")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Establishment of Sunitinib-Resistant Xenograft Model of Renal Cell Carcinoma and the Identification of Drug-Resistant Hub Genes and Pathways
Authors Xie Y, Shangguan W, Chen Z, Zheng Z, Chen Y, Zhong Q, Zhang Y, Yang J, Zhu D, Xie W
Received 19 October 2021
Accepted for publication 6 December 2021
Published 16 December 2021 Volume 2021:15 Pages 5061—5074
DOI https://doi.org/10.2147/DDDT.S343718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yingwei Xie,1,2,* Wentai Shangguan,1,2,* Zhiliang Chen,1,2,* Zaosong Zheng,3 Yuqing Chen,4 Qiyu Zhong,1,2 Yishan Zhang,1,2 Jingying Yang,1,2 Dingjun Zhu,1,2,5 Wenlian Xie1,2,5
1Department of Urology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, 510120, People’s Republic of China; 2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, 510120, People’s Republic of China; 3Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China; 4Department of Pathology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, 510120, People’s Republic of China; 5Guangdong Provincial Clinical Research Center for Urological Diseases, Guangzhou, 510120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenlian Xie Email [email protected]
Introduction: Sunitinib is the first-line targeted drug for the treatment of advanced renal cell carcinoma (RCC), but its therapeutic potential is limited by premature drug resistance. In an attempt to overcome this limitation, a sunitinib-resistant cell-derived xenograft (CDX) model of clear cell renal cell carcinoma (ccRCC) in vivo was constructed in this study. The molecular mechanism of drug resistance was analyzed using sequencing and bioinformatics tools.
Methods: First, mice were injected subcutaneously with tumor cells 786-O to create tumors and were simultaneously treated with sunitinib. After three consecutive passages, a drug-resistant xenograft model was obtained. Then, key pathways and genes were identified via second-generation sequencing of the tissue and the examination of differentially expressed genes (DEGs) with bioinformatics tools.
Results: Analysis of sequencing data revealed that 646 DEGs were upregulated and 465 were downregulated in the drug-resistant tissues when compared with the sensitive tissues. GO showed that the DEGs were significantly enriched in angiogenesis, cell hypoxia response, and apoptosis. KEGG analysis demonstrated that the main pathways were PI3K-Akt, HIF-1, NF-kappa B, and MAPK. Modular analysis of the PPI network indicated that the GO and KEGG analyses of module 1 with the highest ranking were mainly related to ubiquitinase activity. Similarly, the GO and KEGG analyses of the top 10 hub genes were also chiefly linked to ubiquitinase activity. Then, comprehensive expression analysis of the hub genes, and finally, the genes BTRC and TRIM32 were identified, which were consistent in all observations.
Conclusion: In this study, through the construction of in vitro models and bioinformatics analysis, the important pathways and key genes related to ccRCC sunitinib resistance were discovered. Among them, ubiquitinase may play an important role in drug resistance and may be a potential therapeutic target and biomarker.
Keywords: ccRCC, sunitinib, drug resistance, xenograft model, ubiquitinase
Introduction
Renal cell carcinoma (RCC) accounts for approximately 2–3% of all malignant tumors, and its prevalence is rising. Metastatic RCC accounts for 25–30% of all RCC cases, and has an exceedingly poor prognosis.1 In 2020, among approximately 430,000 newly discovered cases of RCC, 179,000 died.2 Clear cell renal cell carcinoma (ccRCC) is the most prevalent form of the disease, and accounts for approximately 70–80% of all RCC cases.3 RCC is insensitive to radiotherapy and chemotherapy. Surgical resection is the main method for the treatment of localized RCC, but nearly 30% of patients have tumor recurrence and metastasis after surgery.3 Therefore, a new treatment method, targeted therapy, was introduced.
At present, receptor tyrosine kinase inhibitors (TKIs), such as sunitinib, constitute the first-line treatment for advanced RCC.4 However, sunitinib use poses challenges when significant efficacy is achieved. According to statistics, approximately 20% of patients will show innate resistance when receiving sunitinib for the first time, and most patients will develop secondary resistance to the drug after 6–11 months of treatment.5,6 Sunitinib-resistant RCC tissues are difficult to obtain for ethical reasons; hence, there are few studies on the mechanism of sunitinib resistance. Therefore, the molecular mechanism of drug resistance is yet to be elucidated.
To screen and evaluate new antitumor drugs and to study the mechanism of tumor resistance, researchers have established tumor xenograft models. The commonly used models are cell-derived xenograft (CDX) model7 and patient-derived xenograft (PDX) model.8 While the CDX model has the characteristics of fast modeling and high success rate, the PDX model has the advantages of retaining the patient’s molecular phenotype and genotype and can, therefore, predict the clinical outcome well.
Bioinformatics is an emerging interdisciplinary subject that has been developed on the basis of life science, computer science, and mathematics. This field utilizes mathematics and computer science to collect, process, store, disseminate, and analyze biological data to understand their significance.9 Nowadays, more and more researchers are using microarray technology combined with bioinformatics tools to identify new genes related to cancer progression, diagnosis, prognosis, and drug resistance.
In this study, a CDX model of sunitinib-resistant ccRCC was first constructed, the gene expression profile of the drug-resistant tissue was sequenced, and then the sequencing data were analyzed using bioinformatics tools to identify the potential genes responsible for sunitinib resistance in ccRCC.
Materials and Methods
Establishment of the CDX-Resistance Model
All animal experiments were performed following the protocol approved by the Animal Care and Use Institutional Committee of Sun Yat-sen Memorial Hospital, Sun Yat-sen University (Guangzhou, China). The human ccRCC cell lines 786-O was sourced from the American Type Culture Collection (ATCC; Manassas, VA, USA). To obtain the ccRCC tissues resistant to sunitinib, approximately 5.0×106 of 786-O cells were subcutaneously injected into the upper back of 5-week-old female BALB/c nude mice. When the volume of the xenograft reached 200 mm3, the mice were treated orally with saline or sunitinib (APExBIO). The treatment course followed the 4/2 plan (40 mg/kg/day, 4 weeks of treatment, followed by 2 weeks of rest). After the treatment course, the xenotransplanted ccRCC tissues were separated, and a 3-mm-diameter tissue was transplanted into the mice again, and the administration was continued. At the end of the treatment, the above step was repeated to obtain the third generation of ccRCC tissues that were resistant to sunitinib; the subsequent ccRCC tissues were identified by the pathologist at our hospital. The tumor volume (V) was calculated using the following formula, every 3 days: V = (W2 × L)/2.
Acquisition of Drug-Resistant ccRCC Cells
In order to isolate the ccRCC cells from xenografts, the xenografts were first mechanically decomposed, washed with cold-PBS containing penicillin (500 U/mL) and streptomycin (500 μg/mL), and then transferred to the DMEM medium containing type II/IV collagenase (1 mg/mL) and digested in a constant temperature oscillator at 37°C for 1 h. Finally, the cell suspension was passed through 80-μM filter (BD Biosciences, USA) and centrifuged at 1000 rpm for 5 min. The precipitate was suspended and inoculated in a Petri dish. After passage 3 generations, the half inhibitory concentration (IC50) of sunitinib on the isolated tumor cells was detected.
IC50 Detection
The tumor cells (1 × 103; 100 µL/well) were seeded into a 96-well-plate and then placed in the incubator for 24 h, sunitinib was added to make the final concentration of 0, 1.5, 3, 6, 12, 25, 50, and 100 µM. There were 3 replicate wells in each group. After 48 h, the liquid was aspirated, media containing 20% MTS was added, and the solution was incubated in the dark for 2 h; the optical density of the resultant cells was measured at 492 nm.
Sequencing and DEGs Identification
The xenograft tumor tissues (3 resistant tissues and 3 control tissues) were sent to the Lianchuan Biological Company for human transcriptome mRNA sequencing. The “limma” package in the R software was used to screen the differentially expressed genes (DEGs) between drug-resistant and control tissues in the sequencing results. The threshold of DEGs was as follows: log2|(FC)|> 2, P < 0.05, the false discovery rate (FDR) < 0.05.
Functional Enrichment Analyses
In order to evaluate the function of DEGs in the drug-resistance tumor, “Cluster Profiler” package in the R software was used to conduct GO annotation and KEGG pathway enrichment analyses.10 The ruling standard was set as P < 0.05.
PPI Network Construction and Module Function Analyses
In order to evaluate the interaction between DEGs, the online search tool STRING database was employed to construct a protein-protein interaction (PPI) network, which can provide a comprehensive interaction between proteins and genes.11 The PPI network was visualized using the Cytoscape.12 The mcode plug-in of Cytoscape was used to extract important modules from the PPI network for analyzing the interaction relationship of DEGs. The default threshold included a degree cut-off: 2, node score cut-off: 0.2, k-core: 2, and max depth: 100.13,14
Identification of Hub Genes
Hub gene is a gene with high connectivity. The Maximum Group Centrality (MCC) algorithm in cytohubba plug-in was used to identify hub genes, and the top 10 hub genes with the highest MCC score were subsequently analyzed.15
Verify the mRNA and Protein Expression Levels of Hub
We downloaded the ccRCC cohort data from the TCGA database in July 2021, which included the patient gene expression data and clinical information. We then used the “limma” and “beeswarm” packages in the R software to extract the mRNA expression of the Hub genes and compared the results between cancerous tissues and the adjacent tissues. In addition, the HPA database was referred to conduct an online immunohistochemical analysis to determine the distribution of the hub genes and the protein levels.16
Prognostic Analysis
The prognosis of hub genes was analyzed by the “survival” package in the R software. A total of 500 ccRCC patients followed up for >90 days in the TCGA database were selected. The survival curve was drawn by Kaplan-Meier method. P < 0.05 was considered to indicate statistical significance.
Results
Construction and Identification of Sunitinib-Resistant ccRCC CDX Model
In this study, a CDX model of ccRCC was established by injecting 786-O subcutaneously into mice and treating them with sunitinib to obtain sunitinib-resistant tumors in the third generation. The specific operation process is depicted in Figure 1A. The growth curves of the tumor in the CDX model of the control group, the first generation experimental group, and the third generation experimental group are shown in Figure 1B. The results indicated that the third-generation tumor had developed resistance to sunitinib. The tumor tissues were then dispersed to obtain isolated tumor cells, and the IC50 test of sunitinib was performed, which suggested that the IC50 of the third generation tumor cells was 12.5μM. This value was significantly higher than that of the control group (4.0μM), which indicated that the isolated ccRCC cells had also developed resistance to sunitinib in vitro (Figure 1C). In summary, our established CDX model of sunitinib-resistant ccRCC was successful and reliable.
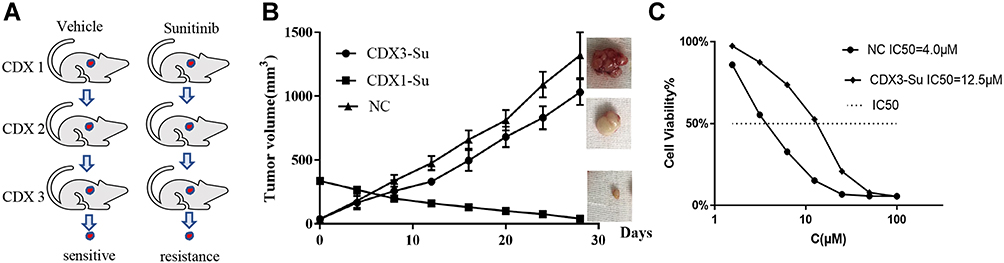 |
Figure 1 Construction and identification of the CDX model of ccRCC sunitinib resistance. (A) Flow chart of ccRCC sunitinib-resistance model construction. (B) The curve of tumor volume with time in the control group, the CDX1-Su group, and the CDX3-Su group. (C) The half inhibitory concentration (IC50) of sunitinib on tumor cells in the control and CDX3-Su groups. |
Identification of DEGs
The R package was used to analyze the sequencing data of sunitinib-resistant and sensitive tumors in ccRCC. The volcano map of differential gene expression is presented in Figure 2A (all differential genes are shown in Supplementary Excel S1: Sheet1). Based on the thresholds of P < 0.05 and log2|FC| > 2, there were 646 upregulated and 465 downregulated genes in the resistant tissues (Figure 2B).
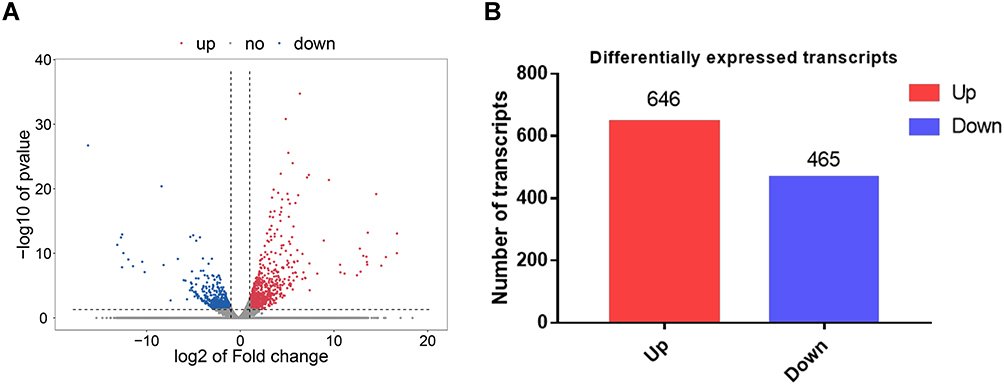 |
Figure 2 Identification of genes that were differentially expressed in drug-resistant tissues and the relatively sensitive tissues. (A) The volcano map reflecting the differentially expressed genes. Red color represents upregulated genes, while the blue color represents downregulated genes. The cutoff threshold was |logFC|>2.0 and adj. P < 0.05. (B) The histogram depicts that, among the genes with differential expression, there were 646 upregulated genes and 465 downregulated genes. |
PPI Network Construction, GO Annotation, and KEGG Pathway Enrichment Analysis
To comprehend the interaction between the DEGs, the STRING database was used to determine the PPI pairs among the 1011 DEGs. As shown in Figure 3A, the constructed PPI network included 613 nodes (genes) and 4056 edges (interactions) (threshold: interaction score > 0.4). To obtain a deeper understanding of the biological effects of these DEGs, the “Cluster Profiler” package in R was used to perform GO annotation and KEGG pathway enrichment analysis (for detailed results, see Supplementary Excel S1: Sheet2 and Excel S1: Sheet3). The top 20 results are shown in the figure. GO analysis revealed that these DEGs were significantly enriched in the collagen-containing extracellular matrix, angiogenesis, cellular response to hypoxia, and positive regulation of apoptotic process (Figure 3B). In addition, the KEGG pathways were significantly enriched in the PI3K-Akt signaling pathway, HIF-1 signaling pathway, NF-kappa B signaling pathway, and MAPK signaling pathway (Figure 3C). Our results are consistent with the current understanding of sunitinib resistance, and bypass activation may be involved in sunitinib resistance.
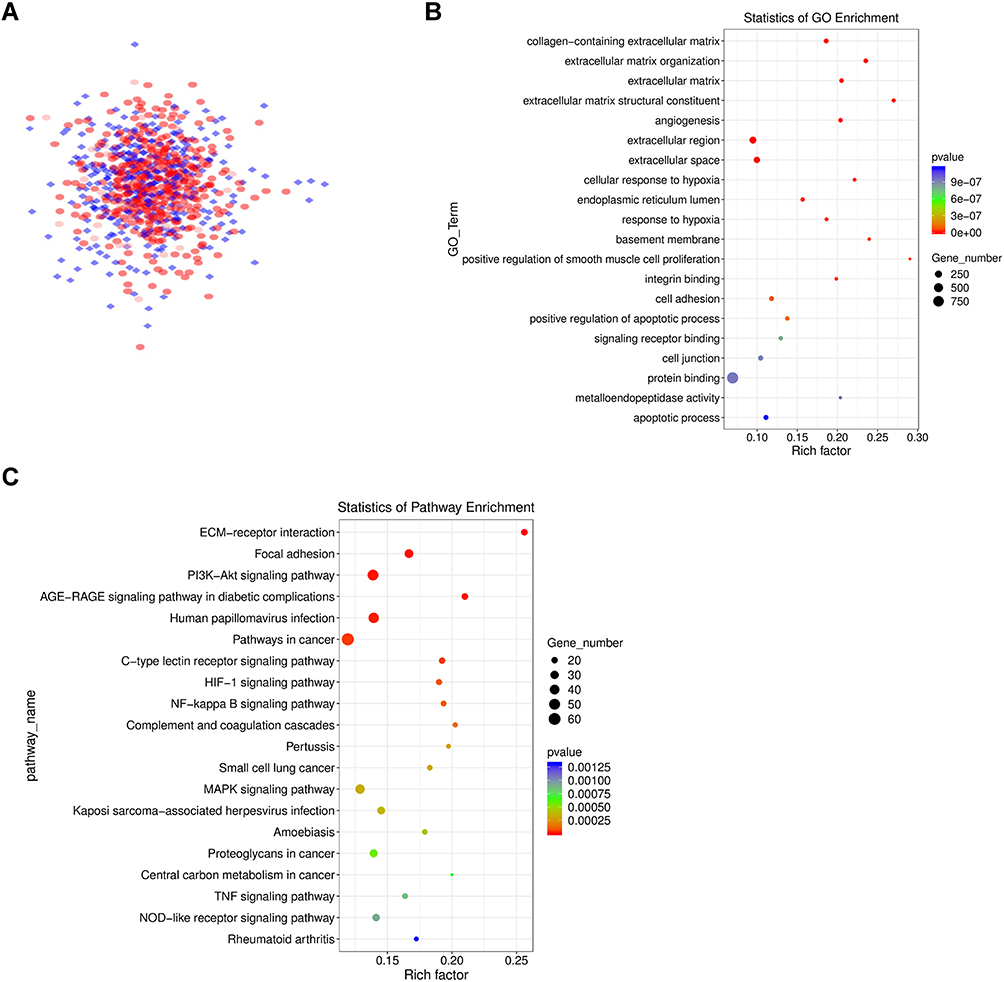 |
Figure 3 PPI network diagram of differentially expressed genes, as well as GO and KEGG analysis. (A) The PPI network diagram is composed of 613 genes and is visualized by Cytoscape. The interaction score > 0.4. (B) GO enrichment analysis results of genes in the PPI network. (C) KEGG analysis results of genes in the PPI network. |
PPI Network Module Analysis
Since the PPI network constructed by us was relatively large, the MCODE plugin was used to calculate the modules with an MCODE score of >5 and identify the most important modules. Finally, a total of 6 modules (for detailed results, see Supplementary Excel S1: Sheet4 and Figure S1) was identified, out of which, two (modules 1 and 3) were rich in biological processes and pathways related to tumor progression. These modules were considered to be functionally important. There were 23 genes in module 1 (12 upregulated genes and 11 downregulated genes), as shown in Figure 4A. There existed a total of 17 genes in module 3 (13 upregulated genes and 4 downregulated genes), as shown in Figure 4B. GO analysis of module 1 signified that the main enrichment was in ubiquitin–protein transferase activity. KEGG analysis showed that the main enrichment pathway was ubiquitin-mediated proteolysis (Figure 4C). The GO analysis of module 3 implied that it was mainly enriched in cell adhesion molecule binding, insulin-like growth factor I binding, and growth factor binding. KEGG analysis alluded that the main enriched pathway was the p53 signaling pathway, and PI3K-Akt signaling pathway (Figure 4D). According to module analysis, it appears that ubiquitinase may play an important role in sunitinib resistance.
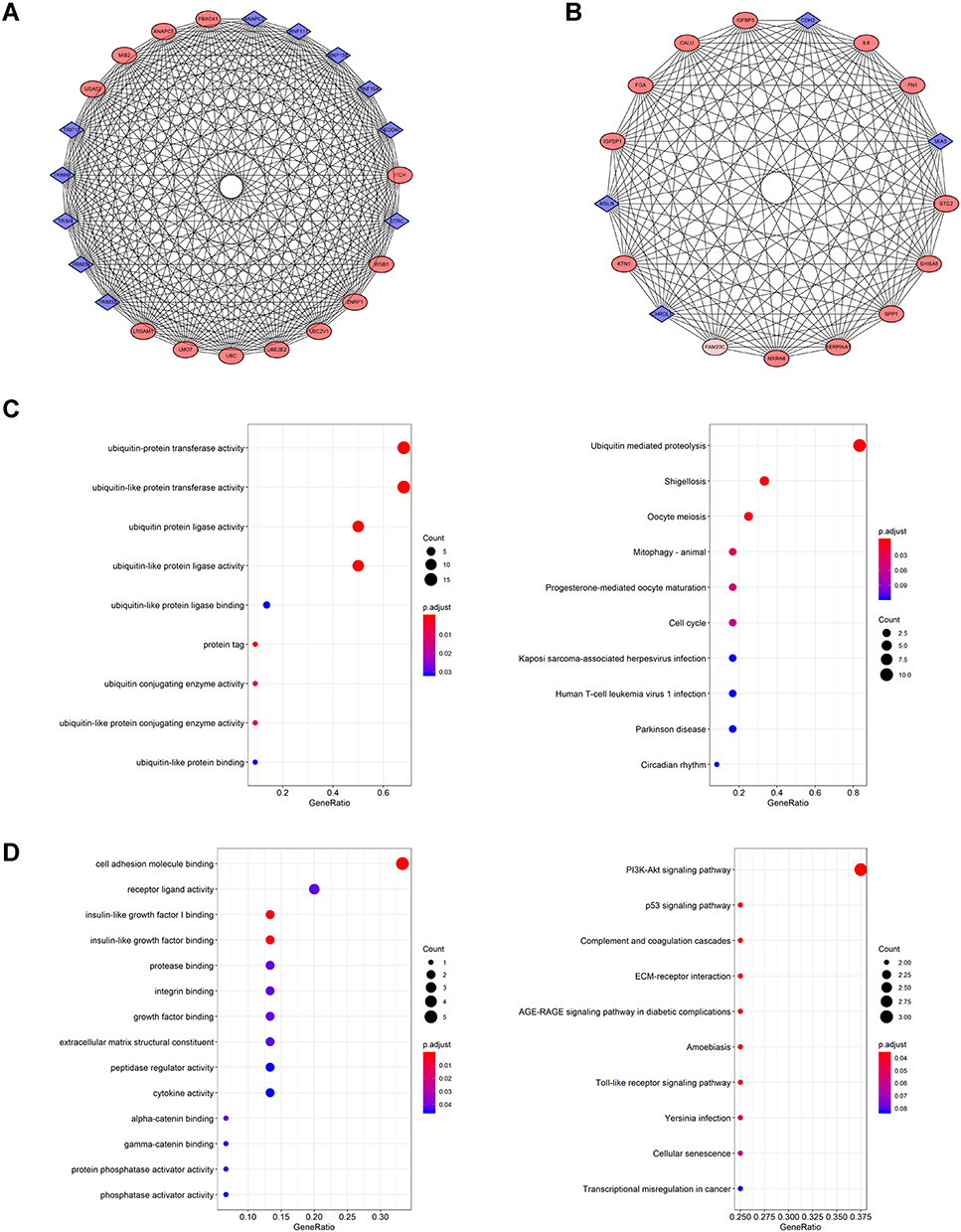 |
Figure 4 Modular analysis of PPI network (A) Module 1 contains 23 genes. (B) Module 3 contains 17 genes. (C) GO and KEGG analysis of genes in Module 1. (D) GO and KEGG analysis of genes in Module 3. |
Hub Gene Identification
To identify the hub genes in the interaction network, the CytoHubba plugin was used to determine the top 10 hub genes based on the MCC score. The results included UBC, TRIP12, ZNRF1, UBA52, BTRC, NEDD4L, ITCH, TRIM32, ANAPC5 and ANAPC7 (Figure 5A) (upregulated genes: UBC, ZNRF1, UBA52, ITCH, and ANAPC5; downregulated genes: TRIP12, BTRC, NEDD4L, TRIM32, and ANAPC7). Interestingly, the hub genes determined based on the MCC score were consistent with the highest score module 1 from MCODE analysis. The GO analysis revealed that the hub genes were highly enriched in ubiquitin–protein transferase activity, protein phosphatase binding, and CXCR chemokine receptor binding (Figure 5B). The KEGG pathway enrichment included ubiquitin-mediated proteolysis and Cell cycle (Figure 5C). Based on the above analyses, the hub gene function identified from the PPI network was mainly found to be related to the ubiquitinating enzyme activity. This result suggests that the ubiquitinating enzyme system may play an important role in sunitinib resistance.
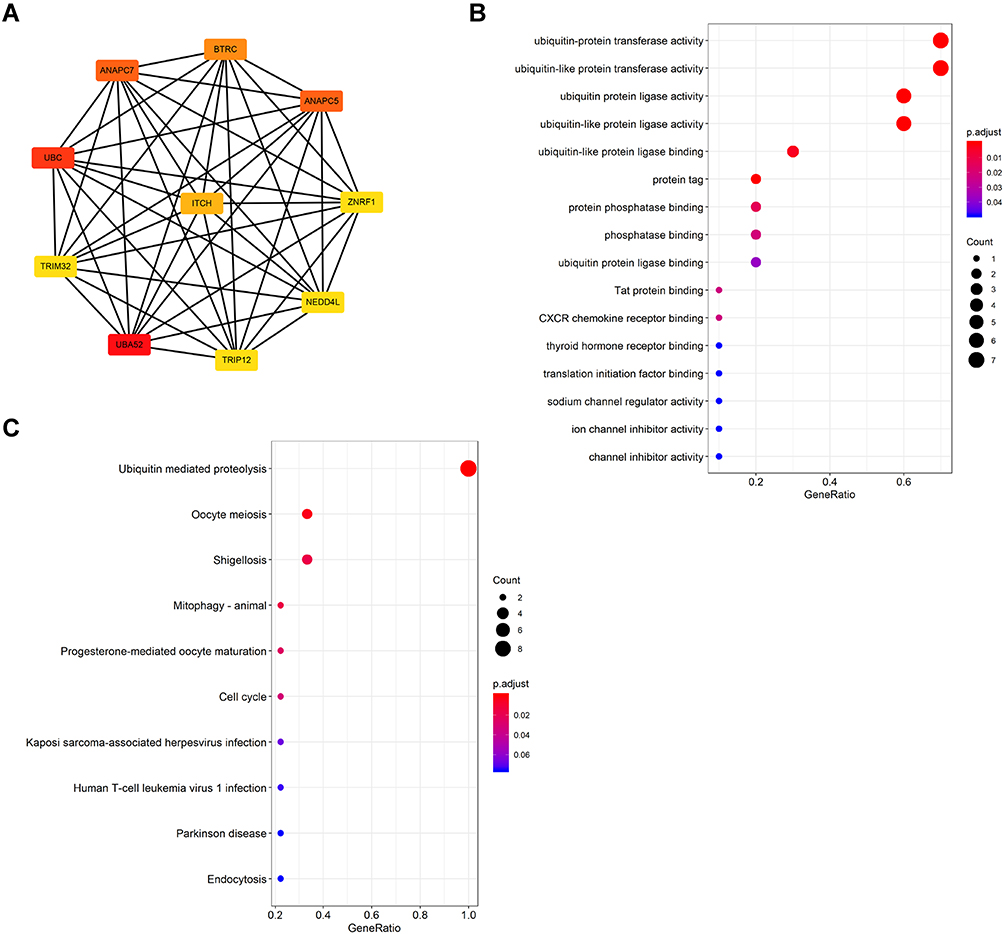 |
Figure 5 Hub gene analysis of PPI network (A) The top ten genes calculated by the MCC algorithm, the darker the color, the higher the ranking. (B) GO enrichment results of hub genes. (C) KEGG analysis result of Hub genes. |
Comprehensive Expression Analysis of the Hub Genes at the mRNA and Protein Levels and Prognosis in ccRCC
The transcriptome data of ccRCC was downloaded from the TCGA database, which included 539 cases of cancer tissues and 72 cases of adjacent tissues. As shown in Figure 6, the mRNA expression of UBC, UBA52, ANAPC5 and ANAPC7 in the ccRCC tissues were significantly higher than that in the adjacent tissues (P<0.05). The mRNA expression of TRIP12, ZNRF1, BTRC, NEDD4L, ITCH and TRIM32 in the ccRCC tissues were significantly lower than the adjacent tissues (P<0.05).
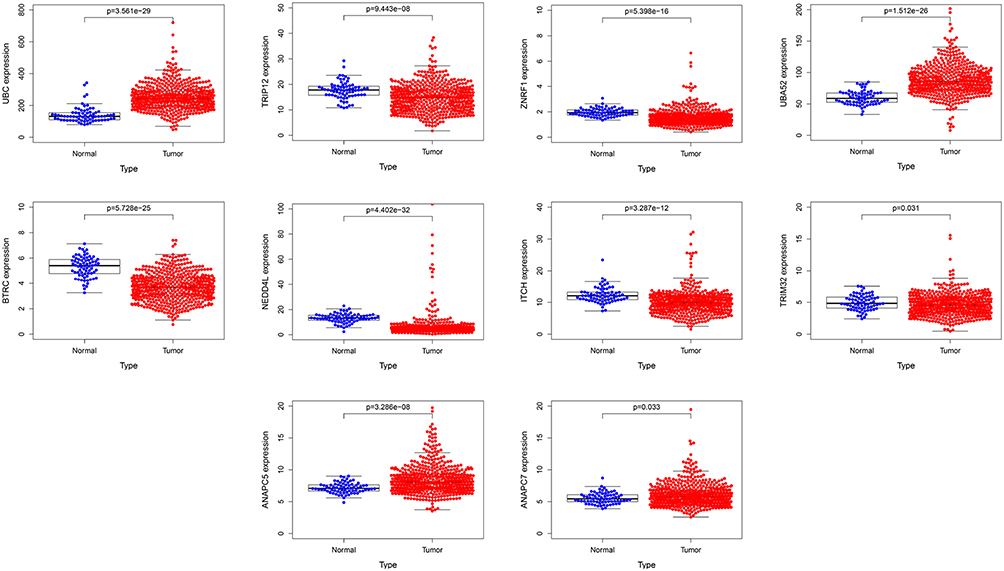 |
Figure 6 TCGA database was used to analyze the difference mRNA expression of hub genes between ccRCC and adjacent tissues. |
The protein expressions of the hub genes in ccRCC were analyzed through the online HPA database, as shown in Figure 7. The protein expression of UBC, TRIP12, UBA52, NEDD4L and ANAPC7 in the ccRCC tissues were significantly higher than that in normal tissues. On the other hand, the protein expression of ZNRF1, BTRC, ITCH, TRIM32 and ANAPC5 in the ccRCC tissues were significantly lower than that in the normal tissues.
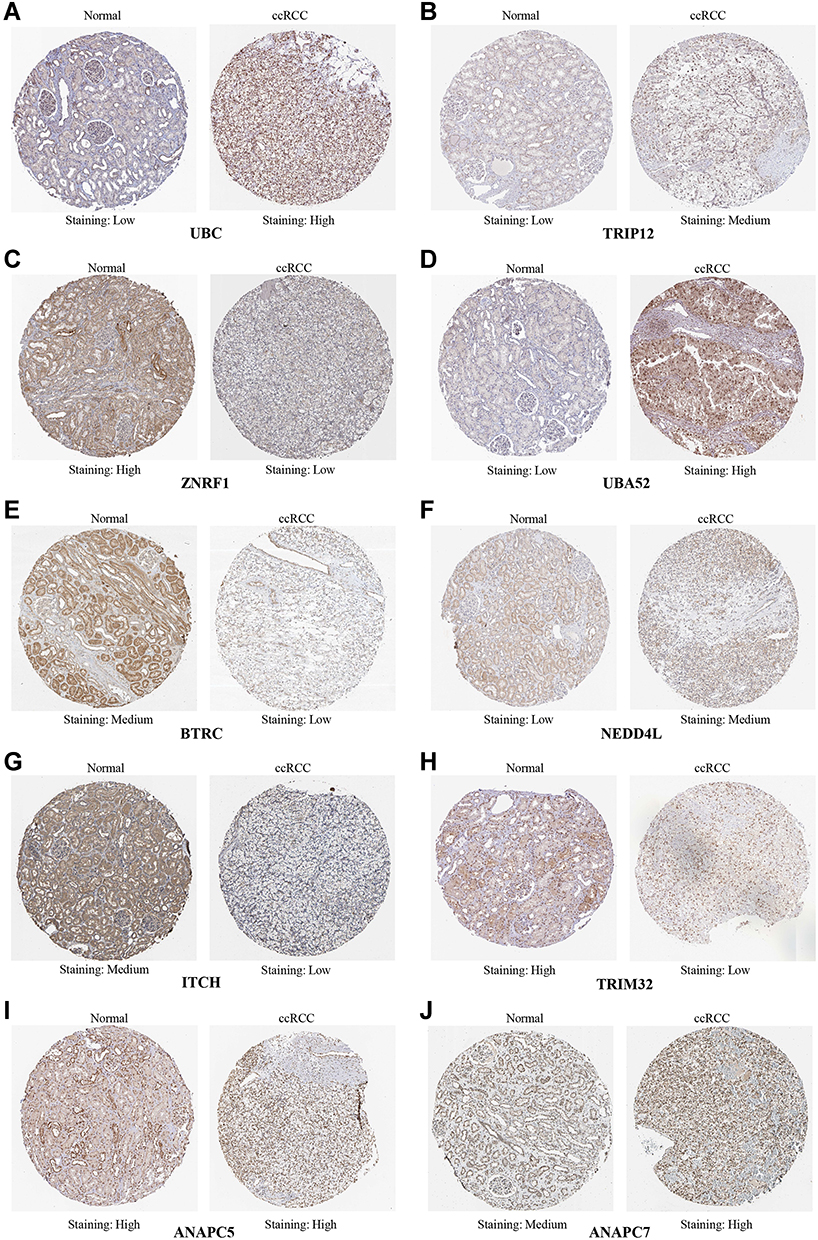 |
Figure 7 HPA database was used to analyze the difference protein expression of hub genes between ccRCC tissues and normal tissues. (A) UBC protein is highly expressed in ccRCC. (B) TRIP12 protein is highly expressed in ccRCC. (C) ZNRF1 protein is low expressed in ccRCC. (D) UBA52 protein is highly expressed in ccRCC. (E) BTRC protein is low expressed in ccRCC. (F) NEDD4L protein is highly expressed in ccRCC. (G) ITCH protein is low expressed in ccRCC. (H) TRIM32 protein is low expressed in ccRCC. (I) ANAPC5 protein is highly expressed in ccRCC tissues and normal tissues. (J) ANAPC7 protein is highly expressed in ccRCC. |
The clinical information of patients with ccRCC was downloaded from the TCGA database, and the prognosis based on the hub genes was analyzed. The results indicated that the high expression of TRIP12, BTRC, NEDD4L, ITCH, and TRIM32 improved the prognosis of patients with ccRCC, while the high expressions of ANAPC5 and ANAPC7 suggested a poor prognosis (Figure 8).
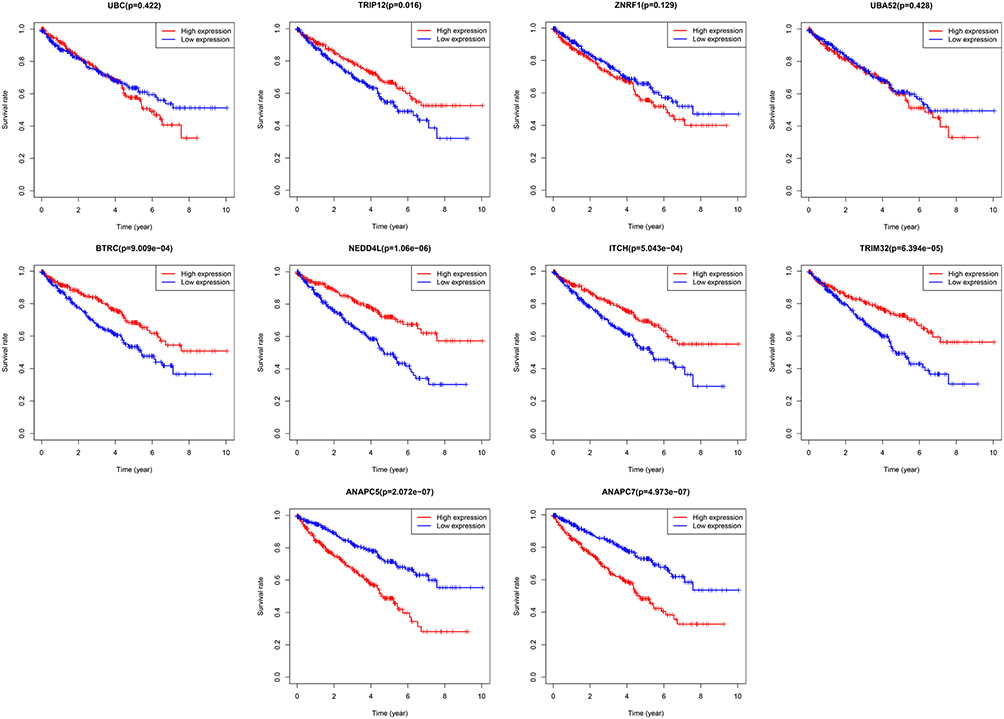 |
Figure 8 TCGA database was used to analyze the effect of Hub genes expression on the prognosis of ccRCC. |
The Venn diagram showed that there were no common genes in the intersection of hub genes with upregulated mRNA expression in the drug-resistant CDX model, hub genes with up-regulated mRNA and protein expressions in ccRCC, and hub genes related to poor prognosis (Figure 9A). On the other hand, there were two genes (BTRC and TRIM32) in the intersection of hub genes with the downregulated mRNA expression in the drug-resistant CDX model, hub genes with downregulated mRNA and protein expressions in ccRCC, and hub genes related to good prognosis (Figure 9B). The details of BTRC and TRIM32 genes are presented in Table 1. It was found that BTRC and TRIM32 belonged to E3 ubiquitinase. In conclusion, we believe that BTRC and TRIM32, as E3 ubiquitinases, may play key roles in the sunitinib resistance in RCC.
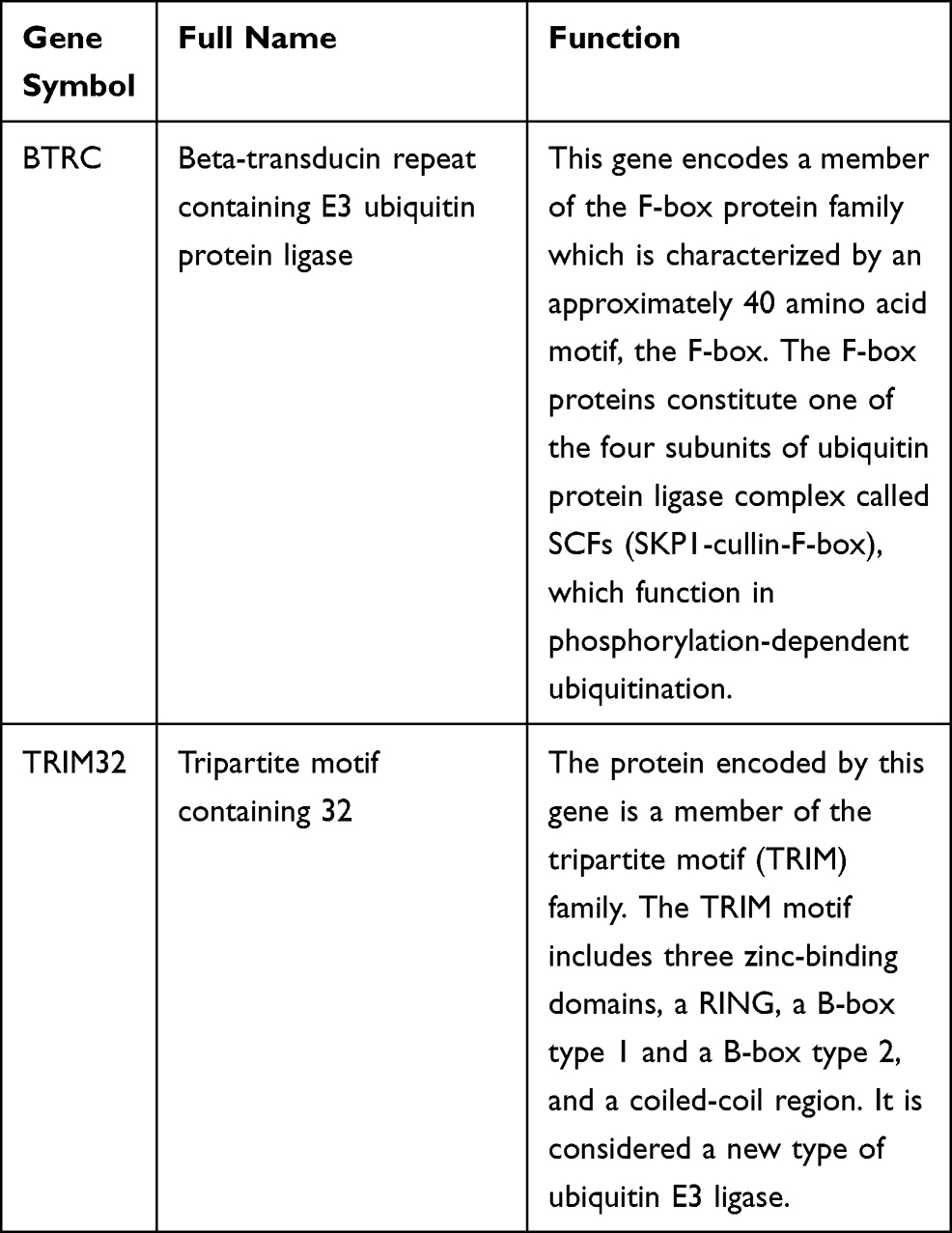 |
Table 1 Summary of the Functions of 2 Hub Genes |
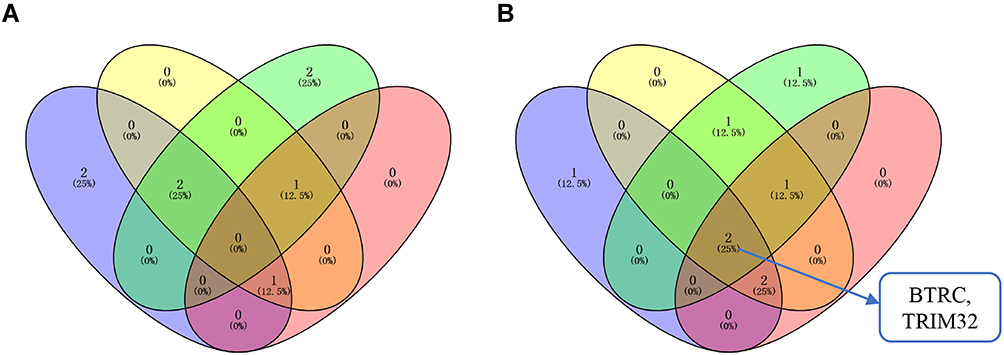 |
Figure 9 Venn diagram. (A) The intersection of the upregulated hub genes and the hub genes with poor prognosis. Blue circles represent hub genes with upregulated mRNA expression in drug-resistant tissues; yellow circles represent hub genes with upregulated mRNA expression in ccRCC; green circles represent hub genes with upregulated protein expression in ccRCC; red circles represents hub genes related to poor prognosis. (B) The intersection of downregulated hub genes and hub genes with good prognosis. The blue circles represent hub genes with downregulated mRNA expression in drug-resistant tissues; the yellow circles represent hub genes with downregulated mRNA expression in ccRCC; the green circles represent hub genes with downregulated protein expression in ccRCC; and the red circles represent hub genes related to good prognosis. |
Discussion
RCC is a highly vascularized tumor. In the early stages of tumor formation, RCC acquires an ability to synthesize angiogenic factors and form neovascularization.17 Since the advent of antiangiogenic therapy, the overall survival (OS) rate of patients with RCC has increased drastically from 13 months in the cytokine era 10 years ago18 to 30 months in 2014.19,20 Sunitinib is the first generation of multitarget TKI-targeted drugs, and can non-covalently bind to several tyrosine kinase receptors and inhibit their phosphorylation. In a mice model of RCC tumors, sunitinib has been shown to reduce vascular permeability, inhibit neovascularization, and destroy the generated tumor vessels.21
The application of targeted drugs has improved the prognosis of patients with RCC. However, with the application of targeted drugs, drug resistance has gradually emerged and has lowered the clinical effects. Although combined medication can help overcome drug resistance, it is fraught with the disadvantages of high cost, drug toxicity, and side effects, which have posed several hurdles in the treatment of patients.22
The mechanisms of primary and secondary drug resistance of sunitinib often overlap each other. The drug resistance mechanisms can be divided into activation of angiogenesis signaling pathway, alteration of tumor microenvironment, increase of tumor invasion and metastasis, role of non-coding RNA, and activation of other signaling pathways.23–25 Some reports have been published on the mechanism of targeted drug resistance in RCC; however, the underlying molecular mechanism is not clear. Furthermore, there is a lack of effective biomarkers to predict targeted drug resistance. Also, there are no effective treatments to reverse or delay drug resistance after it occurs.
To decipher the molecular mechanism of sunitinib resistance in ccRCC, a CDX mouse model of sunitinib resistance in ccRCC was established in this study. By sequencing the drug-resistant and sensitive tissues, DEGs were identified and their expression data were analyzed using bioinformatics tools. The results suggested the following: (1) When compared with the sensitive tissues, sunitinib-resistant tissues had 646 upregulated genes and 465 downregulated genes. GO analysis showed that these DEGs were mainly enriched in angiogenesis, cellular response to hypoxia, and positive regulation of apoptotic process. KEGG analysis implied that phosphatidylinositol-3-kinase-(PI3K)-Akt, hypoxia-inducible factor (HIF)-1, nuclear factor kappa-B (NF-kappa B), and MAPK signaling pathways were significantly enriched. (2) Through the construction of PPI network and module analysis, six important modules were identified. The GO and KEGG analyses of module 1 showed that the network was mainly enriched in ubiquitin-mediated proteolysis, which indicates that ubiquitin may play an important role in sunitinib resistance. The GO and KEGG analyses of module 3 showed that the p53 and PI3K-Akt signaling pathways were significantly enriched, which suggests that these pathways may have key functions in regulating sunitinib resistance. (3) Ten hub genes were identified in the DEGs. By analyzing the expressions of mRNA and protein and their impacts on the prognosis of patients with ccRCC, BTRC and TRIM32 were finally found to be consistent in expression and prognosis. It is speculated that these two genes may be chiefly involved in the sunitinib resistance in ccRCC.
At present, the research on sunitinib resistance in RCC mainly includes cell lines, CDX models, and PDX models. However, it is difficult to obtain drug-resistant tissues from patients owing to ethical reasons. Studies on animal models of sunitinib resistance in RCC are limited. Zhang established a CDX model of sunitinib resistance in ccRCC and found that LncARSR promotes the expressions of AXL and c-MET and the activation of downstream STAT3, AKT, and ERK signaling pathways, which leads to sunitinib resistance in ccRCC.26 Diaz-Montero created a sunitinib-resistant PDX model for ccRCC. They discovered that during single-agent TKI treatment, the tumor showed reduced MEK phosphorylation. However, after combined TKI/MEKi treatment, it effectively inhibited phosphorylated MEK and phosphorylated ERK, which can prolong the antitumor effect of sunitinib.27 In this study, resistance-related DEGs were found to be enriched in PI3K-Akt, HIF-1, NF-kappa B, and MAPK signaling pathways. However, network module analysis revealed that the GO and KEGG analyses results of the highest ranked module 1 (containing 23 genes) were mainly enriched in ubiquitin–protein transferase activity and ubiquitin-mediated proteolysis. Our finding suggests that the ubiquitinase system may play an important role in the resistance of ccRCC to sunitinib.
Furthermore, the hub genes in the PPI network were calculated using the MCC algorithm. The top 10 genes were UBC, TRIP12, ZNRF1, UBA52, BTRC, NEDD4L, ITCH, TRIM32, ANAPC5, and ANAPC7. It is worth noting that these 10 genes completely overlapped with the genes in module 1. The GO and KEGG analyses of these hub genes were predominantly enriched in ubiquitin–protein transferase activity and ubiquitin-mediated proteolysis. Module and hub gene analyses further proved that ubiquitinase may play an important role in sunitinib resistance. Additionally, the mRNA and protein expressions of the hub genes in ccRCC and their influence on the prognosis of patients with ccRCC were analyzed. As a result of these analyses, BTRC and TRIM32 were finally identified to be the key genes, with consistent results in all studies. Hence, we speculate that these two genes may play a pertinent role in sunitinib resistance.
Beta-transducing repeat containing E3 ubiquitin protein ligase (BTRC) encodes F-box protein, which contains multiple WD-40 repeat sequences. On the one hand, BTRC has been shown to ubiquitinate phosphorylated NFKBIA and target its degradation, thereby activating NF-κB, promoting its translocation to the nucleus, and activating the transcription of metastasis-related genes.28,29 For instance, Zhang found that TSPAN15 interacts with BTRC to promote phosphorylated-inhibitor of NF-κB alpha (p-IκBα) ubiquitination and proteasomal degradation, thereby triggering NF-κB nuclear translocation and the subsequent transcriptional activation of several metastasis-related genes. These events promote the metastasis of esophageal squamous cell carcinoma.30 Similarly, in triple-negative breast cancer, Lim found that WBP2 induces the stability BTRC, promotes its targeting of IκBα for ubiquitination and degradation, and enhances cancer progression.31 On the other hand, BTRC has also been shown to play a vital role in the epithelial–mesenchymal transition (EMT) process because it facilitates the ubiquitination of Snail in cancer. The inhibition of BTRC leads to the upregulation of Snail, thereby inducing EMT.32,33 For example, Zheng identified that miR-224 inhibits the expression of its target BTRC protein and promotes the associated Wnt/β-catenin pathway to cause the migration and invasion of rectal cancer cells.34 Zhou discovered that miR-193a-3p is upregulated in patients with glioma and promotes the invasion, migration, and EMT of glioma by inhibiting the expression of BTRC protein.35
Tripartite motif containing 32 (TRIM32) is a new type of ubiquitin E3 ligase. The protein encoded by this gene is a member of the tripartite motif (TRIM) family. The TRIM motif includes three zinc binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. Studies have demonstrated that TRIM32 plays an important role in regulating cell death induced by oxidative stress. TRIM32 activates the caspase-mediated apoptosis pathway under conditions of oxidative stress.36 A recent study has shown that TRIM32 enhances X-linked inhibitor-of-apoptosis protein (XIAP) turnover during TNF-mediated apoptosis.37 A decrease in the level of TRIM32 may increase the level of XIAP. Furthermore, XIAP has been proven to act as an antioxidant by regulating the expressions of several antioxidant genes, including superoxide dismutase type 1 (SOD1), SOD2, NAD (P) H: quinone oxidoreductase 1, heme-oxygenase-1, and thioredoxin 2.38
The findings of this study showed that BTRC and TRIM32 belong to the same E3 ubiquitin ligase. Although some functions have been studied in tumors, there is no research on angiogenesis or antiangiogenesis resistance. Moreover, another gene of the same E3 ubiquitin ligase, von Hippel–Lindau (VHL), was noted to be involved in tumor angiogenesis. The activation of hypoxia signaling pathway in tumors is closely related to the increase in vascular endothelial growth factor (VEGF) synthesis and angiogenesis. The core of the hypoxia pathway is the formation of the HIF complex. The HIF-1 complex can bind to the hypoxia response elements (HRE) on the promoter of the target gene to regulate downstream gene transcription.39 The HIF-1 complex consists of two subunits, namely HIF-1α and HIF-1β. Among them, HIF-1α undergoes proline hydroxylation under the action of proline hydroxylase and then combines with VHL E3 ligase to be degraded via the ubiquitinated proteasome pathway.40 In ccRCC, the deletion of the VHL gene results in the inability to degrade HIF-1α, and its accumulation enhances the expression of its downstream products (VEGF, transforming growth factor-α, CXC motif chemokine receptor type 4, etc.) and promotes angiogenesis.41 Whether BTRC and TRIM32 have functions similar to those of VHL remains to be investigated.
Conclusion
In this study, a CDX model of ccRCC sunitinib resistance was successfully constructed, and a series of bioinformatics analyses were performed on the DEGs related to the resistance via sequencing analysis. Finally, some key hub genes that may be related to the resistance of ccRCC to sunitinib treatment were identified. Among the identified genes and their related pathways, ubiquitin ligase activity was established for the first time to play a key role in drug resistance. The dysregulation of ubiquitin ligase expression was predicted to be a potential therapeutic target or biomarker for ccRCC sunitinib resistance. However, further molecular and biological experiments are required to confirm the functions of the key genes in ccRCC sunitinib resistance.
Data Sharing Statement
The data presented in this study are available in the Supplementary Materials.
Ethics Statement
The Institutional Animal Care and Use Committee (IACUC), Sun Yat-Sen University evaluated and approved the experimental protocols under approval number SYSU-IACUC-2019-B822. All animal procedures comply with the general requirements of laboratory animals-General requirements for animal experiment (GB/T35823-2018) and laboratory animals-Guideline for ethical review of animal welfare (GB/T35892-2018).
Acknowledgments
The authors would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by grants from Guangdong Basic and Applied Basic Research Foundation (Grant No.2019A1515012199), National Natural Science Foundation of China (Grant No.81672534), Guangdong Science and Technology Department (Grant No.2020B1212060018), and Guangdong Provincial Clinical Research Center for Urological Diseases (Grant No.2020B1111170006).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kabaria R, Klaassen Z, Terris MK. Renal cell carcinoma: links and risks. Int J Nephrol Renovasc Dis. 2016;9:45–52. doi:10.2147/IJNRD.S75916
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70(1):93–105. doi:10.1016/j.eururo.2016.02.029
4. Ljungberg B, Bensalah K, Canfield S, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol. 2015;67(5):913–924. doi:10.1016/j.eururo.2015.01.005
5. Oudard S, Rt E. Sequential therapy with targeted agents in patients with advanced renal cell carcinoma: optimizing patient benefit. Cancer Treat Rev. 2012;38:981–987. doi:10.1016/j.ctrv.2011.12.009
6. Molina AM, Lin X, Korytowsky B, et al. Sunitinib objective response in metastatic renal cell carcinoma: analysis of 1059 patients treated on clinical trials. Eur J Cancer. 2014;50:351–358. doi:10.1016/j.ejca.2013.08.021
7. Brennecke P, Arlt MJ, Campanile C, et al. CXCR4 antibody treatment suppresses metastatic spread to the lung of intratibial human osteosarcoma xenografts in mice. Clin Exp Metastasis. 2014;31(3):339–349. doi:10.1007/s10585-013-9632-3
8. Tentler JJ, Tan AC, Weekes CD, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–350. doi:10.1038/nrclinonc.2012.61
9. Deo RC. Machine Learning in Medicine. Circulation. 2015;132(20):1920–1930. doi:10.1161/CIRCULATIONAHA.115.001593
10. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
11. Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–15. doi:10.1093/nar/gks1094
12. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
13. Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302(5643):249–255. doi:10.1126/science.1087447
14. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
15. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl4):S11. doi:10.1186/1752-0509-8-S4-S11
16. Pontén F, Schwenk JM, Asplund A, Edqvist PH. The human protein atlas as a proteomic resource for biomarker discovery. J Intern Med. 2011;270(5):428–446. doi:10.1111/j.1365-2796.2011.02427.x
17. Qian CN, Huang D, Wondergem B, Teh BT. Complexity of tumor vasculature in clear cell renal cell carcinoma. Cancer. 2009;115(10 Suppl):2282–2289. doi:10.1002/cncr.24238
18. Motzer RJ, Bacik J, Murphy BA, Russo P, Mazumdar M. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol. 2002;20(1):289–296. doi:10.1200/JCO.2002.20.1.289
19. Motzer RJ, Barrios CH, Kim TM, et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2014;32(25):2765–2772. doi:10.1200/JCO.2013.54.6911
20. Choueiri TK, Figueroa DJ, Fay AP, et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: results from COMPARZ, a randomized controlled trial. Clin Cancer Res. 2015;21(5):1071–1077. doi:10.1158/1078-0432.CCR-14-1993
21. Christensen JG. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Ann Oncol. 2007;18(Suppl 10):x3–10. doi:10.1093/annonc/mdm408
22. Xie Y, Chen Z, Zhong Q, Chen Y, Shangguan W, Xie W. Efficacy and safety of immunological checkpoint inhibitors combined with anti-angiogenic drugs in first-line treatment of metastatic renal cell carcinoma: a systematic review and meta-analysis. Transl Androl Urol. 2021;10(1):300–309. doi:10.21037/tau-20-969
23. Joosten SC, Hamming L, Soetekouw PM, et al. Resistance to sunitinib in renal cell carcinoma: from molecular mechanisms to predictive markers and future perspectives. Biochim Biophys Acta. 2015;1855(1):1–16. doi:10.1016/j.bbcan.2014.11.002
24. Rausch M, Rutz A, Allard PM, et al. Molecular and functional analysis of sunitinib-resistance induction in human renal cell carcinoma cells. Int J Mol Sci. 2021;22(12):6467. doi:10.3390/ijms22126467
25. Chen SK, Wang YC, Lin TY, Wu HJ, Huang CJ, Ku WC. G-protein-coupled estrogen receptor 1 agonist G-1 perturbs sunitinib resistance-related phosphoproteomic signatures in renal cell carcinoma. Cancer Genomics Proteomics. 2021;18(3):207–220. doi:10.21873/cgp.20253
26. Qu L, Ding J, Chen C, et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29(5):653–668. doi:10.1016/j.ccell.2016.03.004
27. Diaz-Montero CM, Mao FJ, Barnard J, et al. MEK inhibition abrogates sunitinib resistance in a renal cell carcinoma patient-derived xenograft model. Br J Cancer. 2016;115(8):920–928. doi:10.1038/bjc.2016.263
28. Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–294. doi:10.1101/gad.13.3.284
29. Hatakeyama S, Kitagawa M, Nakayama K, et al. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc Natl Acad Sci U S A. 1999;96(7):3859–3863. doi:10.1073/pnas.96.7.3859
30. Orian A, Gonen H, Bercovich B, et al. SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J. 2000;19(11):2580–2591. doi:10.1093/emboj/19.11.2580
31. Zhang B, Zhang Z, Li L, et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-κB signaling. Nat Commun. 2018;9(1):1423. doi:10.1038/s41467-018-03716-9
32. Xinyi Lim Y, Lin H, Chu T, Pin Lim Y. WBP2 promotes BTRC mRNA stability to drive migration and invasion in triple-negative breast cancer via NFκB activation. Mol Oncol. 2021. doi:10.1002/1878-0261.13048
33. Zhong J, Ogura K, Wang Z, Inuzuka H. Degradation of the transcription factor Twist, an oncoprotein that promotes cancer metastasis. Discov Med. 2013;15(80):7–15. doi:10.1038/s41419-018-0657-z
34. Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi:10.1038/ncb1173
35. Zheng Q, Yu JJ, Li C, Li J, Wang J, Wang S. miR-224 targets BTRC and promotes cell migration and invasion in colorectal cancer. 3 Biotech. 2020;10(11):485. doi:10.1007/s13205-020-02477-x
36. Zhou DD, Li HL, Liu W, et al. miR-193a-3p promotes the invasion, migration, and mesenchymal transition in glioma through regulating BTRC. Biomed Res Int. 2021;2021:8928509. doi:10.1155/2021/8928509
37. Berthelet J, Dubrez L. Regulation of apoptosis by inhibitors of apoptosis (IAPs). Cells. 2013;2(1):163–187. doi:10.3390/cells2010163
38. Lazzari E, Meroni G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: from muscular dystrophy to tumours. Int J Biochem Cell Biol. 2016;79:469–477. doi:10.1016/j.biocel.2016.07.023
39. Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A. 1997;94(15):8104–8109. doi:10.1073/pnas.94.15.8104
40. Maynard MA, Ohh M. Von Hippel-Lindau tumor suppressor protein and hypoxia-inducible factor in kidney cancer. Am J Nephrol. 2004;24(1):1–13. doi:10.1159/000075346
41. Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruïne AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010;221(2):125–138. PMID: 20225241. doi:10.1002/path.2689
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.