")
Back to Journals » Vascular Health and Risk Management » Volume 11
Epoprostenol sodium for treatment of pulmonary arterial hypertension
Authors Saito Y, Nakamura K , Akagi S, Sarashina T, Ejiri K, Miura A, Ogawa A, Matsubara H, Ito H
Received 5 January 2015
Accepted for publication 18 March 2015
Published 14 May 2015 Volume 2015:11 Pages 265—270
DOI https://doi.org/10.2147/VHRM.S50368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Daniel Duprez
Yukihiro Saito,1 Kazufumi Nakamura,1 Satoshi Akagi,1 Toshihiro Sarashina,1 Kentaro Ejiri,1 Aya Miura,1 Aiko Ogawa,2 Hiromi Matsubara,2 Hiroshi Ito1
1Department of Cardiovascular Medicine, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan; 2Division of Cardiology, National Hospital Organization Okayama Medical Center, Okayama, Japan
Abstract: The release of endogenous prostacyclin (PGI2) is depressed in patients with pulmonary arterial hypertension (PAH). PGI2 replacement therapy by epoprostenol infusion is one of the best treatments available for PAH. Here, we provide an overview of the current clinical data for epoprostenol. Epoprostenol treatment improves symptoms, exercise capacity, and hemodynamics, and is the only treatment that has been shown to reduce mortality in patients with idiopathic PAH (IPAH) in randomized clinical trials. We have reported that high-dose epoprostenol therapy (>40 ng/kg/min) also results in marked hemodynamic improvement in some patients with IPAH. High-dose epoprostenol has a pro-apoptotic effect on PAH-PASMCs via the IP receptor and upregulation of Fas ligand (FasL) in vitro. However, long-term intravenous administration of epoprostenol is sometimes associated with catheter-related infections and leads to considerable inconvenience for the patient. In the future, the development of new routes of administration or the development of powerful PGI2 analogs, IP-receptor agonists, and gene and cell-based therapy enhancing PGI2 production with new routes of administration is required.
Keywords: pulmonary arterial hypertension, prostacyclin, apoptosis
Background
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by progressive elevation of pulmonary vascular resistance (PVR) and pulmonary artery pressure (PAP) that leads to right heart failure and death. Increased PVR is induced by pulmonary vasoconstriction, vascular remodeling, and thrombosis.1,2 Those factors are associated with many molecules, substrates, and signaling pathways. Three pathways, namely prostacyclin (PGI2), nitric oxide (NO), and endothelin pathways, are critical for the pathogenesis and progression of PAH.3,4 Impaired production of vasodilators, such as PGI2 and NO, along with over-expression of vasoconstrictors, such as endothelin-1, play an important part in the pathogenesis of idiopathic PAH (IPAH).3 Drugs targeting the three pathways are currently available, and have been shown to be useful. In this review, we focus on the efficacy of high-dose epoprostenol sodium, a PGI2, for treatment of PAH.
Deficient endogenous PGI2 in PAH
An increase in the release of the vasoconstrictor thromboxane A2, suggesting the activation of platelets, occurs in patients with the primary form as well as those with the secondary form of pulmonary hypertension (PH). In contrast, the release of PGI2 is depressed in these patients.5 PGI2 synthase expression has been reported to be decreased in lung tissues from patients with severe PH.6 Whether the imbalance in the release of these mediators is a cause or a result of PH is unknown, but this imbalance may play a part in the development and maintenance of both forms of the disorder.5
Protocol of epoprostenol therapy
We start epoprostenol therapy at a low dose (0.25–0.5 ng/kg/min) and increase the dose daily by 0.25–2.0 ng/kg/min. When the dose of epoprostenol exceeds 10 ng/kg/min, we increase the dose weekly as previously described.7 Thereafter, we gradually increase the dose weekly or monthly to the maximal tolerated dose based on clinical symptoms and side effects in individual cases. We adjust the dose to match the change in body weight. Epoprostenol has a short half-life, and the in vivo half-life of epoprostenol in humans is expected to be no greater than 6 minutes. Continuous intravenous administration by means of an infusion pump and a permanent tunneled catheter is needed for long-term treatment.8
When the patient develops right heart failure before the start of epoprostenol therapy, we initiate treatment with dobutamine and/or dopamine in our centers as previously described.9 We initiate dobutamine infusion at a low dose (3 μg/kg/min) when the patient’s mixed venous oxygen saturation (SvO2) is <60% or cardiac index (CI) is <2.0 L/min/m2 or when right ventricular (RV) failure is clinically suspected before the start of epoprostenol therapy. Clinical RV failure is defined as leg edema and jugular venous distention, heart enlargement in a chest radiograph (cardiothoracic ratio >50%), and a high level of brain natriuretic peptide (>100 pg/mL). If the value of SvO2 does not increase over 60% or CI does not increase over 2.0 L/min/m2 or if RV failure is not improved, the dose of dobutamine is titrated up. We initiate dopamine infusion at a low dose (3 μg/kg/min) when the patient’s systolic blood pressure (BP) is <90 mmHg or urine volume is <20 mL/h before the start of epoprostenol therapy. If systolic BP cannot be kept over 90 mmHg, the dose of dopamine is titrated up.
Efficacy of epoprostenol therapy
The efficacy of continuous intravenous epoprostenol therapy has been tested in three unblinded randomized clinical trials (RCTs) in patients with IPAH10,11 and in patients with PH due to the scleroderma spectrum of disease, WHO-functional class (WHO-FC) III or IV despite optical medical therapy.12 Epoprostenol treatment improves symptoms, exercise capacity, and hemodynamics, and is the only treatment that has been shown to reduce mortality of patients with IPAH in RCTs.11,13 Meta-analysis for total mortality in the three RCTs performed with the Mantel–Haenszel and Peto fixed-effect methods showed relative risk reductions of 70% and 68%, respectively.13 The fifth World Symposium on Pulmonary Hypertension (WSPH) in Nice, France in 2013 recommended continuous intravenous epoprostenol as first-line therapy for WHO-FC IV patients because of the survival benefit in this subset.13 McLaughlin et al reported the long-term efficacy of epoprostenol therapy in 162 patients with primary PH.14 After 17 months, mean pulmonary pressure (mPAP) had decreased from 61±13 mmHg to 53±13 mmHg, and observed survival with epoprostenol therapy at 1–3 years was significantly greater than the expected survival based on historical data. Additionally, combination therapy with epoprostenol and bosentan improved the hemodynamics, functional class, and exercise capacity of IPAH, and anorexigen-associated PAH patients compared with those in matched controls who received epoprostenol monotherapy.15
In patients with PH due to the scleroderma spectrum of disease, continuous intravenous epoprostenol improves exercise capacity, hemodynamics, and survival compared with conventional therapy.12,16 Additionally, epoprostenol might improve the hemodynamics and survival in patients with PAH associated with connective tissue diseases (CTD), including mixed CTDs, CREST syndrome, systemic lupus erythematosus, scleroderma, and Sjogren syndrome.16–19
Rosenzweig et al reported the results of long-term epoprostenol therapy in patients with PH associated with congenital heart diseases (CHD-PAH).20 Twenty patients (eleven patients: previous cardiac surgery, eleven patients: residual systemic to pulmonary shunt, 15±14 years old) were treated with epoprostenol. mPAP had decreased from 77±20 mmHg to 61±15 mmHg one year after the start of treatment with epoprostenol at a dose of 82±37 ng/kg/min. Thomas et al also reported that long-term continuous PGI2 therapy in adult patients with CHD-PAH (37±10.5 years old) resulted in hemodynamic and clinical improvements.21
Epoprostenol therapy in patients with PH associated with portal hypertension improved hemodynamics but did not improve long-term survival.22–24
Long-term epoprostenol therapy in the dose range of 21–40 ng/kg/min reduced mPAP by 12%–22% and reduced PVR by 32%–53% compared with baseline values (Table 1).14,25,26 The dosage of epoprostenol is adjusted upward on the basis of symptoms of PAH and side effects of the drug. Since a chronic overdose of epoprostenol could lead to a high cardiac output state,27 the appropriate dose range of epoprostenol is thought to be 25–40 ng/kg/min on the basis of results of previous studies.3,14,25,26,28 However, treatment with epoprostenol at doses less than 40 ng/kg/min sometimes cannot improve hemodynamics in patients with severe PAH. We reported that high-dose epoprostenol therapy (>40 ng/kg/min) caused marked hemodynamic improvement in 14 patients with IPAH.7 Compared with the baseline state, high-dose epoprostenol therapy reduced mPAP by 30% and PVR by 68% (Table 1), and increased CI by 89% and SvO2 by 19%. Further studies are needed to clarify the efficacy of high-dose epoprostenol therapy (>40 ng/kg/min) in patients with PAH associated with CTD and CHD.
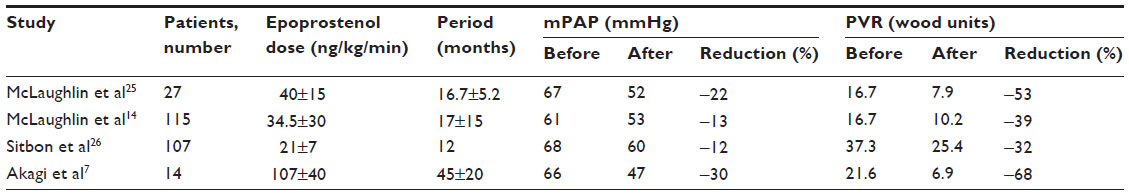 | Table 1 Hemodynamics before and after epoprostenol therapy |
Reverse remodeling of pulmonary arteries by high-dose epoprostenol therapy
Increased PVR is induced by pulmonary vasoconstriction, vascular remodeling, including pulmonary vascular intimal and medial thickening, and thrombosis.1,2 Intima and media thickening is largely composed of smooth muscle cells and myofibroblasts.29,30 Most cases of severe PH also exhibit a disorganized growth of primitive endothelial cells that form plexiform lesions.30 Enhanced proliferation and impaired apoptosis of pulmonary artery smooth muscle cells (PASMCs) cause an inappropriate increase in PASMCs and induce pulmonary vascular medial hypertrophy in PAH.31–35
Medical agents that have anti-proliferative and pro-apoptotic effects on PASMCs are required for effective treatment that achieves reverse remodeling.35,36 PGI2 analogs have anti-proliferative effects on PASMCs in vitro.37 We have also reported that high-dose epoprostenol has a pro-apoptotic effect on PAH-PASMCs via the IP receptor and upregulation of Fas ligand (FasL) in vitro (Figure 1).38 In a case series, we examined the reverse pulmonary vascular remodeling effects of epoprostenol in lung tissues obtained from an IPAH patient treated with high-dose epoprostenol and an IPAH patient not treated with epoprostenol.39 Apoptotic cells were detected in small pulmonary arteries of the IPAH patient treated with high-dose epoprostenol (115 ng/kg/min) but not in those from the IPAH patient not treated with epoprostenol. The media of peripheral pulmonary arteries was thick in the IPAH patient not treated with epoprostenol. On the other hand, the media of peripheral pulmonary arteries was thin in the IPAH patient treated with high-dose epoprostenol. These results indicate that high-dose epoprostenol therapy has the potential for reverse pulmonary vascular remodeling by reduction of medial hypertrophy.
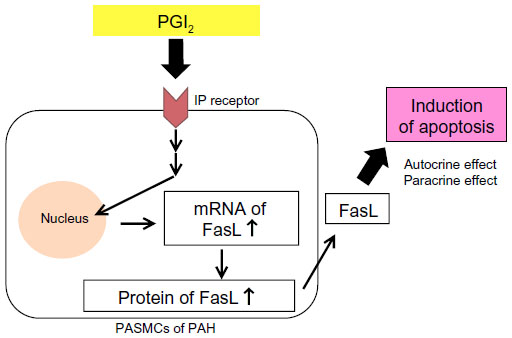 | Figure 1 PGI2 induces apoptosis via upregulation of Fas ligand (FasL) in pulmonary artery smooth muscle cells (PASMCs) from patients with pulmonary arterial hypertension (PAH). |
However, advanced proliferative vasculopathy after long-term (18 years) and high-dose (60 ng/kg/min) epoprostenol therapy in a patient with IPAH was reported.40 There were frequent plexiform lesions adjacent to arterial branch points. Achcar et al also reported PGI2-treated cases that showed increased perivascular inflammation.41 Of note is the Stacher et al study on the impact of modern treatments of PAH, including treatment with PGI2 and its analogs, endothelin receptor antagonist (ERA), and phosphodiesterase type 5 (PDE-5) inhibitors, on pulmonary vascular pathology.30 Morphometric intima and media fractional thicknesses were significantly larger in the PAH group than in control lungs. Media fractional thickness was largely superimposable with the range of media fractional thickness measured in normal pulmonary arteries. However, the overlap was less in intima fractional thickness, and marked perivascular inflammation was present in a large number of PAH lungs. Furthermore, the number of profiles of plexiform lesions was significantly smaller in the lungs of patients who had never been treated with PGI2 or its analogs. These results indicate that epoprostenol therapy might reverse pulmonary vascular medial thickening but might not ameliorate intima thickening, perivascular inflammation, and plexiform lesions. Further studies to assess the reversal vascular remodeling effects of high-dose epoprostenol therapy are needed.
Acute vasoreactivity testing
Acute vasoreactivity testing is usually performed to predict a good prognosis and identify acute responders who are more likely to have a sustained beneficial response to oral calcium channel blockers (CCBs) and can be treated with these less-expensive drugs. Acute responders have been defined as patients who show a decrease in mPAP of at least 10 mmHg to an absolute level below 40 mmHg with preserved or increased cardiac output.42
Acute vasoreactivity testing is most commonly performed using intravenous epoprostenol,43,44 intravenous adenosine,45 or inhaled nitric oxide.44 Acute testing using intravenous epoprostenol was shown to be useful for identifying patients with good prognosis; however, a good response to epoprostenol does not indicate that all patients will have a long-term response to CCBs.44,46,47 On the other hand, occurrence of life-threatening hemodynamic compromise has often been documented in CCB testing (nifedipine and verapamil).48–51 Therefore, it is accepted that CCBs should not be used for acute testing.3,42 In contrast, intravenous CCB nicardipine might be useful for acute testing,52 because the drug is short-acting compared to nifedipine and more vasoselective than other CCBs.53,54 We have reported that in 65 PAH patients administered low-dose nicardipine, there was no hemodynamic instability requiring additional inotropic agents or death during the testing.52
Storage and stability
Unopened vials of epoprostenol sodium with glycine-mannitol excipients (epoprostenol GM) are stable when stored at 15°C−25°C (59°F–77°F), and protected from light in a carton. However, epoprostenol GM in solution has limited stability at room temperature. Prior to use, reconstituted solutions of epoprostenol GM must be protected from light, and must be refrigerated at 2°C−8°C (36°F–46°F) if not used immediately. During use, a single reservoir of reconstituted solution of epoprostenol GM can be administered at room temperature for a total duration of 8 hours or it can be used with a cold pouch and administered up to 24 hours with the use of two frozen 6-oz gel packs in a cold pouch. The need for refrigeration or the use of frozen gel packs during long-term administration leads to considerable inconvenience for the patient. Recently, epoprostenol sodium with arginine excipient has improved thermal stability. It might provide patients with an increased sense of treatment convenience.55,56
Adverse reactions
The most common adverse events (occurring in ≥10% of patients) during dose initiation were flushing, headache, nausea/vomiting, hypotension, anxiety, and chest pain. The most common adverse events (occurring in ≥10% of patients) during chronic administration were dizziness, headache, nausea/vomiting, jaw pain, myalgia, flushing, diarrhea, nonspecific musculoskeletal pain, tachycardia, chills/fever/sepsis/flu-like symptoms, anxiety, and hypesthesia.
Chronic infusion of epoprostenol is delivered using a small, portable infusion pump through an indwelling central venous catheter. It results in limited improvement in quality of life. Catheter-related infections are problematic during long-term treatment. During long-term follow-up, sepsis was reported at a rate of 0.3 infections/patient per year in patients treated with epoprostenol. We have reported that the use of a closed hub system reduces catheter-related infections in patients with PAH receiving continuous therapy with epoprostenol at home.8 The development of new routes of administration is required in the future. Powerful PGI2 analogs, IP-receptor agonists, or gene and cell-based therapy enhancing PGI2 production with new routes of administration are needed.57
Anticoagulation therapy is associated with a survival benefit in patients with IPAH and has been recommended.13,58 Epoprostenol has antiplatelet activity, and has been reported to reduce the levels of plasma tissue-type plasminogen activator and plasminogen activator inhibitor.59 We previously reported that many hemorrhagic complications occur in patients with IPAH who receive both anticoagulation and epoprostenol treatment.60 Among 31 patients, nine had eleven bleeding episodes, nine (81.8%) of which were alveolar hemorrhages. Therefore, we have stopped using anticoagulation in patients receiving high-dose epoprostenol.
Conclusion
PGI2 replacement therapy by epoprostenol infusion is one of the best treatments available for PAH. High-dose epoprostenol therapy (>40 ng/kg/min) causes marked hemodynamic improvement in some patients with PAH.
Disclosure
Drs Nakamura, Akagi, Ogawa, Matsubara, and Ito have received lecturer fees from GlaxoSmithKline and Actelion Pharmaceuticals, Japan. The authors report no other conflicts of interest in this work.
References
Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “work in progress”. Circulation. 2000;102(22):2781–2791. | |
Miura A, Nakamura K, Kusano KF, et al. Three-dimensional structure of pulmonary capillary vessels in patients with pulmonary hypertension. Circulation. 2010;121(19):2151–2153. | |
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–1436. | |
Nakamura K, Shimizu J, Kataoka N, et al. Altered nano/micro-order elasticity of pulmonary artery smooth muscle cells of patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2010;140(1): 102–107. | |
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | |
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | |
Akagi S, Nakamura K, Miyaji K, et al. Marked hemodynamic improvements by high-dose epoprostenol therapy in patients with idiopathic pulmonary arterial hypertension. Circ J. 2010;74(10):2200–2205. | |
Akagi S, Matsubara H, Ogawa A, et al. Prevention of catheter-related infections using a closed hub system in patients with pulmonary arterial hypertension. Circ J. 2007;71(4):559–564. | |
Akagi S, Ogawa A, Miyaji K, Kusano K, Ito H, Matsubara H. Catecholamine support at the initiation of epoprostenol therapy in pulmonary arterial hypertension. Ann Am Thorac Soc. 2014;11(5):719–727. | |
Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112(7):485–491. | |
Barst RJ, Rubin LJ, Long WA, et al. Primary Pulmonary Hypertension Study Group. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The primary pulmonary hypertension study group. N Engl J Med. 1996;334(5):296–302. | |
Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434. | |
Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl): D60–D72. | |
McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002; 106(12):1477–1482. | |
Kemp K, Savale L, O’Callaghan DS, et al. Usefulness of first-line combination therapy with epoprostenol and bosentan in pulmonary arterial hypertension: an observational study. J Heart Lung Transplant. 2012;31(2):150–158. | |
Badesch DB, McGoon MD, Barst RJ, et al. Longterm survival among patients with scleroderma-associated pulmonary arterial hypertension treated with intravenous epoprostenol. J Rheumatol. 2009;36(10): 2244–2249. | |
Humbert M, Sanchez O, Fartoukh M, et al. Short-term and long-term epoprostenol (prostacyclin) therapy in pulmonary hypertension secondary to connective tissue diseases: results of a pilot study. Eur Respir J. 1999;13(6):1351–1356. | |
Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167(4): 580–586. | |
Shirai Y, Yasuoka H, Takeuchi T, Satoh T, Kuwana M. Intravenous epoprostenol treatment of patients with connective tissue disease and pulmonary arterial hypertension at a single center. Mod Rheumatol. 2013;23(6):1211–1220. | |
Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99(14):1858–1865. | |
Thomas IC, Glassner-Kolmin C, Gomberg-Maitland M. Long-term effects of continuous prostacyclin therapy in adults with pulmonary hypertension associated with congenital heart disease. Int J Cardiol. 2013;168(4):4117–4121. | |
Kuo PC, Johnson LB, Plotkin JS, Howell CD, Bartlett ST, Rubin LJ. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation. 1997;63(4):604–606. | |
Krowka MJ, Frantz RP, McGoon MD, Severson C, Plevak DJ, Wiesner RH. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): a study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology. 1999;30(3):641–648. | |
Fix OK, Bass NM, De Marco T, Merriman RB. Long-term follow-up of portopulmonary hypertension: effect of treatment with epoprostenol. Liver Transpl. 2007;13(6):875–885. | |
McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338(5): 273–277. | |
Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780–788. | |
Rich S, McLaughlin VV. The effects of chronic prostacyclin therapy on cardiac output and symptoms in primary pulmonary hypertension. J Am Coll Cardiol. 1999;34(4):1184–1187. | |
McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114(13):1417–1431. | |
Tuder RM. Pathology of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30(4):376–385. | |
Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3): 261–272. | |
Fujio H, Nakamura K, Matsubara H, et al. Carvedilol inhibits proliferation of cultured pulmonary artery smooth muscle cells of patients with idiopathic pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2006;47(2):250–255. | |
Ogawa A, Nakamura K, Matsubara H, et al. Prednisolone inhibits proliferation of cultured pulmonary artery smooth muscle cells of patients with idiopathic pulmonary arterial hypertension. Circulation. 2005;112(12):1806–1812. | |
Geraci MW, Moore M, Gesell T, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88(6):555–562. | |
Zhang S, Fantozzi I, Tigno DD, et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L740–L754. | |
Nakamura K, Akagi S, Ogawa A, et al. Pro-apoptotic effects of imatinib on pdgf-stimulated pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2012; 159(2):100–106. | |
Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by pdgf inhibition. J Clin Invest. 2005;115(10): 2811–2821. | |
Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic amp generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26(2):194–201. | |
Akagi S, Nakamura K, Matsubara H, et al. Prostaglandin i2 induces apoptosis via upregulation of fas ligand in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2013;165(3):499–505. | |
Akagi S, Nakamura K, Matsubara H, et al. Reverse remodeling of pulmonary arteries by high-dose prostaglandini2 therapy. A case report. J Cardiol Cases. 2013;9(5):173–176. | |
Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138(5): 1234–1239. | |
Achcar RO, Yung GL, Saffer H, Cool CD, Voelkel NF, Yi ES. Morphologic changes in explanted lungs after prostacyclin therapy for pulmonary hypertension. Eur J Med Res. 2006;11(5):203–207. | |
Galie N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (esc) and the european respiratory society (ers), endorsed by the international society of heart and lung transplantation (ishlt). Eur Heart J. 2009;30(20):2493–2537. | |
Rubin LJ, Groves BM, Reeves JT, Frosolono M, Handel F, Cato AE. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation. 1982;66(2):334–338. | |
Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–3111. | |
Zuo XR, Zhang R, Jiang X, et al. Usefulness of intravenous adenosine in idiopathic pulmonary arterial hypertension as a screening agent for identifying long-term responders to calcium channel blockers. Am J Cardiol. 2012;109(12):1801–1806. | |
Tonelli AR, Alnuaimat H, Mubarak K. Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension. Respir Med. 2010;104(4):481–496. | |
Raffy O, Azarian R, Brenot F, et al. Clinical significance of the pulmonary vasodilator response during short-term infusion of prostacyclin in primary pulmonary hypertension. Circulation. 1996;93(3): 484–488. | |
Crevey BJ, Dantzker DR, Bower JS, Popat KD, Walker SD. Hemodynamic and gas exchange effects of intravenous diltiazem in patients with pulmonary hypertension. Am J Cardiol. 1982;49(3): 578–583. | |
Young TE, Lundquist LJ, Chesler E, Weir EK. Comparative effects of nifedipine, verapamil, and diltiazem on experimental pulmonary hypertension. Am J Cardiol. 1983;51(1):195–200. | |
Packer M, Medina N, Yushak M. Adverse hemodynamic and clinical effects of calcium channel blockade in pulmonary hypertension secondary to obliterative pulmonary vascular disease. J Am Coll Cardiol. 1984;4(5):890–901. | |
Packer M, Medina N, Yushak M, Wiener I. Detrimental effects of verapamil in patients with primary pulmonary hypertension. Br Heart J. 1984;52(1):106–111. | |
Saito Y, Nakamura K, Miyaji K, et al. Acute vasoreactivity testing with nicardipine in patients with pulmonary arterial hypertension. J Pharmacol Sci. 2012;120(3):206–212. | |
Terai M, Takenaka T, Maeno H. Inhibition of calcium influx in rabbit aorta by nicardipine hydrochloride (yc-93). Biochem Pharmacol. 1981;30(4):375–378. | |
Bristow MR, Ginsburg R, Laser JA, McAuley BJ, Minobe W. Tissue response selectivity of calcium antagonists is not due to heterogeneity of [3h]-nitrendipine binding sites. Br J Pharmacol. 1984;82(2): 309–320. | |
Chin KM, Badesch DB, Robbins IM, et al. Two formulations of epoprostenol sodium in the treatment of pulmonary arterial hypertension: Epitome-1 (epoprostenol for injection in pulmonary arterial hypertension), a phase iv, open-label, randomized study. Am Heart J. 2014;167(2):218–225. e211. | |
Sitbon O, Delcroix M, Bergot E, et al. Epitome-2: an open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. 2014;167(2):210–217. | |
Ruan CH, Dixon RA, Willerson JT, Ruan KH. Prostacyclin therapy for pulmonary arterial hypertension. Tex Heart Inst J. 2010;37(4): 391–399. | |
Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the comparative, prospective registry of newly initiated therapies for pulmonary hypertension (compera). Circulation. 2014;129(1):57–65. | |
Boyer-Neumann C, Brenot F, Wolf M, et al. Continuous infusion of prostacyclin decreases plasma levels of t-pa and pai-1 in primary pulmonary hypertension. Thromb Haemost. 1995;73(4):735–736. | |
Ogawa A, Matsubara H, Fujio H, et al. Risk of alveolar hemorrhage in patients with primary pulmonary hypertension – anticoagulation and epoprostenol therapy. Circ J. 2005;69(2):216–220. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.