")
Back to Archived Journals » Hypoxia » Volume 4
Erythropoietin and the use of a transgenic model of erythropoietin-deficient mice
Authors Pichon A, Jeton F, El Hasnaoui-Saadani R, Hagström L, Launay T, Beaudry M, Marchant D, Quidu P, Macarlupu J, Favret F, Richalet J, Voituron N
Received 11 September 2015
Accepted for publication 28 December 2015
Published 7 April 2016 Volume 2016:4 Pages 29—39
DOI https://doi.org/10.2147/HP.S83540
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Roland Wenger
Video abstract presented by Jeton F, Richalet JP, Voituron N.
Views: 618
Aurélien Pichon,1–3 Florine Jeton,1,2 Raja El Hasnaoui-Saadani,4 Luciana Hagström,5 Thierry Launay,6 Michèle Beaudry,1 Dominique Marchant,1 Patricia Quidu,1 Jose-Luis Macarlupu,7 Fabrice Favret,8 Jean-Paul Richalet,1,2 Nicolas Voituron1,2
1Laboratory “Hypoxia and Lung” EA 2363, University Paris 13, Sorbonne Paris Cité, Bobigny Cedex, 2Laboratory of Excellence GR-Ex, Paris, 3Laboratory MOVE EA 6314, FSS, Poitiers University, Poitiers, France; 4Research Unit, College of Medicine, Princess Noura University, Riyadh, Saudi Arabia; 5Laboratório Interdisciplinar de Biociências, Universidade de Brasília, Brasília, Brazil; 6Unité de Biologie Intégrative des Adaptations à l'Exercice, University Paris Saclay and Genopole®, University Sorbonne-Paris-Cité, Paris, France; 7High Altitude Unit, Laboratories for Research and Development, Universidad Peruana Cayetano Heredia, Lima, Peru; 8Laboratory “Mitochondrie, Stress Oxydant et Protection Musculaire” EA 3072, University of Strasbourg, Strasbourg, France
Abstract: Despite its well-known role in red blood cell production, it is now accepted that erythropoietin (Epo) has other physiological functions. Epo and its receptors are expressed in many tissues, such as the brain and heart. The presence of Epo/Epo receptors in these organs suggests other roles than those usually assigned to this protein. Thus, the aim of this review is to describe the effects of Epo deficiency on adaptation to normoxic and hypoxic environments and to suggest a key role of Epo on main physiological adaptive functions. Our original model of Epo-deficient (Epo-TAgh) mice allowed us to improve our knowledge of the possible role of Epo in O2 homeostasis. The use of anemic transgenic mice revealed Epo as a crucial component of adaptation to hypoxia. Epo-TAgh mice survive well in hypoxic conditions despite low hematocrit. Furthermore, Epo plays a key role in neural control of ventilatory acclimatization and response to hypoxia, in deformability of red blood cells, in cerebral and cardiac angiogenesis, and in neuro- and cardioprotection.
Keywords: Epo-TAgh mice, mouse model, physiological functions, hypoxia
Introduction
Life is dependent upon transport and utilization of oxygen (O2) for the metabolic conversion of nutrients into energy,1 making O2 homeostasis an essential process for survival. Indeed, an inadequate level of O2 is detrimental for the tissues, and complex mechanisms serve to maintain in vivo the cellular O2 concentration within a physiological range.1 An imbalance between O2 delivery and requirement, such as at high altitude, activates a variety of specific mechanisms at molecular, cellular, and systemic levels. High altitude is accompanied by low atmospheric O2 pressure, which sequentially leads to insufficient O2 uptake and reduced tissue oxygenation. Hypoxic exposure can be intermittent (obstructive sleep apnea) or continuous (high altitude, cardiorespiratory failure), leading to different strategies to address these stresses. Indeed, acute hypoxia triggers rapid and transient compensatory mechanisms, while chronic hypoxia (CHx) leads to more durable changes with gene expression modifications.1 The hypoxia-inducible factor-1 (HIF-1) is the most important protein regulating homeostasis when O2 is lacking.2 Under hypoxia, stabilization of HIF-1 modulates the expression of hypoxia-regulated genes such as vascular endothelial growth factor (VEGF), glucose transporters, or erythropoietin (Epo).1–5 Epo is a hematopoietic growth factor and represents the main regulator of erythropoiesis. In low O2 conditions, the number of erythrocytes increases to sustain O2 delivery.6 This polycythemia, secondary to an increase in Epo release, is one of the key factors of acclimatization to CHx, through the increase in O2-carrying capacity.7–9 However, despite this well-known role, it was suggested that Epo plays a key role in other physiological functions. Thus, Epo is also known to be fundamental for embryonic development,10 and it is also largely involved in the normal functioning of most organs, including brain, heart, and muscles by regulating numerous cell functions such as calcium flux or cell survival.11,12 To assess these pleiotropic actions of Epo on organs and cells, transgenic models of mice have been developed.13,14
The aim of this review is to provide a brief presentation of the physiological effects of Epo and describe the effects of Epo deficiency on adaptation to normoxic and hypoxic environments in an original model of Epo-deficient (Epo-TAgh) mice. From these observations, we suggest a key role of Epo on multiple physiological adaptive functions, especially in response to hypoxia.
Epo and Epo-receptor expression in organs
Epo is a glycoprotein that is synthesized by peritubular fibroblasts in the kidneys of adults and in hepatocytes of fetal liver.15 Epo was originally believed to play a role that restricted to stimulation of early erythroid precursor proliferation, inhibition of apoptosis, and differentiation of the erythroid lineage. Currently, in addition to its well-known role in red blood cell production, a diverse group of cells has been identified to produce Epo and/or Epo receptor (Epo-R) including endothelial cells, smooth muscle cells, retina tissues, testis, and cells of the central nervous system.12,16–19 Moreover, Epo mRNA was also detected in lungs, testis, heart, and brain but not in skeletal muscles, intestine, or bone marrow of rodents.20,21
Epo and the brain
Epo mRNA is constitutively expressed in the mice brain,22,23 and Epo-R mRNA and protein are also expressed in the brain of rodents.23,24 Indeed, Epo and its receptors are found in the nervous system, including in neurons, astrocytes, and endothelial cells.16,18,22,23,25–27 Epo/Epo-R couple is involved in neuroprotection,28 promotes neural plasticity,29 and could have a potentially antidepressor effect.30,31 Furthermore, Epo/Epo-R pathway is required for normal brain development.32 Indeed, Epo is required for neural progenitor cell proliferation,33 and it avoids neural apoptosis by maintaining Bcl-2 and Bcl-xl expression.34,35 Indirectly, Epo improves sensory, cognitive, and endocrine functions of the central nervous system through its erythropoiesis-stimulating effect, because it increases the O2 supply to the brain. The direct effects of Epo are independent of erythropoiesis.36 Epo has been demonstrated to have neuroprotective effects after ischemic, hypoxic, metabolic, neurotoxic, and excitotoxic stress in the nervous system. Epo operates at several levels within the central nervous system, including limiting the production of reactive O2 species and glutamate, neurotransmission modulation, promotion of angiogenesis, prevention of apoptosis, reduction in inflammation, and recruitment of stem cells.37–40 Moreover, Epo-R was localized in both brainstem respiratory centers and carotid bodies,24 and it is involved in ventilatory regulations during hypoxic challenges.
Epo and the heart
As in the brain, animal models of ischemia and acute myocardial infarction have shown that Epo reduces infarct size and improves left ventricular (LV) function. These effects are mediated mostly through apoptosis inhibition by activating pro-survival pathways in the myocardium, mobilization of endothelial progenitor cells, and inhibition of migration of inflammatory cells as well as potent pro-angiogenic properties.41 Moreover, recent studies reported that the heart could be a site of Epo production42–44 and, in particular, that cardiac tissue reveals Epo gene and protein expression.20,45,46 Epo-R expression is high in cardiac progenitor cells.47 Epo has angiogenic and antiapoptotic effects in the heart,48,49 and intraperitoneal injection of Epo promotes the differentiation of cardiac progenitor cells into endothelial cells.45 Overall, Epo is known to have a cardioprotective effect through antiapoptotic, anti-inflammatory, and angiogenic effects,50 and it could act on the oxidative stress.51 For example, Epo may enhance protein kinase B or Akt and protect the heart from ischemia–reperfusion injuries.52 This effect of Epo on Akt could regulate cardiomyocyte mitochondrial biogenesis,53 by acting on the Akt/endothelial nitric oxide (NO)-synthase pathway.54 Recently, it has also been shown that acute Epo injection can efficiently improve resuscitation and survival rates in a model of cardiac arrest in pigs.55,56
Epo and the muscle
Epo-R has been identified in muscle biopsies suggesting a potential but undefined role of Epo on muscle metabolism or function.57,58 In humans, these effects were suggested by excessive intake of Epo during endurance exercise in order to improve performance.59 In some athletes, Epo increases performance during endurance exercises more than expected on the only basis of an increase in hematocrit. This suggests a potentially direct effect of Epo on muscle metabolism,60 even if this effect remains to be validated.
Epo could induce angiogenesis and accumulation of VEGF in skeletal muscles submitted to ischemia–reperfusion experiments61 and act as a promoter of growth factors in skeletal muscles.57 In vitro, Epo-R mRNA and protein expression were identified in the primary muscle cells of mammals.57,62,63 However, the effects of administration of human recombinant Epo (rhu-Epo) and the role of Epo-R in culture media remain controversial. Some authors showed a positive effect,57 and some others showed no effect,62,63 on primary myoblast proliferation. In vivo, although mRNA-encoding Epo-R and Epo-R proteins were detected in rats,64 skeletal muscle treatment by rhu-Epo have shown controversial results. However, Epo is suspected to induce a shift in muscle fiber metabolism toward a more oxidative phenotype65,66 and prevent the impairment of mitochondrial structure and function.67 Moreover, some authors observed a positive effect of rhu-Epo on myoblast apoptosis,64,68 proteolysis, glycolysis, and mitochondrial functions.69 The authors suggested70 that indirect effects of Epo treatment through the amelioration of O2 supply could explain the observed effects of Epo on skeletal muscles during endurance exercises.
Epo underproduction and clinical disorders
In adults, insufficient Epo production results mostly from direct damage to Epo-producing cells in the kidneys or to a lesser extent from the suppression of Epo production by inflammatory cytokines.71 Indeed, in patients suffering from rheumatoid arthritis, cancer, and acquired immune deficiency syndrome (AIDS), inflammatory cytokines suppress Epo gene expression.71–75 In contrast, patients with renal failure generally develop anemia due to the suppression of erythropoiesis and to a moderate reduction in red cell life span.71 Furthermore, the main reason of anemia in patients with uremia is an insufficient Epo production.71,76,77 Diabetic nephropathy can also lead to Epo deficiency and anemia.78 Moreover, exposure to metals such as cadmium or platinum results in a modification of the structure and function of renal proximal tube, resulting in the suppression of Epo production.71 Patients with increased plasma viscosity due to monoclonal dysproteinemias have an inappropriate Epo production.71,79
Mouse model of Epo deficiency
A mouse model of Epo-TAgh mice was used for the first time for the identification of the renal Epo-producing cells.80 We then developed this model in 2006 in our laboratory to assess the potential roles of Epo and/or anemia in the adaptation processes to hypoxia.
The transgenic construct contains an SV40 sequence in the five untranslated region of the mouse Epo gene, which is flanked on each side by 9 and 7.5 kb of DNA from the mouse Epo locus.80 Anemia-inducible Epo expression was observed in the kidneys.80 Thus, these mice present a severe reduction in Epo gene expression,13,80 leading to low plasmatic level of Epo (122±16 pg/mL in wild type [WT] vs 53±18 pg/mL in Epo-TAgh mice) and thus chronic anemia with low hemoglobin concentration (17.1±0.3 g in WT vs 6.9±0.3 in Epo-TAgh mice; Table 1). However, Epo-TAgh mice have a good survival rate in CHx (14 days, 4,500 m) through an increase in ventilation and cardiac output.81,82 They also develop cerebrovascular adaptations to chronic anemia and hypoxia.83
 | Table 1 Characteristics of wild-type and Epo-TAgh male mice in normoxic conditions |
Characteristics of Epo-TAgh mice in normoxic and hypoxic conditions: effect of Epo deficiency and/or anemia
Blood characteristics
Red blood cells are responsible for the transport of >98% of the O2 into the blood. In order to deliver O2 to the tissues, red blood cells deform as they enter capillaries. In Epo-TAgh mice, hematocrit and hemoglobin are ~60% lower (Table 1) than those observed in WT mice.82 Furthermore, Epo deficiency decreases blood viscosity and slightly reduces red blood cell deformability.84 After CHx, hemoglobin concentration remained 60% lower in Epo-TAgh mice as compared with that of WT mice,81 despite a proportional increase in Epo concentration as compared with WT animals.85 Nevertheless, in Epo-TAgh mice, Epo concentration following acute hypoxia increased to reach only the normoxic WT value83 and returned to basal value after CHx.20
Cardiac characteristics
Epo-TAgh mice show right ventricular and LV hypertrophy,82 leading to increased stroke volume and cardiac output (Table 1). This adaptive process, already described,86 allows to offset the fall in arterial O2 content due to anemia. Echocardiographic data confirmed compensatory LV hypertrophy, higher myocardial chamber volumes, and a higher cardiac output,20 which could be explained by an increase in preload without change in left ventricle afterload as depicted by unchanged blood pressure. Although cardiac output was increased in Epo-TAgh mice, O2 delivery remained lower than in control WT animals. Furthermore, Epo/Epo-R pathway is known to be involved in the transcription of target genes that mainly involved in the inhibition of apoptosis and cell proliferation87 through the phosphorylation of Jak2 and STAT-5. Because of unchanged P-STAT-5/STAT-5 ratio, we could not confirm the activation of this cardioprotective pathway in response to chronic Epo deficiency.20 In addition to its angiogenic function, VEGF may also activate pathways associated with NO synthesis and thus induce vasodilation, improving blood supply to cardiac cells.88 Thus, we demonstrated that chronic Epo deficiency induces a cardiac angiogenesis probably mediated by HIF-1α/VEGF (Figure 1) and Epo-R pathways, which could optimize O2 supply and limit the consequences of chronic anemia on cardiac cells.
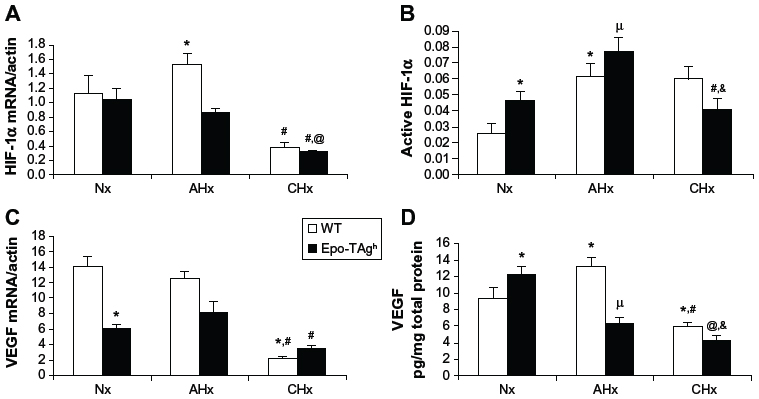 | Figure 1 Cardiac angiogenesis in Epo-TAgh mice. |
The effects of both chronic Epo deficiency and hypoxia on myocardial contractile function and cardioprotective processes were investigated using Epo-TAgh mice.20 The initiation of cardioprotective mechanisms was estimated through Epo-R/P-STAT5 signalization as well as by a change in the P-STAT5/STAT5 ratio in response to chronic Epo deficiency and/or hypoxia. When 14 days of CHx were added to Epo deficiency, the expected cardiac hypertrophy89 was reduced and cardiac output could not catch up with the O2 demand. Systolic blood pressure did not increase indicating that systemic afterload was not responsible for the decrease in cardiac output. Moreover, CHx did not significantly affect right ventricular hemodynamics, and Epo-TAgh mice did not develop pulmonary hypertension. Therefore, the decrease in cardiac output was not the consequence of right ventricular failure. After CHx, Epo-TAgh mice displayed a lower LV hypertrophy than the normoxic anemic mice, which could account for the decrease in cardiac output and O2 delivery. Furthermore, our data showed a mild alteration of diastolic and systolic LV function. These results suggested that altered myocardial function in Epo-TAgh mice exposed to CHx could participate in the failure of cardiac adaptation in this severe condition.
Although hypoxia or Epo-deficiency leads to an overexpression of HIF-1α, VEGF (Figure 1), Epo, and Epo-R, we did not observe a synergic effect of these combined constraints on the heart of Epo-TAgh mice exposed to CHx, except for P-STAT-5/STAT-5 ratio. However, this ratio was lower in Epo-TAgh than in WT mice exposed to CHx, suggesting that the activation of the cardioprotective pathways downstream the Epo/Epo-R system may represent a limiting step. As we found a decrease in LV hypertrophy and functional LV adaptation, a depressed HIF-1α/VEGF pathway (Figure 1) as well as a reduced O2 delivery, we suggested that cardiac adaptive mechanisms that take place with chronic Epo deficiency and hypoxia might require extensive Epo effects (angiogenesis, cardioprotection) on the heart.
Ventilatory and metabolic characteristics
Our first publication on the Epo-TAgh mice model showed a greater ventilation in normoxia in these anemic mice as compared to WT animals.82 This difference was mainly due to a larger tidal volume. However, more recently, we have not observed a baseline ventilatory adaptation in Epo-TAgh male mice in normoxia (Table 1, Figure 2) when compared with that of WT mice.90 Some explanations could be proposed to explain this difference. First, in the study of Macarlupu et al, the control animals were classical WT animals and not littermates, and it is well known that the environmental conditions during the first day of life could induce significant changes in ventilatory variables.91 Second, the method used to measure ventilatory variables can induce slight differences, according to the plethysmograph used, for example. Third, it could be possible that Epo-TAgh mice could have exhibited some epigenetic adjustments between 2006 and 2014. Indeed, more than 30 generations of mice have lived between the first and the last experiment, and it is not impossible that epigenetic changes would improve the whole O2 transport steps, and thus, ventilatory adaptation could appear in the last generations of mice.92
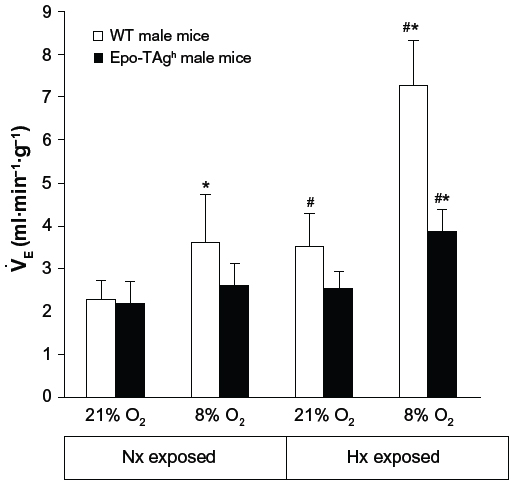 | Figure 2 Ventilatory response to hypoxia in Epo-TAgh mice. |
In female Epo-TAgh mice, we observed a difference in respiratory frequency and minute ventilation with larger values for the Epo-TAgh mice as compared with those of WT mice (unpublished data). There is no change in resting O2 consumption ( O2) in Epo-TAgh mice, while O2 max is only 30% reduced (Table 1) as compared with that of WT mice,82 despite a 60% reduction in hemoglobin concentration. The normal resting O2 in anemic Epo-TAgh mice could also be explained by the elevated cardiac output associated with better tissue extraction of O2,86 which could compensate for the decrease in O2 transport capacity. Severe anemia also generally induces a reduction in physical performance.93–95 In Epo-TAgh mice, the reduction in O2max was only moderate, suggesting compensatory mechanisms such as an increase in maximal cardiac output, augmented capillarization, and better O2 extraction.
O2) in Epo-TAgh mice, while O2 max is only 30% reduced (Table 1) as compared with that of WT mice,82 despite a 60% reduction in hemoglobin concentration. The normal resting O2 in anemic Epo-TAgh mice could also be explained by the elevated cardiac output associated with better tissue extraction of O2,86 which could compensate for the decrease in O2 transport capacity. Severe anemia also generally induces a reduction in physical performance.93–95 In Epo-TAgh mice, the reduction in O2max was only moderate, suggesting compensatory mechanisms such as an increase in maximal cardiac output, augmented capillarization, and better O2 extraction.
The normal ventilatory response to acute hypoxia is characterized in adult mammals by a hyperventilation followed by a relative ventilatory decline named “roll off”.96,97 If hypoxia persists, an increase in ventilation occurs (ventilatory acclimatization to CHx),98 which is accompanied by an increase in the sensitivity of the respiratory control system.99–101 Epo-TAgh mice displayed neither ventilatory response to acute hypoxia nor ventilatory acclimatization to CHx (Figure 2).90 However, after 14 days of exposure to chronic hypoxia, Epo-TAgh mice increased their ventilation when exposed acutely to a hypoxic stress (Figure 2; 8% O2, 5 minutes).90 These results differ from those previously published.82 We cannot exclude the fact that our transgenic mice, along generations, developed adaptation strategies to cope with Epo deficiency and/or chronic anemia.
Brain adaptations
Epo deficiency in Epo-TAgh mice leads to cerebral adaptations (Figures 3 and 4).83 Indeed, in the brain of these normoxic mice, we observed an increase in the transcript and the protein levels of HIF-1α, VEGF (Figure 3), Epo-R (Figure 4), and P-STAT-5/STAT-5 ratio accompanied with an increase in cerebral capillary density. Taken together, these data suggest that Epo-TAgh mice have developed cerebral angiogenesis, probably via the HIF-1α/VEGF pathway (Figure 3), optimizing O2 diffusion as previously described.83,102 Furthermore, the increase in P-STAT-5/STAT-5 ratio in the brain suggests neuroprotective mechanisms and angiogenesis with a decrease in apoptosis and an increase in cell proliferation.23,103 Overall, these results illustrate the direct and indirect effects of Epo in terms of O2 delivery improvement and the activation of neuroprotective mechanism to counteract the lack of Epo in the brain.
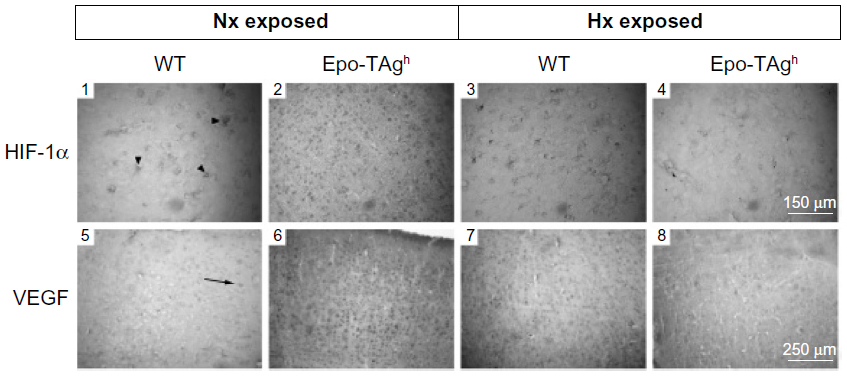 | Figure 3 Cerebral angiogenesis in Epo-TAgh mice. |
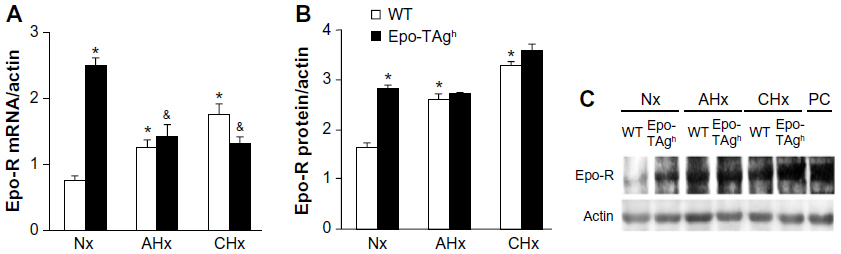 | Figure 4 Effect of Epo deficiency on Epo-R expression in cerebral cortex. |
Skeletal muscles
Our model of transgenic Epo-TAgh mice has allowed us to study the role of Epo on skeletal muscle development, angiogenesis, and acclimatization to hypoxia. Our main results displayed a developmental adaptation to Epo deficiency and/or chronic anemia by an improvement of microvessel network (Table 2) in both fast and slow skeletal muscles.85 This adaptation of Epo-TAgh mice was not accompanied by any difference in skeletal muscles for contractile structure, metabolism, maximal strength, fatigability, contraction time, and relaxation time. Moreover, we never observed any overexpression of HIF-1α or VEGF protein. The discrepancy between our results and those of Mille-Hamard et al’s team69 could be explained by a genetic developmental adaptation to chronic anemia in our transgenic model. In the Mille-Hamard et al’s transgenic model, Epo deficiency is induced by vaccination of adult mice, so that developmental adaptation has not occurred. When exposed to severe chronic hypobaric hypoxia (4,300 m) during 14 days, skeletal muscles of Epo-TAgh mice were not submitted to deconditioning. Neither skeletal muscle phenotype nor skeletal muscle functions were altered compared with those of WT mice.104 In the skeletal muscles of our Epo-TAgh model, we did not detect Epo mRNA in both normoxia and hypoxia. Moreover, the hypoxia-induced elevation of circulating Epo was not correlated with an increase in Epo concentration in skeletal muscle. Overall, these results favor the hypothesis of an indirect effect of Epo on skeletal muscles. We clearly show that, in our model, the deficit in oxygenation caused by anemia is responsible for the main change in skeletal muscles such as the improvement of microvessel network (Table 3). Unfortunately, we cannot study the regeneration process of the Epo-TAgh model, because in this mutant, Epo has been replaced by antigen T, which has been shown to alter skeletal muscle regeneration.105
 | Table 2 Main characteristics of Epo-TAgh mice when compared with those of wild-type mice |
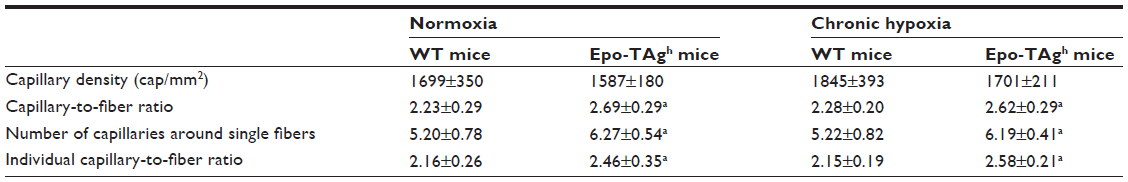 | Table 3 Indices of the microvascular supply of the deep region of gastrocnemius muscle fibers in normoxia and after chronic hypoxia |
Limits of our models
Experiments were performed on whole-body Epo-TAgh mice13 that display a very low hematocrit (20%) and did not develop polycythemia after CHx.81 Therefore, it is important to note that our model of transgenic mice combines the effects of chronic anemia (low O2 content) and the effects of chronic Epo deficiency. To differentiate the respective effect of each constraint is rather difficult since chronic anemia itself is a consequence of chronic Epo deficiency. However, it could be speculated from the activation of HIF/VEGF systems that the reduction in tissue O2 delivery (and therefore chronic anemia) is the main trigger of the observed adaptations.
Conclusion
For many years, Epo was mainly considered a growth factor for erythropoiesis and a determinant factor for the acclimatization to CHx only through an increase in O2 transport capacity. It appears from recent studies that it may also participate in the acute and chronic responses to hypoxia (Table 2) through the activation of Epo-Rs in various organs (brain, heart, muscle, chemoreceptors). However, there is still a lot of debates and uncertainties about the presence and functionalities of these receptors. Our model of Epo-TAgh mice may help to unravel a possible key role of Epo in O2 homeostasis. Indeed, our studies demonstrate that high levels of Epo are not necessary for survival in chronic moderate hypoxia. Moreover, we showed that Epo could play a key-regulating role in the neural control of ventilatory acclimatization to hypoxia and hypoxic ventilatory response probably via a catalyzing role on the NO central pathway. We also demonstrated that chronic Epo deficiency induced cerebral and cardiac angiogenesis, which could have synergic effects not only in neuro- and cardioprotection but also in O2 supply optimization, in order to limit the consequences of chronic anemia on cerebral and cardiac tissues. However, under both constraints (chronic Epo deficiency and hypoxia), angiogenesis, neuroprotective, and cardioprotective pathways along with a functional LV adaptation failed to occur, showing the limits of these adaptive processes in heart and brain, but more importantly suggesting a crucial role of Epo in main physiological functions.
Some future areas of research could focus on the role of Epo as a global regulator of the cardioventilatory adaptations from erythrocytes synthesis, blood hemorheology, blood volume regulation, and ventilatory control.106 Epo seems to be able to protect tissues (cardiomyocytes, lung, neurons) from various aggressions such as hypoxia, ischemia–reperfusion, or inflammation.107 Moreover, there are few data on the possible effect of Epo on the ventilatory response to hypercapnia in contrary to the response to hypoxia. The role of Epo/Epo-R on the oxidative stress needs also to be studied later as this is central on tissue antiapoptosis properties.
Acknowledgments
This work was supported by the University Paris 13, Sorbone Paris Cité, the “Bonus Qualité Recherche”, and “Institut Fédératif de Recherche Biomédicale” programs of the University Paris 13. FJ was supported by a Laboratory of Excellence GR-Ex fellowship. The Laboratory of Excellence GR-Ex (ANR-11-LABX-0051) was funded by the program “Investissement d’avenir” of the French National Research agency (ANR-11-IDEX-0005-02).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2(3):336–361. | |
Gassmann M, Soliz J. Erythropoietin modulates the neural control of hypoxic ventilation. Cell Mol Life Sci. 2009;66(22):3575–3582. | |
Fandrey J, Gorr TA, Gassmann M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc Res. 2006;71(4):642–651. | |
Yeo EJ, Chun YS, Park JW. New anticancer strategies targeting HIF-1. Biochem Pharmacol. 2004;68(6):1061–1069. | |
Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. | |
Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122–1128. | |
Favret F, Richalet JP, Henderson KK, Germack R, Gonzalez NC. Myocardial adrenergic and cholinergic receptor function in hypoxia: correlation with O(2) transport in exercise. Am J Physiol Regul Integr Comp Physiol. 2001;280(3):R730–R738. | |
Gonzalez NC, Clancy RL, Moue Y, Richalet JP. Increasing maximal heart rate increases maximal O2 uptake in rats acclimatized to simulated altitude. J Appl Physiol. 1998;84(1):164–168. | |
Robach P, Fulla Y, Westerterp KR, Richalet JP. Comparative response of EPO and soluble transferrin receptor at high altitude. Med Sci Sports Exerc. 2004;36(9):1493–1498. [discussion 1492]. | |
Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. | |
Ogunshola OO, Bogdanova AY. Epo and non-hematopoietic cells: what do we know? Methods Mol Biol. 2013;982:13–41. | |
Gassmann M, Heinicke K, Soliz J, Ogunshola OO. Non-erythroid functions of erythropoietin. Adv Exp Med Biol. 2003;543:323–330. | |
Binley K, Askham Z, Iqball S, et al. Long-term reversal of chronic anemia using a hypoxia-regulated erythropoietin gene therapy. Blood. 2002;100(7):2406–2413. | |
Wiessner C, Allegrini PR, Ekatodramis D, Jewell UR, Stallmach T, Gassmann M. Increased cerebral infarct volumes in polyglobulic mice overexpressing erythropoietin. J Cereb Blood Flow Metab. 2001;21(7):857–864. | |
Fisher JW. Erythropoietin: physiology and pharmacology update. Exp Biol Med. 2003;228(1):1–14. | |
Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J Biol Chem. 1994;269(30):19488–19493. | |
Marti HH, Gassmann M, Wenger RH, et al. Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney Int. 1997;51(2):416–418. | |
Bernaudin M, Marti HH, Roussel S, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19(6):643–651. | |
Ammarguellat F, Gogusev J, Drueke TB. Direct effect of erythropoietin on rat vascular smooth-muscle cell via a putative erythropoietin receptor. Nephrol Dial Transplant. 1996;11(4):687–692. | |
El Hasnaoui-Saadani R, Marchant D, Pichon A, et al. Epo deficiency alters cardiac adaptation to chronic hypoxia. Respir Physiol Neurobiol. 2013;186(2):146–154. | |
Tan CC, Eckardt KU, Firth JD, Ratcliffe PJ. Feedback modulation of renal and hepatic erythropoietin mRNA in response to graded anemia and hypoxia. Am J Physiol. 1992;263(3 pt 2):F474–F481. | |
Digicaylioglu M, Bichet S, Marti HH, et al. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci USA. 1995;92(9):3717–3720. | |
Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207(pt 18):3233–3242. | |
Soliz J, Joseph V, Soulage C, et al. Erythropoietin regulates hypoxic ventilation in mice by interacting with brainstem and carotid bodies. J Physiol. 2005;568(pt 2):559–571. | |
Siren AL, Knerlich F, Poser W, Gleiter CH, Brück W, Ehrenreich H. Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. 2001;101(3):271–276. | |
Bernaudin M, Bellail A, Marti HH, et al. Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia. 2000;30(3):271–278. | |
Brines ML, Ghezzi P, Keenan S, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA. 2000;97(19):10526–10531. | |
Juul S. Neuroprotective role of erythropoietin in neonates. J Matern Fetal Neonatal Med. 2012;25(suppl 4):105–107. | |
Almaguer-Melian W, Merceron-Martinez D, Pavon-Fuentes N, et al. Erythropoietin promotes neural plasticity and spatial memory recovery in fimbria-fornix-lesioned rats. Neurorehabil Neural Repair. 2015;29(10):979–988. | |
Miskowiak KW, Vinberg M, Harmer CJ, Ehrenreich H, Kessing LV. Erythropoietin: a candidate treatment for mood symptoms and memory dysfunction in depression. Psychopharmacology. 2012; 219(3):687–698. | |
Osborn M, Rustom N, Clarke M, et al. Antidepressant-like effects of erythropoietin: a focus on behavioural and hippocampal processes. PLoS One. 2013;8(9):e72813. | |
Yu X, Shacka JJ, Eells JB, et al. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129(2):505–516. | |
Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem. 2007;282(35):25875–25883. | |
Sola A, Rogido M, Lee BH, Genetta T, Wen TC. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57(4):481–487. | |
Silva M, Grillot D, Benito A, Richard C, Nuñez G, Fernández-Luna JL. Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood. 1996;88(5):1576–1582. | |
Jelkmann W. Effects of erythropoietin on brain function. Curr Pharm Biotechnol. 2005;6(1):65–79. | |
Wang L, Zhang Z, Wang Y, Zhang R, Chopp M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35(7):1732–1737. | |
Ribatti D, Vacca A, Roccaro AM, Crivellato E, Presta M. Erythropoietin as an angiogenic factor. Eur J Clin Invest. 2003;33(10):891–896. | |
Marti HH, Bernaudin M, Petit E, Bauer C. Neuroprotection and angiogenesis: dual role of erythropoietin in brain ischemia. News Physiol Sci. 2000;15:225–229. | |
Jaquet K, Krause K, Tawakol-Khodai M, Geidel S, Kuck KH. Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc Res. 2002;64(2):326–333. | |
Roubille F, Prunier F, Barrere-Lemaire S, et al. What is the role of erythropoietin in acute myocardial infarct? Bridging the gap between experimental models and clinical trials. Cardiovasc Drugs Ther. 2013; 27(4):315–331. | |
Bin-Jaliah I, Ammar HI, Mikhailidis DP, et al. Cardiac adaptive responses after hypoxia in an experimental model. Angiology. 2010;61(2):145–156. | |
Cai Z, Manalo DJ, Wei G, et al. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108(1):79–85. | |
Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109(17):2050–2053. | |
Hoch M, Fischer P, Stapel B, et al. Erythropoietin preserves the endothelial differentiation capacity of cardiac progenitor cells and reduces heart failure during anticancer therapies. Cell Stem Cell. 2011;9(2):131–143. | |
Miro-Murillo M, Elorza A, Soro-Arnaiz I, et al. Acute Vhl gene inactivation induces cardiac HIF-dependent erythropoietin gene expression. PLoS One. 2011;6(7):e22589. | |
Zafiriou MP, Noack C, Unsold B, et al. Erythropoietin responsive cardiomyogenic cells contribute to heart repair post myocardial infarction. Stem Cells. 2014;32(9):2480–2491. | |
Kertesz N, Wu J, Chen TH, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. 2004;276(1):101–110. | |
Marzo F, Lavorgna A, Coluzzi G, et al. Erythropoietin in heart and vessels: focus on transcription and signalling pathways. J Thromb Thrombolysis. 2008;26(3):183–187. | |
Sanchis-Gomar F, Garcia-Gimenez JL, Pareja-Galeano H, Romagnoli M, Perez-Quilis C, Lippi G. Erythropoietin and the heart: physiological effects and the therapeutic perspective. Int J Cardiol. 2014;171(2):116–125. | |
Burger D, Xenocostas A, Feng QP. Molecular basis of cardioprotection by erythropoietin. Curr Mol Pharmacol. 2009;2(1):56–69. | |
Ong SB, Hall AR, Dongworth RK, et al. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. Thromb Haemost. 2015;113(3):513–521. | |
Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res. 2010;106(11):1722–1730. | |
Qin C, Zhou S, Xiao Y, Chen L. Erythropoietin enhances mitochondrial biogenesis in cardiomyocytes exposed to chronic hypoxia through Akt/eNOS signalling pathway. Cell Biol Int. 2014;38(3):335–342. | |
Borovnik-Lesjak V, Whitehouse K, Baetiong A, Artin B, Radhakrishnan J, Gazmuri RJ. High-dose erythropoietin during cardiac resuscitation lessens postresuscitation myocardial stunning in swine. Trans Res. 2013;162(2):110–121. | |
Vasileiou PV, Xanthos T, Barouxis D, et al. Erythropoietin administration facilitates return of spontaneous circulation and improves survival in a pig model of cardiac arrest. Am J Emerg Med. 2014;32(8):871–877. | |
Ogilvie M, Yu X, Nicolas-Metral V, et al. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J Biol Chem. 2000;275(50):39754–39761. | |
Rundqvist H, Rullman E, Sundberg CJ, et al. Activation of the erythropoietin receptor in human skeletal muscle. Eur J Endocrinol. 2009;161(3):427–434. | |
Lundby C, Olsen NV. Effects of recombinant human erythropoietin in normal humans. J Physiol. 2011;589(pt 6):1265–1271. | |
Lundby C, Hellsten Y, Jensen MB, Munch AS, Pilegaard H. Erythropoietin receptor in human skeletal muscle and the effects of acute and long-term injections with recombinant human erythropoietin on the skeletal muscle. J Appl Physiol. 2008;104(4):1154–1160. | |
Contaldo C, Meier C, Elsherbiny A, et al. Human recombinant erythropoietin protects the striated muscle microcirculation of the dorsal skinfold from postischemic injury in mice. Am J Physiol Heart Circ Physiol. 2007;293(1):H274–H283. | |
Launay T, Hagstrom L, Lottin-Divoux S, et al. Blunting effect of hypoxia on the proliferation and differentiation of human primary and rat L6 myoblasts is not counteracted by Epo. Cell Prolif. 2010;43(1):1–8. | |
Lamon S, Zacharewicz E, Stephens AN, Russell AP. EPO-receptor is present in mouse C2C12 and human primary skeletal muscle cells but EPO does not influence myogenesis. Physiol Rep. 2014;2(3):e00256. | |
Rotter R, Menshykova M, Winkler T, et al. Erythropoietin improves functional and histological recovery of traumatized skeletal muscle tissue. J Orthop Res. 2008;26(12):1618–1626. | |
Cayla JL, Maire P, Duvallet A, Wahrmann JP. Erythropoietin induces a shift of muscle phenotype from fast glycolytic to slow oxidative. Int J Sports Med. 2008;29(6):460–465. | |
Hojman P, Brolin C, Gissel H, et al. Erythropoietin over-expression protects against diet-induced obesity in mice through increased fat oxidation in muscles. PLoS One. 2009;4(6):e5894. | |
Nouette-Gaulain K, Bellance N, Prevost B, et al. Erythropoietin protects against local anesthetic myotoxicity during continuous regional analgesia. Anesthesiology. 2009;110(3):648–659. | |
Jia Y, Warin R, Yu X, Epstein R, Noguchi CT. Erythropoietin signaling promotes transplanted progenitor cell survival. FASEB J. 2009;23(9):3089–3099. | |
Mille-Hamard L, Billat VL, Henry E, et al. Skeletal muscle alterations and exercise performance decrease in erythropoietin-deficient mice: a comparative study. BMC Med Genomics. 2012;5:29. | |
Lamon S, Russell AP. The role and regulation of erythropoietin (EPO) and its receptor in skeletal muscle: how much do we really know? Front Physiol. 2013;4:176. | |
Bunn HF. Erythropoietin. Cold Spring Harb Perspect Med. 2013;3(3): a011619. | |
Means RT Jr. Clinical application of recombinant erythropoietin in the anemia of chronic disease. Hematol Oncol Clin North Am. 1994;8(5):933–944. | |
Miller CB, Jones RJ, Piantadosi S, Abeloff MD, Spivak JL. Decreased erythropoietin response in patients with the anemia of cancer. N Engl J Med. 1990;322(24):1689–1692. | |
Spivak JL. Recombinant human erythropoietin and the anemia of cancer. Blood. 1994;84(4):997–1004. | |
Fischl M, Galpin JE, Levine JD, et al. Recombinant human erythropoietin for patients with AIDS treated with zidovudine. N Engl J Med. 1990;322(21):1488–1493. | |
Caro J, Brown S, Miller O, Murray T, Erslev AJ. Erythropoietin levels in uremic nephric and anephric patients. J Lab Clin Med. 1979;93(3):449–458. | |
Cotes PM. Physiological studies of erythropoietin in plasma. In: Jelkmann W, Gross AJ, editors. Erythropoietin. Berlin: Springer; 1989:57–79. | |
Bosman DR, Winkler AS, Marsden JT, Macdougall IC, Watkins PJ. Anemia with erythropoietin deficiency occurs early in diabetic nephropathy. Diabetes Care. 2001;24(3):495–499. | |
Singh A, Eckardt KU, Zimmermann A, et al. Increased plasma viscosity as a reason for inappropriate erythropoietin formation. J Clin Invest. 1993;91(1):251–256. | |
Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993;44(5):1149–1162. | |
Macarlupu JL, Buvry A, Morel OE, León-Velarde F, Richalet JP, Favret F. Time course of ventilatory acclimatisation to hypoxia in a model of anemic transgenic mice. Respir Physiol Neurobiol. 2006;153(1):14–22. | |
Macarlupu JL, Buvry A, Morel OE, León-Velarde F, Richalet JP, Favret F. Characterisation of the ventilatory response to hypoxia in a model of transgenic anemic mice. Respir Physiol Neurobiol. 2006;150(1):19–26. | |
El Hasnaoui-Saadani R, Pichon A, Marchant D, et al. Cerebral adaptations to chronic anemia in a model of erythropoietin-deficient mice exposed to hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296(3):R801–R811. | |
Pichon A, Lamarre Y, Voituron N, et al. Red blood cell deformability is very slightly decreased in erythropoietin deficient mice. Clin Hemorheol Microcirc. 2014;56(1):41–46. | |
Hagstrom L, Agbulut O, El-Hasnaoui-Saadani R, et al. Epo is relevant neither for microvascular formation nor for the new formation and maintenance of mice skeletal muscle fibres in both normoxia and hypoxia. J Biomed Biotechnol. 2010;2010:137817. | |
Deem S, Hedges RG, McKinney S, Polissar NL, Alberts MK, Swenson ER. Mechanisms of improvement in pulmonary gas exchange during isovolemic hemodilution. J Appl Physiol. 1999;87(1):132–141. | |
Lopez TV, Lappin TR, Maxwell P, et al. Autocrine/paracrine erythropoietin signalling promotes JAK/STAT-dependent proliferation of human cervical cancer cells. Int J Cancer. 2011;129(11):2566–2576. | |
Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125(pt 11):2549–2557. | |
Favret F, Henderson KK, Clancy RL, Richalet JP, Gonzalez NC. Exercise training alters the effect of chronic hypoxia on myocardial adrenergic and muscarinic receptor number. J Appl Physiol. 2001;91(3):1283–1288. | |
Voituron N, Jeton F, Cholley Y, et al. Catalyzing role of erythropoietin on the nitric oxide central pathway during the ventilatory responses to hypoxia. Physiol Rep. 2014;2(2):e00223. | |
D’Amato FR, Zanettini C, Lampis V, et al. Unstable maternal environment, separation anxiety, and heightened CO2 sensitivity induced by gene-by-environment interplay. PLoS One. 2011;6(4):e18637. | |
Ivy CM, Scott GR. Control of breathing and the circulation in high-altitude mammals and birds. Comp Biochem Physiol A Mol Integr Physiol. 2015;186:66–74. | |
Gregg SG, Willis WT, Brooks GA. Interactive effects of anemia and muscle oxidative capacity on exercise endurance. J Appl Physiol. 1989;67(2):765–770. | |
Hogan MC, Bebout DE, Wagner PD. Effect of hemoglobin concentration on maximal O2 uptake in canine gastrocnemius muscle in situ. J Appl Physiol. 1991;70(3):1105–1112. | |
Koskolou MD, Roach RC, Calbet JA, Rådegran G, Saltin B. Cardiovascular responses to dynamic exercise with acute anemia in humans. Am J Physiol. 1997;273(4 pt 2):H1787–H1793. | |
Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev. 2010;90(2):675–754. | |
Maxova H, Vizek M. Biphasic ventilatory response to hypoxia in unanesthetized rats. Physiol Res. 2001;50(1):91–96. | |
Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112(2):123–134. | |
Olson EB Jr, Dempsey JA. Rat as a model for humanlike ventilatory adaptation to chronic hypoxia. J Appl Physiol. 1978;44(5):763–769. | |
Bisgard GE, Neubauer LA. Peripheral and central effects of hypoxia. In: Dempsey JA, Pack AI, editors. Regulation of Breathing. New York, NY: Dekker; 1995:617–618. | |
Powell FL, Huey KA, Dwinell MR. Central nervous system mechanisms of ventilatory acclimatization to hypoxia. Respir Physiol. 2000;121(2–3):223–236. | |
LaManna JC, Chavez JC, Pichiule P. Structural and functional adaptation to hypoxia in the rat brain. J Exp Biol. 2004;207(pt 18):3163–3169. | |
Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36(6):1264–1269. | |
Hagstrom L, Canon F, Agbulut O, et al. Skeletal muscle intrinsic functional properties are preserved in a model of erythropoietin deficient mice exposed to hypoxia. Pflugers Arch. 2010;459(5):713–723. | |
Mouly V, Edom F, Decary S, Vicart P, Barbert JP, Butler-Browne GS. SV40 large T antigen interferes with adult myosin heavy chain expression, but not with differentiation of human satellite cells. Exp Cell Res. 1996;225(2):268–276. | |
Rosario R, Epstein M. Relationship between erythropoietin administration and alterations of renin-angiotensin-aldosterone. J Renin Angiotensin Aldosterone Syst. 2006;7(3):135–138. | |
Rocha J, Eduardo-Figueira M, Barateiro A, et al. Erythropoietin reduces acute lung injury and multiple organ failure/dysfunction associated to a scald-burn inflammatory injury in the rat. Inflammation. 2015;38(1):312–326. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.