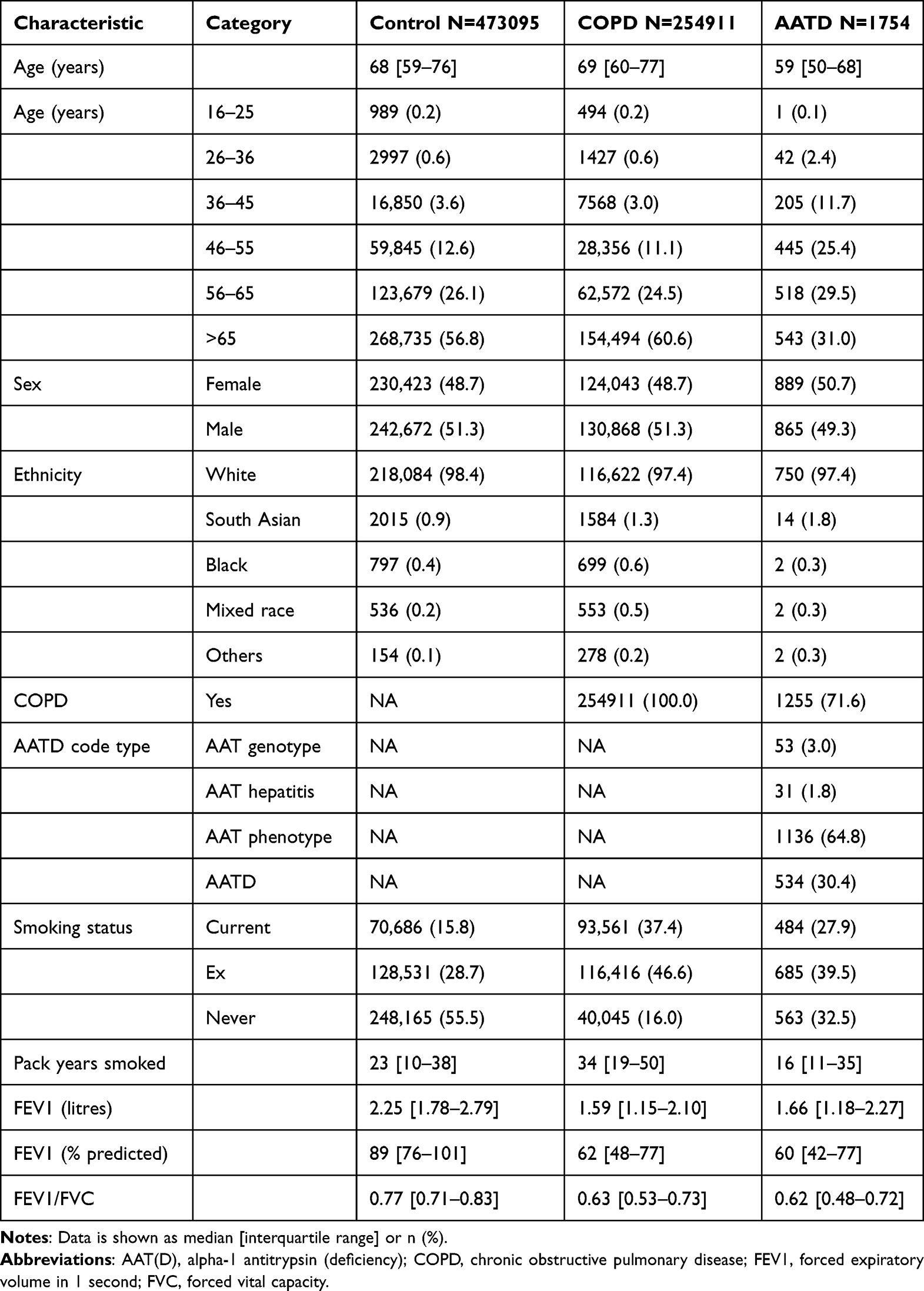 |
Table 1 Characteristics of Patients from the THIN Database |
Birmingham Registry Data
Table 2 shows demographics of 944 included patients. Median time from symptoms to diagnosis was 5 years, and from diagnosis to first assessment by an AATD specialist was 2 years. Age at baseline was generally younger than the coded AATD patients in THIN. Of these patients 998 were of genotype PiZZ/Znull, and after excluding liver index cases the final number of relevant patients was 944. Figure 1 shows the proportion of patients symptomatic and asymptomatic, with or without lung disease according to age and index status; most were symptomatic and had been diagnosed due to lung disease (index case). The proportion of AATD patients with COPD was higher when compared to the THIN data (88% vs 72%), but there were fewer current smokers. Whilst the majority of symptomatic cases had smoked, significant numbers of never smokers had developed lung disease, as shown in Figure 2.
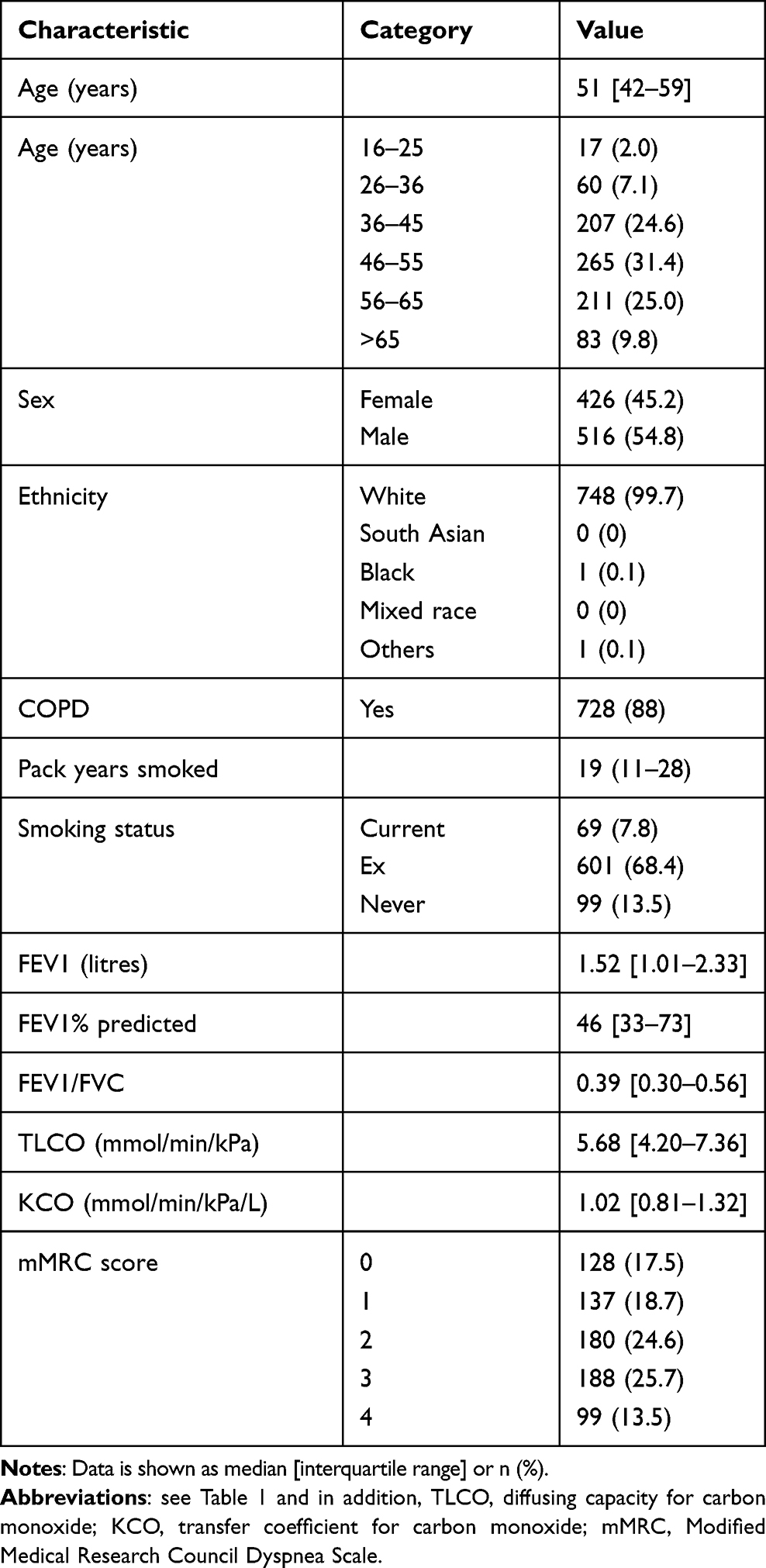 |
Table 2 Characteristics of Patients from the Birmingham Registry |
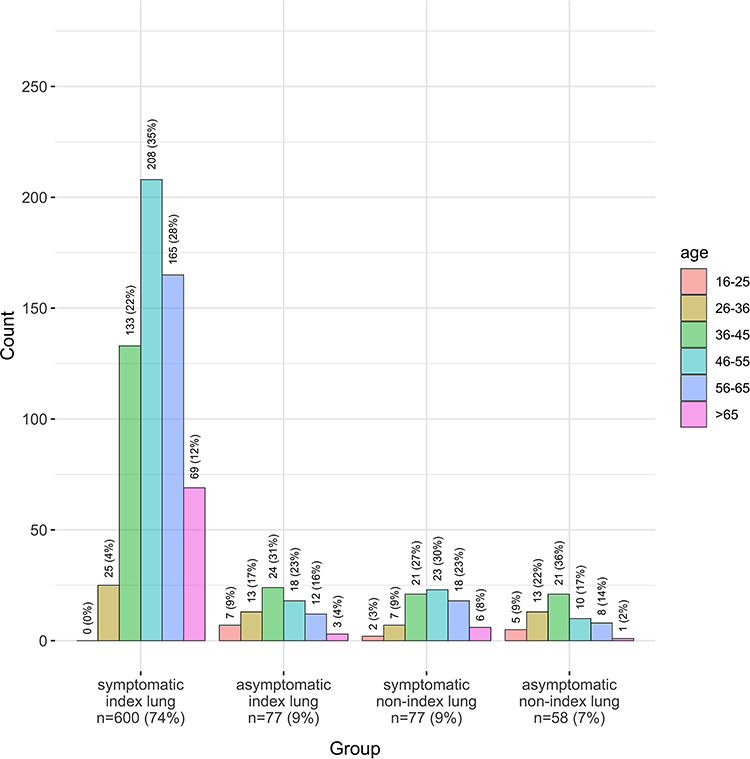 |
Figure 1 Prevalence of symptoms and lung disease according to age in the Birmingham AATD registry. |
 |
Figure 2 Prevalence of smoking status according to age and index status in the Birmingham AATD registry. (a) Symptomatic index lung (n=585); (b) Asymptomatic index lung (n=71); (c) Symptomatic non-index lung (n=74); (d) Asymptomatic non-index lung (n=58). |
Modelling of Likely Patient Numbers
There were n=56,075,912 recorded in the census with age distribution and comparison with COPD and AATD groups shown in Table 3. AATD patients in both THIN and Birmingham groups were younger than COPD THIN patients, but there were few diagnosed/recorded below the age of 36.
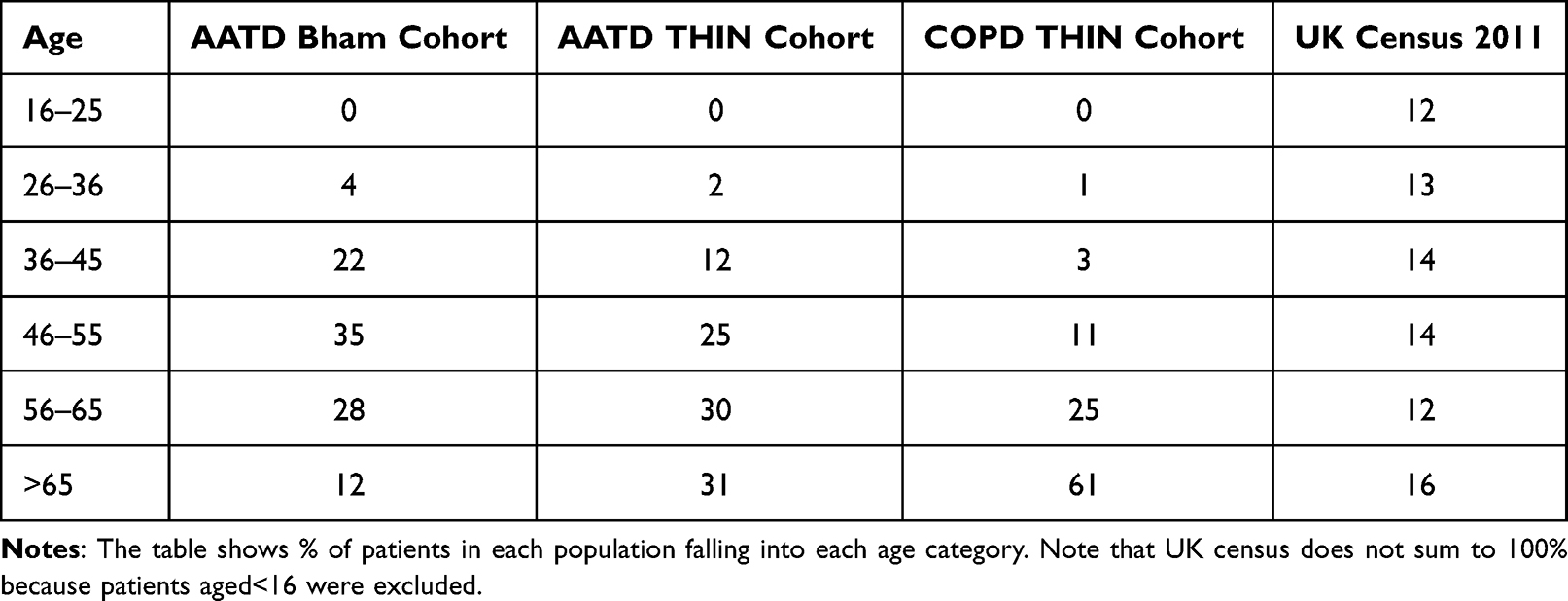 |
Table 3 Age Distribution of Patients in the UK Population, THIN & Birmingham (Bham) Cohorts After Exclusion of Liver Index Cases |
Whilst data on smoking status is available from the UK census it was difficult to categorise smoking status for comparison with the prior groups. 3% of the THIN cohort had COPD, which if extrapolated to the UK resulted in 1,613,607 individuals. Assuming 1:4440 of these have AATD, results in 12,630 AATD cases who have COPD. Since THIN represents 20% of the UK population and AATD prevalence was observed to be 0.005–0.2%, the likely number of diagnosed cases is 2700–8829. Thus undiagnosed patients number 3761–9930 (12,630 – (8829 or 2700). Since 9% of Birmingham patients were asymptomatic index cases, and a further 9% symptomatic non-index cases, if similar proportions are seen nationwide this results in a range of 316–1037 patients across the UK with AATD in each of these groups, and 245–806 asymptomatic non-index cases. Therefore overall the number of symptomatic lung disease patients lies between (2700–8829) + (316–1037) = 3016–9866.
Discussion
We have demonstrated that the majority of symptomatic AATD patients in the UK are likely to be undiagnosed, since the proportion of patients with AATD recorded in primary care is low, and diagnosis appears not to occur until after the age of 36 in most cases. Furthermore, whilst Birmingham is not the only specialist AATD centre in the UK, it seems unlikely that numbers known to specialist AATD services equal even the most conservative projected number, which has implications for specialist service delivery if diagnosis rates were improved. In particular, if planning for augmentation therapy according to its licensed indications, more specialist centres would likely be needed. Additionally, increased testing and diagnosis of AATD could facilitate more targeted COPD treatments including smoking cessation. The median diagnostic delay in our cohort was 5 years, which is similar to previous studies and as this has been associated with worse AATD outcomes, it further reiterates the health impact of underdiagnosis.1,3
Obstacles to early diagnosis of AATD, and indeed other rare diseases, are well recognized and have been reviewed elsewhere.11 The state of readiness of health systems for screening or case finding to be conducted, the availability of easily conducted tests, level of knowledge of patients and healthcare practitioners all play a part. Currently AATD does not meet internationally accepted criteria for screening, largely due to uncertainties about who will develop symptomatic disease, and lack of availability of disease modifying therapies useful in the early stages. Case finding, where tests are conducted in symptomatic patients, would likely be more acceptable, and indeed cost-effective, as in usual COPD.12 If the purpose is to find people eligible for current specific treatment (namely augmentation) then this approach seems relevant, as patients presenting with lung disease would come into contact with many healthcare professionals, over long periods of time hence opportunities for diagnosis via case finding do exist. Our data supports this, with patients under specialist care reporting delays in diagnosis often despite seeing both primary and secondary care clinicians.
Reliable testing methods exist in most developed healthcare systems, either through checking an AAT level and then phenotyping or genotyping,13 or by direct genotyping for common alleles, though in our experience some clinicians are uncertain how to access tests, or worried about costs of conducting them. Low rates of testing (or coding) in THIN might support a lack of knowledge of this type, or could be down to lack of awareness of AATD as a condition, implying that education on both fronts may be valuable. A public health campaign has been shown to increase rate of detection, but the fact that free testing occurred means any effect cannot be attributed to the campaign alone, as a reduced direct cost may have attracted people to be tested too.14 Point of care and patient led testing have also been studied and were associated with a higher detection rate than in general clinics, but most studies did not have a formal control, so effects are uncertain.14–16 Education of physicians was shown to result in an increase in AATD testing of COPD patients after written materials and a quiz to test knowledge were given,17 or after watching an educational video,18 however self-reporting of AAT test rates brings reliability of the data into question. It seems likely that a combination of healthcare policy around case finding, combined with education around AATD and how to test for it would all be required to ensure symptomatic patients were diagnosed reliably.
We found a relatively low prevalence of AATD within the THIN cohort; this is consistent with other data from UK primary care, suggesting that testing of COPD patients is uncommon, occurring in around 2% of COPD cases occurring at an age of <60 years (the population most likely to be tested in a targeted case finding approach).19 It has been suggested that up to 10% of Caucasian COPD patients may have AATD,20 but our data implies this may be an overestimate. The frequency of Read Codes for likely severe deficiency in our study was less than 1%, and was consistent with case finding work in Spain16 and the USA.21 Whilst we recognize the limits of coding as a means of diagnosis, our strategy of calculating likely case numbers from permissive and stringent code choices minimized risks of underdiagnosis, and it is notable that our permissive code strategy gave very similar results to prospective screening of a COPD population (0.5% v 0.63%21 of COPD cases). We used the more conservative codes for projection to nationwide numbers, hence if anything real numbers may be higher.
Our study has some strengths; by use of a large primary care dataset broadly representative of the UK as a whole alongside a specialist centre cohort we have been able to show that diagnostic delay for pulmonary patients appears consistent, in that over 80% of THIN patients were recorded over the age of 46. Birmingham data agreed with this in that patients reported diagnostic delay of several years from onset of symptoms; this is concordant with other AATD cohorts worldwide.22,23 However, THIN data is reliant on diagnostic coding, and it is possible that codes could be misused, in particular if practices are less familiar with them, as may well be the case for AATD. For example, patients that have less severe AAT genotypes (eg, PiMZ) may have been misclassified as AATD or alternatively patients with AATD may have been insufficiently captured by Read Codes and whilst using upper and lower bounds may have mitigated this it remains a potential limitation. Our modeling to the whole UK was reliant on a series of assumptions, and as such the range of possible cases was broad; whilst this is a weakness we do not believe it can be overcome with current methods of data collection about AATD. A centralized record of AAT test results across the UK could provide data on diagnoses, but not on symptoms or COPD diagnoses unless linked to other parts of the routine care record. Finally, some results may be explained by ascertainment bias – for example we excluded cases that were diagnosed due to liver disease, which has a bimodal peak of incidence,24 and theoretically could account for younger cases within the Birmingham cohort being excluded; whole cohort data in Table 1, and liver excluded age ranges in Table 2 demonstrate there was a change in demographics after exclusion, though not in the direction expected because more of our liver patients appear to have been diagnosed in later life. However, this does not apply to the THIN data where we would expect younger cases to remain coded, since exclusion on the basis of index status for liver disease was not possible. Exclusion by liver code alone would not be desirable as it could remove patients where liver disease was identified after a diagnosis of AATD.
In conclusion, AATD is underdiagnosed in the UK and increased testing and diagnosis could help to improve targeted COPD treatments. Projected case numbers suggest an expansion in AATD services is required to adequately manage patients according to current guidance.4
Acknowledgments
We would like to thank K Niranthakumar and T Marshall for access to THIN, as well as training in and use of the data extraction in epidemiological research (DExtER) tool.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by CSL Behring.
Disclosure
Dr Michael Newnham reports grants from CSL Behring, during the conduct of the study. Professor Alice M Turner reports grants from CSL Behring, during the conduct of the study; grants, personal fees from Grifols, grants, personal fees from Vertex, grants, personal fees from AstraZeneca, personal fees from GSK, grants from Chiesi, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Horvath I, Canotilho M, Chlumsky J, et al. Diagnosis and management of α 1 -antitrypsin deficiency in Europe: an expert survey. ERJ Open Res. 2019;5(1):00171–2018. doi:10.1183/23120541.00171-2018
2. de Serres FJ, Blanco I. Prevalence of alpha1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis. 2012;6(5):277–295. doi:10.1177/1753465812457113
3. Meischl T, Schmid-Scherzer K, Vafai-Tabrizi F, et al. The impact of diagnostic delay on survival in alpha-1-antitrypsin deficiency: results from the Austrian Alpha-1 Lung Registry. Respir Res. 2023;24(1):34. doi:10.1186/s12931-023-02338-0
4. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α 1 -antitrypsin deficiency. Eur Respir J. 2017;50(5):1700610. doi:10.1183/13993003.00610-2017
5. Edgar RG, Patel M, Bayliss S, Crossley D, Sapey E, Turner AM. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308. doi:10.2147/COPD.S130440
6. Stockley RA, Turner AM. alpha-1-Antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol Med. 2014;20(2):105–115. doi:10.1016/j.molmed.2013.11.006
7. Tanash HA, Nystedt-Duzakin M, Montero LC, Sveger T, Piitulainen E. The Swedish alpha1-Antitrypsin Screening Study: health Status and Lung and Liver Function at Age 34. Ann Am Thorac Soc. 2015;12(6):807–812. doi:10.1513/AnnalsATS.201410-452OC
8. Blak BT, Thompson M, Dattani H, Bourke A. Generalisability of The Health Improvement Network (THIN) database: demographics, chronic disease prevalence and mortality rates. Inform Prim Care. 2011;19(4):251–255. doi:10.14236/jhi.v19i4.820
9. Pillai AP, Turner AM, Stockley RA. Global Initiative for Chronic Obstructive Lung Disease 2011 symptom/risk assessment in alpha1-antitrypsin deficiency. Chest. 2013;144(4):1152–1162. doi:10.1378/chest.13-0161
10. Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017;12:561–569. doi:10.2147/COPD.S125389
11. Quinn M, Ellis P, Pye A, Turner AM. Obstacles to Early Diagnosis and Treatment of Alpha-1 Antitrypsin Deficiency: current Perspectives. Ther Clin Risk Manag. 2020;16:1243–1255. doi:10.2147/TCRM.S234377
12. Lambe T, Adab P, Jordan RE, et al. Model-based evaluation of the long-term cost-effectiveness of systematic case-finding for COPD in primary care. Thorax. 2019;74(8):730–739. doi:10.1136/thoraxjnl-2018-212148
13. Scott L, Campbell AM, Campbell EJ. Evaluation of Strategies for Comprehensive Diagnostic Testing for Alpha1-Antitrypsin Deficiency that Include Quantitative Isoelectric Focusing and DNA Sequencing. Am Thoracic Society. 2016;1:445.
14. Greulich T, Nell C, Herr C, et al. Results from a large targeted screening program for alpha-1-antitrypsin deficiency: 2003-2015. Orphanet J Rare Dis. 2016;11(1):75. doi:10.1186/s13023-016-0453-8
15. Greulich T, Rodriguez-Frias F, Belmonte I, Klemmer A, Vogelmeier CF, Miravitlles M. Real world evaluation of a novel lateral flow assay (AlphaKit(R) QuickScreen) for the detection of alpha-1-antitrypsin deficiency. Respir Res. 2018;19(1):151. doi:10.1186/s12931-018-0826-8
16. Garcia-Palenzuela R, Timiraos Carrasco R, Gomez-Besteiro MI, Lavia G, Lago Pose M, Lara B. Detection of alpha-1 antitrypsin deficiency: a study on patients diagnosed with chronic obstructive pulmonary disease in primary health care. Semergen. 2017;43(4):289–294. doi:10.1016/j.semerg.2016.05.003
17. de Miguel-Diez J, Jimenez-Garcia R, Lopez de Andres A, Zaragoza Arnaez F. Effectiveness of an Intervention to Improve Management of COPD using the AUDIT Methodology: results of the Neumo-Advance Study. Clin Drug Investig. 2019;39(7):653–664. doi:10.1007/s40261-019-00787-4
18. Nolte JL, Ataya A, Merrill H, Childs M, Brantly M. Alpha1-antitrypsin Deficiency-Increased Knowledge and Diagnostic Testing after Viewing Short Instructional Video. COPD. 2017;14(1):52–55. doi:10.1080/15412555.2016.1245280
19. Soriano JB, Lucas SJ, Jones R, et al. Trends of testing for and diagnosis of α 1 -antitrypsin deficiency in the UK: more testing is needed. Eur Respir J. 2018;52(1):1800360. doi:10.1183/13993003.00360-2018
20. de Serres FJ, Blanco I, Fernandez-Bustillo E. Estimating the risk for alpha-1 antitrypsin deficiency among COPD patients: evidence supporting targeted screening. Copd. 2006;3(3):133–139. doi:10.1080/15412550600829257
21. Rahaghi FF, Sandhaus RA, Strange C, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. Copd. 2012;9(4):352–358. doi:10.3109/15412555.2012.669433
22. Bradi AC, Audisho N, Casey DK, Chapman KR. Alpha-1 antitrypsin deficiency in Canada: regional disparities in diagnosis and management. COPD. 2015;12(Suppl 1):15–21. doi:10.3109/15412555.2015.1021908
23. Campos MA, Alazemi S, Zhang G, et al. Clinical characteristics of subjects with symptoms of alpha1-antitrypsin deficiency older than 60 years. Chest. 2009;135(3):600–608. doi:10.1378/chest.08-1129
24. Townsend S, Newsome P, Turner AM. Presentation and prognosis of liver disease in alpha-1 antitrypsin deficiency. Expert Rev Gastroenterol Hepatol. 2018;12(8):745–747. doi:10.1080/17474124.2018.1477589
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.