Back to Journals » OncoTargets and Therapy » Volume 13
Erdheim–Chester Disease and Acute Myeloid Leukemia with Mutated NPM1 in a Patient with Clonal Hematopoiesis: A Case Report
Authors Papageorgiou SG ![]() , Divane A, Roumelioti M, Kottaridi C, Bouchla A, Georgakopoulos A, Ieremiadou F, Daraki A, Bazani E, Thomopoulos TP, Chatziioannou S, Mavrogenis A
, Divane A, Roumelioti M, Kottaridi C, Bouchla A, Georgakopoulos A, Ieremiadou F, Daraki A, Bazani E, Thomopoulos TP, Chatziioannou S, Mavrogenis A ![]() , Panayiotidis P, Panayiotides IG, Pappa V, Foukas PG
, Panayiotidis P, Panayiotides IG, Pappa V, Foukas PG
Received 14 August 2020
Accepted for publication 20 September 2020
Published 16 November 2020 Volume 2020:13 Pages 11689—11695
DOI https://doi.org/10.2147/OTT.S276497
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Federico Perche
Sotirios G Papageorgiou,1 Aspasia Divane,2 Maria Roumelioti,3 Christine Kottaridi,4 Anthi Bouchla,1 Alexandros Georgakopoulos,5 Fotini Ieremiadou,2 Aggeliki Daraki,2 Efthymia Bazani,1 Thomas P Thomopoulos,1 Sofia Chatziioannou,5,6 Andreas Mavrogenis,7 Panayiotis Panayiotidis,3 Ioannis G Panayiotides,4 Vasiliki Pappa,1,* Periklis G Foukas4,*
1 2nd Department of Internal Medicine and Research Unit, Hematology Unit, University General Hospital “Attikon”, Haidari, Athens, Greece; 2“LIFE CODE” Private Diagnostic Laboratory, Medical Ltd., Athens, Greece; 3 1st Department of Propaedeutic Medicine, Laikon General Hospital, National and Kapodistrian University of Athens, Athens, Greece; 4 2nd Department of Pathology, National and Kapodistrian University of Athens, Medical School, University General Hospital “Attikon”, Haidari, Athens, Greece; 5 2nd Department of Radiology, Nuclear Medicine Section, National and Kapodistrian University of Athens, University General Hospital “Attikon”, Haidari, Athens, Greece; 6Nuclear Medicine Section, Biomedical Research Foundation Academy of Athens, BRFAA, Athens, Greece; 7 1st Department of Orthopaedics, National and Kapodistrian University of Athens, School of Medicine, University General Hospital “Attikon”, Haidari, Athens, Greece
*These authors contributed equally to this work
Correspondence: Sotirios G Papageorgiou
2nd Department of Internal Medicine and Research Unit, University General Hospital “Attikon”, 1 Rimini St., Haidari 12462 Athens, Greece
Tel +30 210-583-2318
Fax +30 210-538-2306
Email [email protected]
Background: Erdheim–Chester Disease (ECD) is a clonal non-Langerhans histiocytosis, classified as a macrophage-dendritic cell neoplasm in the 2016 WHO classification. The exact cell of origin of ECD is unknown, although some limited evidence suggests that it arises from myeloid progenitors.
Case Presentation: A 43-year-old patient, diagnosed with BRAFV600E mutated ECD, developed NPM1+/FLT3+ acute myeloid leukemia (AML) with wild-type BRAF, 15 months after the initial ECD diagnosis. The patient received intensive chemotherapy plus midostaurin, followed by midostaurin maintenance. Six months into maintenance, the patient remains in complete remission with low-level measurable residual disease, whereas ECD shows a sustained partial metabolic response. Molecular karyotype at several distinct timepoints, namely ECD diagnosis, AML diagnosis, and following treatment of AML, highlighted a molecular signature, indicative of a persistent, underlying clonal hematopoiesis.
Conclusion: This case report suggests that ECD and AML might represent an expansion of two distinct clones in a background of clonal hematopoiesis, indicating their shared origin. Moreover, molecular karyotype might serve as a strong, inexpensive tool for revealing clonal hematopoiesis in cases of negative targeted next-generation sequencing. Finally, the moderate response of ECD to midostaurin suggests that kinase inhibition might have a potential role in ECD treatment.
Keywords: Erdheim–Chester disease, acute myeloid leukemia, clonal hematopoiesis, case report, molecular karyotype, midostaurin
Background
Erdheim–Chester Disease (ECD) is a rare clonal non-Langerhans histiocytosis, classified as a macrophage-dendritic cell neoplasm in the 2016 World Health Organization (WHO) classification.1 It is characterized by tissue infiltration by foamy histiocytes with CD68+, CD163+, factor XIIIa+, and fascin+ CD1a-, and Langerin (CD207)- phenotype, as well as fibrosis and inflammation affecting multiple organs including bones, lungs, kidneys, lymph nodes, heart, and brain.2,3 Most recently, consensus recommendations for evaluation, diagnosis, and treatment of ECD have been published.4 Approximately 55–70% of ECD patients harbor the BRAFV600E mutation, while other activating mutations in the MAPK pathway are also demonstrated in the majority of BRAF wild-type ECD patients.2,5 The exact cell of origin of ECD is unknown, although some evidence suggest that it arises from myeloid progenitors.6–11 In addition, Papo et al12 recently showed a high prevalence (10.1%) of concurrent myeloid neoplasms among adult patients with non-Langerhans histiocytosis. We herewith report a patient with clonal hematopoiesis and BRAFV600E mutated ECD who developed BRAFV600E negative acute myeloid leukemia (AML) with mutated NPM1 and FLT3-TKD.
Case Presentation
A 43-year-old man with an unremarkable medical history presented to the Orthopedic Department of Attikon University Hospital in June 2017 with severe right knee joint pain and effusion, resulting in marked functional impairment. The symptoms had initiated 3 months prior to presentation, with progressive aggravation to rest pain and the inability of any weight bearing. He also reported mild pain at his contralateral knee. The remainder of the clinical examination was unremarkable.
X-rays and computed tomography (CT) showed diffuse heterogeneity of the distal femur and proximal tibia with scattered osteolytic and sclerotic areas involving cancellous bone and intramedullary canal. Magnetic resonance imaging (MRI) of both knees showed symmetrical alterations of signal intensity of the bone marrow, with high signal intensity areas in T2-weighted images, without lysis of the cortex nor bone deformity. Bone scan showed increased radioisotope uptake at the distal femurs, proximal and distal tibias, and tarsal bones. Complete blood count (CBC) was WBC: 13.68x109/L (neutrophils: 10.26x109/L, lymphocytes: 1.2x109/L, monocytic cells: 1.44x109/L), Hb: 11.5g/dL, PLT: 486x109/L, and erythrocyte Sedimentation Rate (ESR) was 107 mm in the first hour. Examination of peripheral blood smear was unremarkable. Serum biochemistry, viral testing, and other serological tests, including a comprehensive tumor marker and autoimmunity panel were within the normal range, except from abnormally elevated C-reactive protein (CRP) (68.20 mg/L, normal range=0.00–6.00). The patient was referred to the Hematology Department for further evaluation and treatment. Bone marrow aspiration and trephine biopsy were performed, which did not reveal any significant pathological findings. Molecular testing for BCR-ABL and JAK2 mutations were negative. Chromosome preparation after bone marrow culture revealed a 46, XY, del(2)(p23p25), t(13;14)(q14;q24)15 karyotype in all examined metaphases (Figure 1IA). Subsequently, the patient underwent surgical biopsy of a lesion from the right femur and knee that showed diffuse infiltration by a CD68+, CD163+, CD1a-, S100- histiocytic cell population, many xanthomatous and a few multinucleated cells, histologic features compatible with ECD (Figure 1IIA–E). Pyrosequencing analysis performed on DNA extracted from formalin-fixed, paraffin-embedded tumor sections (“Supplementary methods” section in Supplementary materials) revealed the presence of the BRAFV600E mutation (Figure 1IIF). An 18-fluorodeoxyglucose- (18-FDG-) positron emission tomography (PET-CT) staging approach revealed metabolic active disease in the left occipital lobe of the brain, cervical and external iliac lymph nodes, the sinuses, multiple areas along the skeleton, as well as in soft tissues (Figure 2A and B). Additionally, MRI of the brain showed a soft tissue lesion at the choroid plexus of the left lateral ventricle.
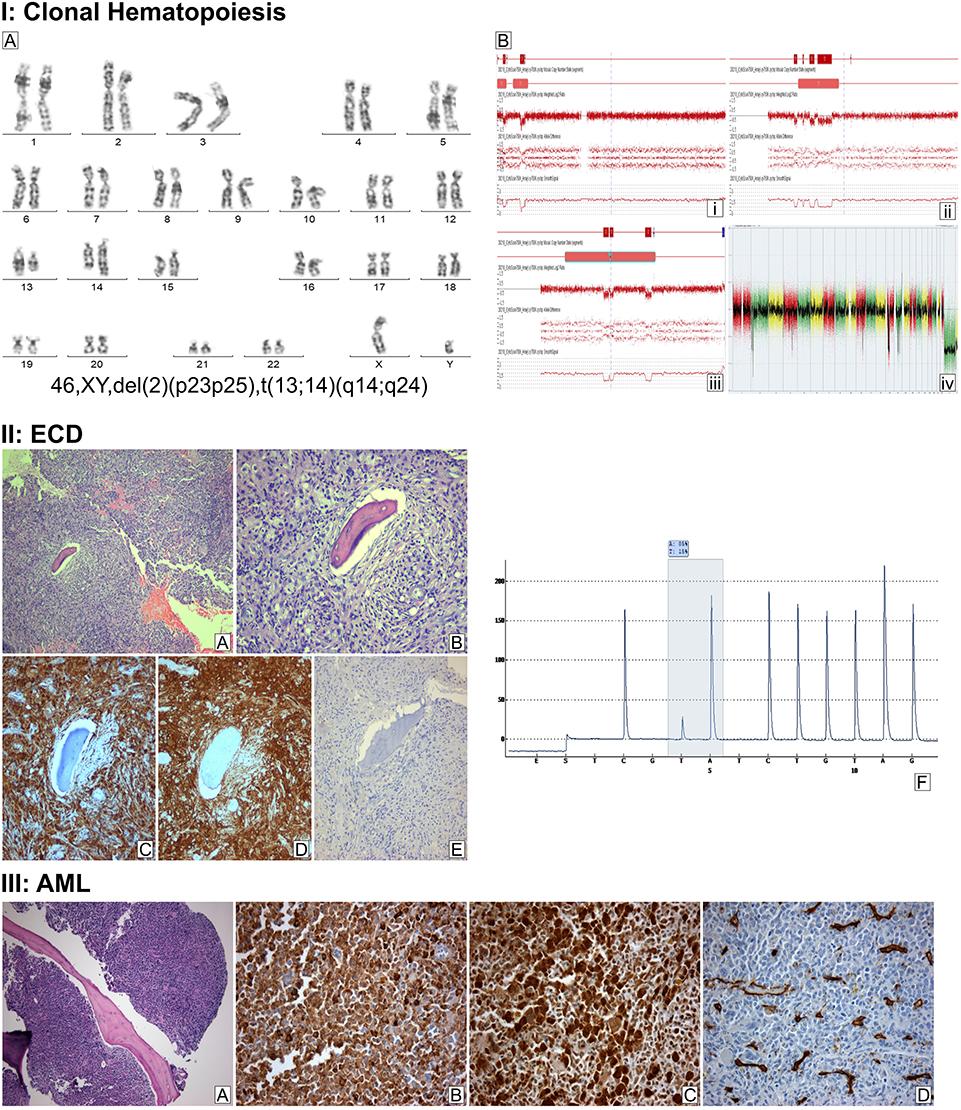 |
Figure 1 I: Clonal hematopoiesis. (Α) G-banded chromosome preparation. The arrows indicate the rearranged chromosomes. (Β) Detailed view of array analysis for chromosomes 2, 13, and 14, respectively (A–C). Regions with a DNA copy number loss are indicated in red, and intervals with gains are highlighted in blue. Whole-genome profiling by microarray identified multiple genomic imbalances (D). Microarray probes are arranged according to their physical map locations on each chromosome from the distal p-arm (on the left) to the distal q-arm (on the right). Chromosomes are plotted in a horizontal fashion and marked with different colors. An average logarithmic ratio (log2) is displayed for all oligonucleotide probes. Probes with a log2 ratio clustered around zero indicate DNA segments with normal copy numbers. A positive log2 ratio (above zero) indicates a gain (extra copy) of the chromosomal region, while intervals with a negative log2 ratio (below zero) represent loss of DNA copy number. II (A–E) Histopathologic findings of Erdheim-Chester disease. Bone tissue section showing intense fibrohistiocytic infiltrate (A), with prominent proliferation of foamy histiocytes (B). Immunohistochemistry revealed immunopositivity of the infiltrate for CD68 (C) and CD163 (D), but not for S100 (E). (A and B) Hematoxylin and eosin stain, 100x and 200x magnification, respectively; (C–E) immunohistochemistry, 200x magnification. (F) Pyrosequencing assay highlighting the presence of the BRAFV600E mutation in tumor cells (nucleotide substitution 1799T>A, frequency 15% units). III (A–D) Histopathologic findings of acute myeloid leukemia with mutated NPM1. Bone marrow tissue section showing abnormally increased (>98%) cellularity (A). Immunohistochemistry revealed immunopositivity of the blastic infiltrate for myeloperoxidase (B) and NPM1 (C, nuclear and cytoplasmic staining) but not for CD34 (D). ((A): hematoxylin and eosin stain, 100x magnification; (B–D) immunohistochemistry, 400x magnification). |
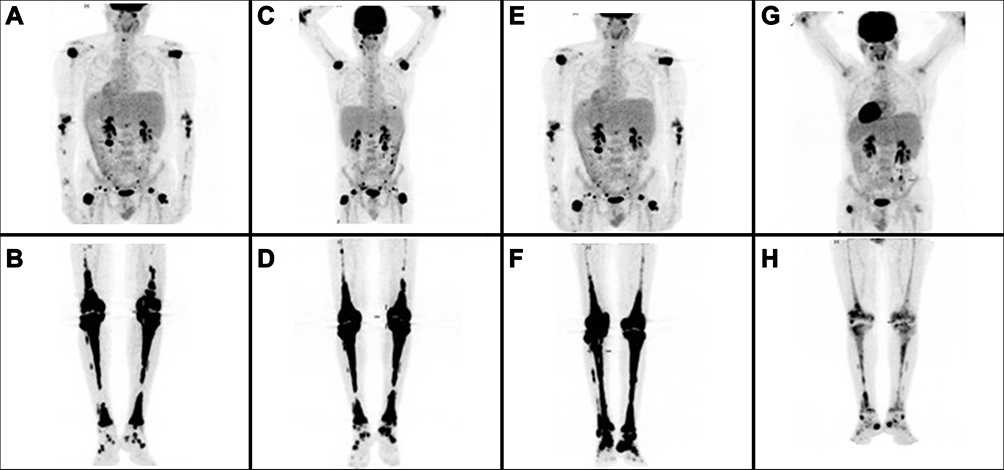 |
Figure 2 Serial follow-up with Maximum Intensity Projection Whole Body Positron Emission Tomography. (A, B) Initial staging at ECD diagnosis with extensive metabolically active disease; (C, D) progressive metabolically active disease after 1 year treatment with INF-a; (E, F) stable metabolically active disease following induction chemotherapy with two cycles of Ara-C and idarubicin plus midostaurin, and (G, H) metabolic response with significant improvement of the previous PET findings 6 months following the initiation of consolidation therapy with three cycles of HiDAC plus midostaurin following maintenance with midostaurin. |
The patient was started on interferon-alfa (INF-a) at a dose of 9.000.000 IU subcutaneously (s.c) 3-times a week. He tolerated the treatment well and showed mild clinical improvement mainly in terms of pain and mobility. However, a complete reassessment of the disease with PET/CT scan a year later (June 2018) showed metabolic progression in the form of both increased uptake of existing lesions and emergence of new ones (Figure 2C and D).
In September 2018, 15 months after the diagnosis of ECD, he presented again with worsening malaise and fatigue, shortness of breath, and fever up to 38.5°C. The complete blood count showed WBC: 24.33x109/L (neutrophils: 36%, lymphocytes: 10%, monocytes: 21%, and blasts: 33%), Hb: 5.0g/dL, PLT: 42x109/L. Routine biochemistry was normal except for LDH that was increased: 933 U/L (normal values 135–225 U/L). Bone marrow trephine biopsy demonstrated almost complete (>98%) infiltration by large blasts that were immunostained for myeloperoxidase, CD68, CD163, and NPM1 (with both nuclear and aberrant cytoplasmic expression) and were negative for CD34, CD3, CD20, CD61, CD138, and S100 (Figure 1IIIA–D). These findings were consistent with the diagnosis of AML with mutated NPM1. Conventional karyotypic analysis with G-banding from bone marrow cells showed the same aberrations as those presented at ECD diagnosis in all examined metaphases, ie, {46, XY, del(2)(p23p25), t(13;14)(q14;q24)[20]}. Furthermore, molecular karyotype on DNA extracted from bone marrow cells (“Supplementary methods” section) revealed clonal hematopoiesis with multiple aberrations, including copy number variations (CNV) and loss of heterozygosity (LOH) affecting in regions of chromosomes 2, 4, 10, 13, 14, and 17 (Figure 1IB–D). These regions include more than 400 genes including ALK, DNMT3A, ASXL2, PTEN, BRCA1, BRCA2, RB1, CDKN3, MAP4K5, STAT5B, STAT5A, and STAT3, which are involved in crucial molecular pathways such as apoptosis, cell cycle regulation, DNA methylation, histone modification, DNA damage repair machinery, and are implicated in hematological or other malignancies. A comprehensive list of the exact CMVs and LOHs of the patients as emerged from the molecular karyotype, as well as the potentially implicated genes is provided in Supplementary Tables 1 and 2). Targeted next generation sequencing (NGS) (“Supplementary methods” section) revealed mutations in the following genes: NPM1 (Variant Allele Frequency, VAF: 43.11%), FLT3-TKD (VAF: 13.84%), FLT3 internal tandem duplication (ITD) (VAF: 1.6%), and three nonsense mutations in CEBPA (VAF: 12.7%, 7.97%, 17.96%), as shown in Table 1. Fragment analysis using capillary electrophoresis confirmed NPM1 and FLT3-ITD mutations. Pyrosequencing testing for BRAFV600E mutation in bone marrow was negative.
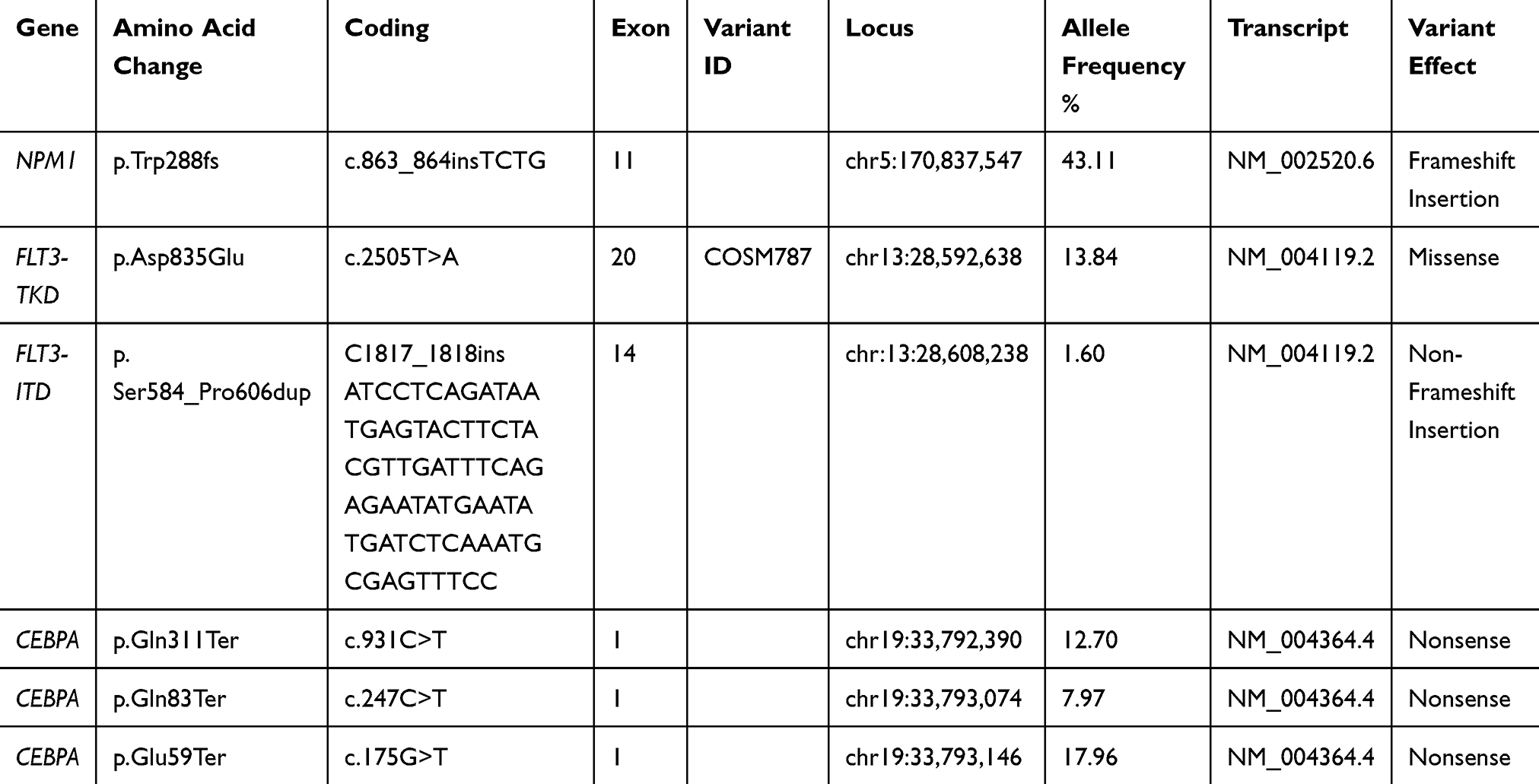 |
Table 1 Mutated Genes Detected by Targeted Next Generation Sequencing at the Time of AML Diagnosis |
Thereafter, with the patient’s written consent, we performed analysis with molecular karyotype and targeted NGS on DNA from stored peripheral blood and bone marrow cells, collected at the time of ECD diagnosis. We found the exact same aberrations in the molecular karyotype, while the targeted NGS revealed no mutations. Moreover, NPM1 staining by immunohistochemistry on the ECD sample did not reveal aberrant cytoplasmic localization of the NPM1 protein.
The patient received two cycles of induction chemotherapy with cytosine-arabinoside (Ara-C) 100 mg/m2/day from days 1 to 7 (continuous infusion) and idarubicin 12 mg/m2/day from days 1 to 3 (7+3) plus midostaurin 50 mg bid from days 8 to 21 achieving complete response (CR). However, the molecular signature as highlighted by the molecular karyotype remained unchanged. Additionally, the patient showed significant clinical improvement with complete pain relief and restoration of his mobility, although reassessment with PET/CT scan showed stable metabolic disease (Figure 2E and F). He subsequently received three cycles of consolidation chemotherapy with high dose Ara-C 3 g/m2 on days 1, 3, and 5, plus midostaurin, and then maintenance treatment with midostaurin, without major complication. Six months after starting maintenance treatment with midostaurin, the patient is clinically well, in CR from AML, with a low burden of measurable residual disease (MRD) (12.64 mutant NPM1 copies/10,000 ABL copies, as detected by Real-Time Quantitative Polymerase Chain Reaction, RQ-PCR), but with persistent clonal hematopoiesis, as shown by the molecular karyotypic analysis. Importantly, reassessment with PET/CT scan showed significant improvement of the ECD lesions (Figure 2G and H).
Discussion
Clonal hematopoiesis is characterized by the presence of acquired cytogenetic rearrangements and/or mutations in hematopoietic cells, in the absence of significant morphological abnormalities and often in the context of normal peripheral blood counts.
We have reported a patient with clonal hematopoiesis, as suggested by the presence of multiple CNVs in molecular karyotype, that persisted from the diagnosis of ECD to the diagnosis of AML and thereafter. In the presence of this clonal hematopoiesis, a hematopoietic clone, harboring the BRAFV600E mutation, might have emerged that gave rise to ECD, at a different time-point a distinct BRAFV600E negative, NPM1+/FLT3+ clone emerged, driving AML. This case is important for three reasons. First, it is suggestive that a molecular relationship of ECD with clonal hematopoiesis could be present, therefore suggesting that the cell of origin might arises from a premalignant hematopoietic clone.13 Although the cellular origin of ECD is not clear, previous studies have suggested that hematopoietic/myeloid progenitor cells might be the precursors of histiocytic neoplastic cells in both ECD and Langerhans cell histiocytosis (LCH).6–11 Additionally, Papo et al12 recently reported that 10% of adult patients with non-LCH had a concomitant myeloid neoplasm, mainly chronic myelomonocytic leukemia (CMML), myeloproliferative disorders and myelodysplastic syndromes (MDS). One study reported an ECD/LCH mixed histiocytic neoplasm with an NRAS mutation in ECD/LCH tissue as well as in bone marrow after diagnosis of CMML.10 Moreover, two recent studies have shown that ECD is associated with AML. The first reported a patient with mixed histiocytosis and BRAFV600E, TET2, and SRSF2 mutations in LCH cells from skin, and concurrent AML with TET2 and SRSF2 mutations, suggesting a clonal relationship between the two malignancies.9 The second described a patient with BRAFV600E mutated ECD who developed BRAFV600E acute monocytic leukemia (AML-M5) and whole exome sequencing confirmed that both pathologies had multiple shared mutations and arose from the same cell of origin.11 Both ECD and AML with mutated NPM1 in our patient might have originated from clonal hematopoiesis, sharing genetic aberrations considered the initiating events for the generation of preleukemic stem cells. In this background, the acquisition of additional cooperative genetic events could have driven the emergence of distinct neoplastic subclones, i.e. BRAFV600E in the case of ECD, and NPM1, FLT3-TKD, and CEBPA in AML. It should be noted that, in the absence of functional genomics studies on purified hematopoietic populations, which represents a limitation of our study, no definitive clonal origin conclusions could be rendered, moreover the results of the molecular karyotype should be interpreted cautiously as not every single one of the CNV should be considered to be associated with clonal hematopoiesis.
Second, our study provides, for the first time to our knowledge, clinical and imaging evidence that the use of a multi-targeted protein kinase inhibitor, such as midostaurin, could be beneficial for ECD and might present a promising therapeutic approach. Therapeutic options in ECD include INF-a and targeted therapies (BRAF inhibitors and/or MEK inhibitors). Since 2012, up to 200 adult patients with ECD or ECD and LCH (mixed histiocytosis) have been treated with targeted therapies. These therapies have shown an overall robust efficacy in ECD, without any acquired resistance reported so far and manageable toxicities.14 Our patient received INF-a at a high dose and showed mild clinical improvement. However, metabolic progression of the disease was observed a year later as documented by PET/CT scan. After administration of midostaurin for AML, additional significant clinical and imaging improvement of ECD was revealed in parallel to the expected improvement of AML. It should be noted that recent case reports suggest the role of intermediate-dose of ARA-C in the treatment of ECD with central nervous system involvement.15 The role of ARA-C in the improvement of our patient could not be excluded; however, the fact that the imaging improvement preceded the consolidation treatment with ARA-C stands against the argument that the improvement should be attributed solely on ARA-C. On the other hand, the improvement followed treatment with two cycles of midostaurin, suggesting a potential role of midostaurin in ECD treatment. Regarding midostaurin, apart from FLT3 it inhibits multiple other kinases, including vascular endothelial growth factor receptor 2 (VEGFR2).16 A recent study has demonstrated increased VEGF levels in over half of the ECD patients, suggesting VEGF inhibition as a potential therapeutic approach in ECD,17 which is investigated in the context of an ongoing Phase I/II trial (NCT01552434).
Finally, this case report illustrates a case where a diagnosis of clonal hematopoiesis could have been overlooked, if based solely on targeted next-generation sequencing, as NGS was negative at diagnosis of ECD; however, detection of CNVs through molecular karyotype in this patient made this diagnosis feasible. This notion highlights the need for incorporation of single‐nucleotide polymorphism (SNP) array analysis for CNVs along with NGS for somatic point mutations in the diagnostic approach of clonal hematopoiesis.18
Conclusion
This case report suggests that ECD might arise from acquisition of a BRAF mutation in a pre-malignant clone with several CNVs in various loci, harboring genes implicated in crucial cellular pathways. The same clone might have the potential to progress in AML after acquisition of AML-specific mutations. Therefore, it provides additional data in support of the previously proposed categorization of ECD as a myeloid malignancy. Moreover, it proposes the use of CNVs by SNP array analysis in ascertainment of clonal haematopoiesis and the utility of kinase inhibition in treatment of ECD.
Ethics Approval and Consent to Participate
Ethical approval is not appropriate. The authors obtained patient’s consent to participate.
Consent for Publication
The authors obtained informed consent from the patient to publish information on his disease and clinical course.
Acknowledgments
The authors acknowledge the assistance of Dr A. Pouliakis, who designed Figure 1.
Author Contributions
S.G.P: designed research, treated the patient, analyzed data, and wrote the paper. A.D: performed cytogenetics research, analyzed data, and wrote part of the paper. M.R: performed molecular research, analyzed data, and wrote part of the paper. C.K: performed molecular research, analyzed data, and wrote part of the paper. S.N.C: performed imaging research and analyzed data. A.B: performed research and analyzed data. A.G: performed imaging research and analyzed data. F.I: performed cytogenetics research and analyzed data. A.D: performed cytogenetics research and analyzed data. E.B: treated the patient and analyzed data. T.T: treated the patient, analyzed data, and wrote part of the paper. A.F.M: treated the patient and analyzed data. P.P: performed molecular research and analyzed data. I.G.P: performed immunostaining research and analyzed data. V.P: designed research, treated the patient, and analyzed data. P.G.F: designed research, performed immunostaining research, analyzed data, and wrote the paper. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, or publication of this article.
Disclosure
The authors declare that they have no competing interests.
References
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
2. Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124:483–492. doi:10.1182/blood-2014-03-561381
3. Mavrogenis AF, Igoumenou VG, Antoniadou T, et al. Rare diseases of bone: Erdheim-Chester and Rosai-Dorfman non-Langerhans cell histiocytosis. EFORT Open Rev. 2018;3:381–390. doi:10.1302/2058-5241.3.170047
4. Goyal G, Heaney ML, Collin M, et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020;135:1929–1945.
5. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672–2681. doi:10.1182/blood-2016-01-690636
6. Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211:669–683. doi:10.1084/jem.20130977
7. Haroche J, Cohen-Aubart F, Charlotte F, et al. The histiocytosis Erdheim-Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol. 2015;11:1033–1042. doi:10.1586/1744666X.2015.1060857
8. Milne P, Bigley V, Bacon CM, et al. Hematopoietic origin of langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017;130:167–175. doi:10.1182/blood-2016-12-757823
9. Durham BH, Roos-Weil D, Baillou C, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017;130:176–180. doi:10.1182/blood-2016-12-757377
10. Bonnet P, Chasset F, Moguelet P, et al. Erdheim-Chester disease associated with chronic myelomonocytic leukemia harboring the same clonal mutation. Haematologica. 2019;104(11):e530–e533. doi:10.3324/haematol.2019.223552
11. Ghobadi A, Miller CA, Li T, et al. Shared cell of origin in a patient with Erdheim-Chester disease and acute myeloid leukemia. Haematologica. 2019;104:e373–e375. doi:10.3324/haematol.2019.217794
12. Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017;130:1007–1013.
13. Capo-Chichi J-M, Michaels P, Tremblay-Le R, Abelson S, Hasserjian RP, Xia D. Emerging patterns in clonal haematopoiesis. J Clin Pathol. 2019;72:453–459. doi:10.1136/jclinpath-2019-205851
14. Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood. 2020;135(16):1311–1318. doi:10.1182/blood.2019002766
15. Wang JN, Qiu Y, Niu N, et al. Successful treatment of central nervous system involved Erdheim-Chester disease by intermediate-dose cytarabine as first-line therapy. Acta Oncol. 2020;59:302–305. doi:10.1080/0284186X.2019.1670355
16. Tvedt TH, Nepstad I, Bruserud Ø. Antileukemic effects of midostaurin in acute myeloid leukemia – the possible importance of multikinase inhibition in leukemic as well as nonleukemic stromal cells. Expert Opin Investig Drugs. 2017;26(3):343–355. doi:10.1080/13543784.2017.1275564
17. Roeser A, Bravetti M, Azoulay L-D, et al. High serum VEGF level in erdheim-chester disease: correlation with cardiovascular involvement and response to treatment. Blood. 2019;134(Supplement_1):2324. doi:10.1182/blood-2019-129984
18. Takahashi K, Wang F, Kantarjian H, et al. Copy number alterations detected as clonal hematopoiesis of indeterminate potential. Blood Adv. 2017;1:1031–1103. doi:10.1182/bloodadvances.2017007922
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.