Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Epithelial–mesenchymal transition is driven by transcriptional and post transcriptional modulations in COPD: implications for disease progression and new therapeutics
Authors Eapen MS ![]() , Sharma P
, Sharma P ![]() , Gaikwad AV
, Gaikwad AV ![]() , Lu W
, Lu W ![]() , Myers S, Hansbro PM, Sohal SS
, Myers S, Hansbro PM, Sohal SS
Received 29 March 2019
Accepted for publication 22 June 2019
Published 18 July 2019 Volume 2019:14 Pages 1603—1610
DOI https://doi.org/10.2147/COPD.S208428
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Mathew Suji Eapen,1 Pawan Sharma,1–3 Archana Vijay Gaikwad,1 Wenying Lu,1 Stephen Myers,1 Philip M Hansbro4,5, Sukhwinder Singh Sohal1
1Respiratory Translational Research Group, Department of Laboratory Medicine, College of Health and Medicine, University of Tasmania, Launceston, TAS 7248, Australia; 2Medical Sciences, University of Technology Sydney, Sydney, NSW 2007, Australia; 3Woolcock Emphysema Centre, Woolcock Institute of Medical Research, University of Sydney, Sydney, NSW 2037, Australia; 4Priority Research Centre for Healthy Lungs, Hunter Medical Research Institute and the University of Newcastle, Newcastle, NSW 2308, Australia; 5Centre for Inflammation, Centenary Institute and University of Technology Sydney, Sydney, NSW 2007, Australia
Abstract: COPD is a common and highly destructive disease with huge impacts on people and health services throughout the world. It is mainly caused by cigarette smoking though environmental pollution is also significant. There are no current treatments that affect the overall course of COPD; current drugs focus on symptomatic relief and to some extent reducing exacerbation rates. There is an urgent need for in-depth studies of the fundamental pathogenic mechanisms that underpin COPD. This is vital, given the fact that nearly 40%–60% of the small airway and alveolar damage occurs in COPD well before the first measurable changes in lung function are detected. These individuals are also at a high risk of lung cancer. Current COPD research is mostly centered around late disease and/or innate immune activation within the airway lumen, but the actual damage to the airway wall has early onset. COPD is the end result of complex mechanisms, possibly triggered through initial epithelial activation. To change the disease trajectory, it is crucial to understand the mechanisms in the epithelium that are switched on early in smokers. One such mechanism we believe is the process of epithelial to mesenchymal transition. This article highlights the importance of this profound epithelial cell plasticity in COPD and also its regulation. We consider that understanding early changes in COPD will open new windows for therapy.
Keywords: epithelial-to-mesenchymal transition, EMT, cancer, fibrosis, inflammation, HuR, EGFR, MMP, TGFβ
Background
COPD is a devastating disease of the lung found mainly to affect cigarette smokers, though fossil-based air pollution in developing nations is now considered an equal contributor. The WHO estimates that there are over 250 million persons affected by the disease, which accounts for at least 3 million deaths annually as of 2015, which roughly averages around 5% of total worldwide reported mortality.1 The disease is characterized clinically as a slowly progressive airway narrowing with only partial reversibility at best. In vulnerable smokers, the entire lung is affected2 which is evident from the development of large airway squamous metaplasia, mucus hypersecretion and smooth muscle hyperplasia along with increases in small airway fibrosis and airway wall thickening.3,4 These changes affect the normal airway architecture, subsequently causing airway narrowing and obliteration, accompanied by bronchitis.5 It is also notable that patients also suffer from varying degrees of emphysematous lung destruction,6,7 which significantly adds to airflow limitation.5
The epithelial lining forms the outer layer of the lung mucosa and thus is the first line of defense for any external insults.10 These cells act as a barrier to infectious microbes and toxic particles emanating from entities such as smoke, via increased mucus secretion and ciliary clearance; however, in chronic conditions, the mucus is often thickened with phlegm and impaired ciliary function.5 Epithelial cells also produce various antibacterial, antioxidant, antiprotease factors and have tight epithelial junctions11 and thus deny entry of foreign entities and stop them from damaging the inner lung mucosa.12 Nevertheless, chronic exposure to these external insults causes observable changes to the normal architecture of the epithelium in both smokers and COPD patients13 with increases in basal cell abnormalities and decreases in epithelial cellular junction integrity.14,15 Ultimately, the changes associated have a direct impact on disease outcome wherein almost half of the small airways are lost at early stages of the disease. Findings by Koo et al8 provided evidence to this effect, demonstrating that both small airways and alveolar structures are damaged in early COPD. Terminal bronchioles number obliterated by 40% in patients with mild-to-moderate COPD patients. Interestingly, transitional bronchioles numbers reduced in both mild and mild-to-moderate patients by 55%, and alveolar surface area decreased by 40%.8 Further, total airway count has also been shown to decrease by 19% in both mild-to-moderate COPD compared to never smokers.9
This devastating effect on the lung of early COPD patients can be attributed to active remodeling, whose impact on disease progression and manifestation needs further recognition. In this commentary, we wish to highlight the factors that contribute to such changes and the role of post-transcriptional regulators such as human antigen R (HuR) as possible modulators to these responses.
In both COPD patients and active smokers, cellular reprogramming in epithelial cells14 consequently could lead to epithelial–mesenchymal transition (EMT),16 a process where epithelial cells progressively lose cellular polarity and, adhesiveness, become migratory and assume a mesenchymal phenotype.17–19 Previous findings have established the presence of EMT in other fibrotic diseases and cancers,20–22 but recent studies have increasingly considered EMT as the possible core pathological factor in the development of COPD as well.23–27 Indeed, studies now show that the epithelium of COPD patients has increased cellular expression of mesenchymal markers such as S100A4 and vimentin,28 which is especially higher in basal cells and is omnipresent in both large and small airways.26,29,30 The other characteristic EMT feature is reticular basement membrane (Rbm) fragmentation which was found to be higher in COPD patients than in nonsmokers, and this has been attributed to increase in epithelial matrix metalloprotease (MMP) activity especially of MMP-9 (collagenase/gelatinase).31 In the same set of patients, increased Rbm vascularity was also observed, which is another prominent feature of EMT leading to the formation of a pro-cancer stroma.32 The increased expression of mesenchymal markers S100A4 and vimentin negatively correlated with lung function, suggesting a physiological consequence.26 COPD ex-smokers had significantly less Rbm S100A4 expression and a similar trend was observed for Rbm vessels.33,34 Appropriate smoking cessation studies are needed to further confirm these findings, but certainly, it seems to be making a difference. Initial proof of concept trial by our group revealed that inhaled corticosteroids (ICS) such as fluticasone propionate were effective in decreasing mesenchymal expression of S100A4 and vimentin, along with significant reduction in Rbm fragmentation, EGFR and MMP-9 expression in the COPD group compared to placebo.35 The study provided sufficient rationale for translational strategies that can be employed with currently available therapies to overcome this pathological aspect.35,36 Epidemiological studies have also revealed that ICS are associated with reduced lung cancer risks in COPD patients, which might be again through the anti-EMT effects of ICS.37 Milara et al38 in their in vitro studies found that cigarette smoke extract (CSE) does induce EMT activity in normal primary bronchial epithelial cells (pHBECs) via ROS generation, which enhanced TGFβ1 expression while decreasing cyclic adenosine monophosphate (cAMP) levels. Further, the group demonstrated that EMT could be attenuated by elevating cAMP levels using a specific phosphodiesterase 4 inhibitor.38 Gohy et al observed that targeting TGF-β1 expression via specific monoclonal antibodies, during in vitro differentiation of pHBECs, reduced mesenchymal vimentin expression and fibronectin release.39 Similar increases in mesenchymal markers N-cadherin and α-smooth muscle actin (αSMA) were reported by Wang et al, with an associated decrease in E-cadherin and α-catenin in small airway epithelial cells when stimulated in vitro with CSE. They further found that urokinase plasminogen activation receptor could be responsible for modulating EMT, positively correlating with an increase in vimentin expression in human small airways.40 Recent in vitro epithelial–fibroblast co-culture studies also examined the probable role of IL-1α in promoting EMT.41 They reported that IL-1α caused aberrant epithelial–fibroblast interactions when stimulated by CSE.42
Although there are now several studies suggesting upregulation of EMT markers in COPD, there have only been few molecular signaling pathway studies undertaken to elucidate the underlying mechanisms of EMT in COPD. Recent studies suggest that EMT in smokers and early (mild to moderate) COPD patients is likely driven by the activation of canonical pathways associated with TGFβ,43 which induces increases in the expression of nuclear transcription factors such as pSMAD2/3 with simultaneous reduction of inhibitory SMAD6/7 expression.44–46 The increased expression of pSMAD2/3 negatively correlated with lung function in patients with COPD, which indicates possible physiological consequences.44 Although pathways triggered by TGFβ and its superfamily, including bone morphogenetic proteins (BMPs) and growth differentiation factors (GDFs), play vital roles,47 other canonical pathways such as Wingless/integrase-1 (Wnt)-mediated epithelial changes could also be crucial in COPD EMT pathology. However, there are few literature evidence in COPD. Our group observed enhanced expression of β-catenin in the large airways of both smokers and COPD, though COPD patients were observed to have increased nuclear to cytoplasmic ratio of β-catenin expression suggesting active translocation and possible transcription of genes associated with mesenchymal proteins.48 Further, in vitro studies with bronchial epithelial cells showed that nicotine, which is a major component of tobacco smoke, could induce EMT in a WNT-3A/β-catenin-dependent manner.49 In the peripheral tissue, it was demonstrated that a decrease in Wnt/β catenin signaling was involved in parenchymal tissue impairment and associated disruption of repair mechanism, leading to an increase in emphysema in COPD patients.50 Again, this decrease may be further augmented by the lack of frizzled (FZD) receptors in the peripheral tissue, effectively enhancing the impairment process in alveolar cells.51 Interestingly, non-canonical pathways of Wnt signaling were found to be active in bronchial epithelial cells of COPD patients compared to smokers and nonsmokers.52 Wnt5B was found to modulate EMT changes independent of β-catenin possibly through the induction of TGFβ/SMAD3 signaling.53 Moreover, evidence also suggests cross-talk between TGFβ/SMADs and Wnt/β-catenin pathways in TGFβ-induced alveolar epithelial cells, and that these interactions synergize to increase the expression of αSMA, a hallmark of EMT.54,55 Transcriptional factors such as SNAIL1/2, ZEB1/2 and TWIST, which are independently activated by canonical or non-canonical pathways also show increased epithelial nuclear translocation in COPD patients.48 Other potential interactive pathways were demonstrated to enhance EMT changes, for example, TGF-β1 and p38 MAPK/PI3K/Akt signaling pathways complement each other and are known to activate SMAD2/3 signaling in in vivo models of COPD and in lung cancer.56 Further, an increase in EGFR ligand HB-EGF in smokers and COPD was also observed to potentiate EMT-related and airway modeling changes possibly by interacting and modulating TGFβ pathways.57,58 Considering the complexity in the number of pathways involved would indeed be hard to pinpoint one pathway which could be specifically targeted to inhibit this profound epithelial plasticity. However, given the published evidence in the literature, it is now almost certain that EMT does play an important role in COPD pathology.
In line with these findings in EMT, recent study by Sun et al59 provided vital initial evidence of the post-transcriptional regulatory pathways that are involved in modulating EMT in COPD patients. Their study especially investigates the role of (HuR, which is an ubiquitously expressed post-transcriptional regulator of RNA synthesis and regulates several proteins including the ones involved in EMT. HuR are members of the ELAV/Hu family of RNA-binding proteins (RBP), specifically bind to AU (SRE) sites of the 3’-end of the mRNA of several growth factors, transcription factor proteins, cell cycle regulators and plays the crucial role in translocating mRNA from the nucleus to the cytoplasm for protein translation purposes.60 Once in the cytoplasm HuR effects mRNA stability and translation of EMT-associated proteins (Figure 1). Sun et al59 demonstrate that the levels of HuR expression are significantly increased in both smokers and COPD patients but more substantially in the latter group suggesting the possibility of a COPD effect. The cytoplasmic HuR expression was also shown to increase in human bronchial epithelial immortalized cell lines such as BEAS-2B when treated with cigarette smoke extracts (CSE). Most interestingly and relevant to EMT was their finding of a close association of cytoplasmic HuR levels and increases in the EMT trans-nuclear transcription factor ZEB1. The finding of increasingly stable mRNA levels of ZEB1 indicates that the translocated cytoplasmic HuR may play a direct role in modulation of ZEB1, which again was evidently higher in the epithelium of smokers and COPD patients compared to nonsmoking controls.59 There are only a few other studies that demonstrate HuR having a direct role in driving EMT in COPD. A recent study by Gu et al has shown that in vitro CSE induces changes in small airway epithelial cells by increasing the expression of EMT-promoting proteins such as vimentin and SNAIL while decreasing the expression of E-cadherin in these cells. This increase in the cellular content of EMT proteins was found to be diminished in cells treated with siRNA targeting HuR.61 This inhibitory action by silencing HuR was also demonstrated by Sun et al59 wherein siRNA against HuR reduced ZEB-1 protein expression and decreased E-cadherin while increasing the expression of mesenchymal marker such as vimentin. Previous studies in cancer showed that HuR modulates EMT regulators such as SNAIL and Wnt/β-Catenin by stabilizing their protein levels in the cytoplasm.60,62 Considering the fact that both these transcription factors and HuR are upregulated in COPD patients, investigating the possible role of HuR in their regulation is important. One of the key comorbidities of COPD patients is lung cancer. Indeed, epithelia with active EMT63 are potentially vulnerable to malignant transformation, and EMT in COPD airways may well be the vital link between airway fibrosis and cancer development.64 Recently, our group showed strong correlations between EMT activity in the leading edge of invasive cancer and EMT activity in the nonmalignant airway areas from which the tumor originated.65 Thus, increased expression of HuR and associated EMT in COPD patients suggest a potential therapeutic target with relevance in early COPD to cancer transition.66
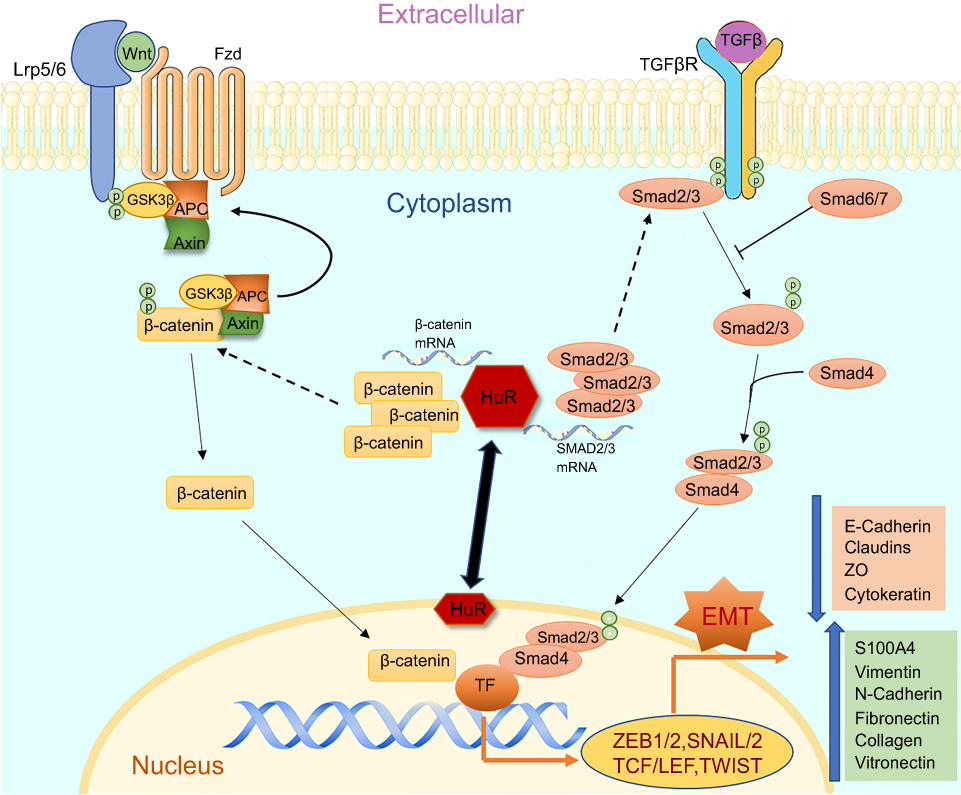 |
Figure 1 The figure illustrates the cellular pathways associated with EMT and HuR as a modulator regulating the changes in smokers and COPD. Both TGFβ and Wnt pathways play a crucial role in inducing EMT and potentially activate their associated receptors TGFβR and Frizzled, respectively. Activation of TGFβ-TGFβR results in phosphorylation of SMAD2/3 which tags with SMAD4 and translocates into the nucleus causing transcriptional changes. Activation of Wnt-Frizzled receptor signaling leads to inhibition of GSK3β and consequential accumulation of cytoplasmic β-catenin. The free β-catenin translocates into the nucleus and regulates target gene transcription. Finally, both pathways could lead to the induction of EMT with increases in mesenchymal and corresponding decreases in epithelial proteins. HuR, under pathological conditions, translocates from peri-nuclear into the cytoplasm and enhances further synthesis and stabilization of these translocating nuclear proteins, thus increasing the potential of EMT. |
Also, EMT as a process could lead to an increase in fibroblastic cell populations that can invade the underlying mucosa, potentially causing airflow limitation in COPD, especially in the small airways wherein tissue reconfiguration and scarring occurs. Further, active fibroblasts can cause aberrant changes to the extracellular matrix (ECM) leading to an increase in airway wall thickening, stiffness and gradual obliteration of the small airways.26,65 Although EMT seems to be a major contributor of fibroblasts, other sources which include fibrocytes, pericytes, residential fibroblasts and mesenchymal cells could also be contributing to the overall population of fibroblasts.67 Interestingly, endothelial cells which are the major cell type in blood vessels and responsible for structure and function possess the propensity to transit into fibroblast, through the process of endothelial-to-mesenchymal transition.18,34,68 During this process, endothelial cells lose endothelial adherence proteins such as VE-cadherins and gain mesenchymal markers similar to EMT.69 Under pathological conditions such as in chronic lung disease, fibroblasts are further transformed to their more active form, myofibroblasts.3 These cells appear to have spindle-shaped morphology and are highly contractile. The migratory potential of myofibroblasts directly relates to the increased expression of αSMA myofilaments, which forms nonmuscle cells (stress fibers). Myofibroblasts are known to secrete a broad array of ECM proteins, which include fibrous proteins such as collagen and elastin, as well as glycoproteins such as fibronectin, tenascin C and various forms of proteoglycans.70 Both canonical, Wnt/β-catenin and TGFβ/SMAD pathways are active in fibroblasts and their induction promotes myofibroblast proliferation and generation of ECM proteins.71,72 Thus, understanding the role of HuR in stabilizing proteins that induce these changes in fibroblast and myofibroblast populations is important and requires further investigation.73
Finally, another major aspect in COPD is identifying the role played by inflammation and infections in driving epithelial and other cellular transformations.3,74–76 Our group recently observed an overall reduction in total airway wall cellularity that further corresponded to either decrease or nonsignificant changes in key innate immune cells such as macrophage and neutrophils in COPD patients.77 On further evaluation of macrophage subpopulation in the bronchoalveolar lavage of COPD patients, we observed increases in M2 compared to M1 macrophage polarization which matched the cytokine profile.78 M2/Th2 cytokines are known to suppress M1-related inflammation, induce epithelial plasticity and promote proliferation of fibroblasts.79–81 Evidence also suggest dysfunctional immunity82 in COPD patients74–76 with poor phagocytic bacterial clearance by macrophage and ineffective killing of viral-infected cells by cytotoxic T cells.83,84 Interestingly, previous studies in colorectal cancer demonstrated that increases in cytoplasmic HuR in macrophages led to the suppression of pro-inflammatory cytokines, dysfunctional immunity and M2 polarisation.85,86 Thus, it would be of intrinsic value to evaluate the role of HuR in regulating inflammation in COPD and other chronic lung diseases.
Conclusions
We consider that EMT has important implications in various aspects of COPD and are crucial to large and small airway pathology, for both fibrosis and lung cancer.87,88 The role of TGFβ and Wnt pathways is implicated in these transformations and could have a potential difference in activity in the lung based on the affected region. This is a novel field in COPD, though could have wider implications in other chronic lung diseases as well. The role of HuR in the regulation of EMT and disease manifestations makes it an attractive target for therapeutic strategies; however, further research in this area is mandated. ICS do prevent against lung cancer risk in COPD, which may be through EMT. However, a safer drug than ICS is needed for long-term use.
Disclosure
SSS is supported by Clifford Craig Foundation Launceston General Hospital, Rebecca L. Cooper Medical Research Foundation, Cancer Council Tasmania, and Thoracic Society of Australia & New Zealand (TSANZ) Boehringer Ingelheim COPD Research Award for investigating mechanisms in COPD. PS is supported by Rebecca L. Cooper Medical Research Foundation, Australia. WL is supported by Cancer Council Tasmania. PMH is supported by an National Health and Medical Research Council Principal Research Fellowship and a Brawn Fellowship, Faculty of Health, University of Newcastle. The authors report no other conflicts of interest in this work.
References
1. WHO. World Health. Organisation, chronic obstructive pulmonary disease (COPD)- key facts. 2016. Available from: http://wwwwhoint/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd).
2. Lee JJ, Liu D, Lee JS, et al. Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J Natl Cancer Inst. 2001;93(14):1081–1088. doi:10.1093/jnci/93.14.1081
3. Eapen MS, Myers S, Walters EH, Sohal SS. Airway inflammation in chronic obstructive pulmonary disease (COPD): a true paradox. Expert Rev Respir Med. 2017;11(10):827–839. doi:10.1080/17476348.2017.1360769
4. Peters EJ, Morice R, Benner SE, et al. Squamous metaplasia of the bronchial mucosa and its relationship to smoking. Chest. 1993;103(5):1429–1432. doi:10.1378/chest.103.5.1429
5. Ojo O, Lagan AL, Rajendran V, et al. Pathological changes in the COPD lung mesenchyme - Novel lessons learned from in vitro and in vivo studies. Pulm Pharmacol Ther. 2014. doi:10.1016/j.pupt.2014.04.004
6. Makita H, Nasuhara Y, Nagai K, et al. Characterisation of phenotypes based on severity of emphysema in chronic obstructive pulmonary disease. Thorax. 2007;62(11):932–937. doi:10.1136/thx.2006.072777
7. American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;152(5 Pt 2):S77–S121.
8. Koo HK, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med. 2018;6(8):591–602.
9. Kirby M, Tanabe N, Tan WC, et al. Total airway count on computed tomography and the risk of chronic obstructive pulmonary disease progression. findings from a population-based study. Am J Respir Crit Care Med. 2018;197(1):56–65.
10. Hallstrand TS, Hackett TL, Altemeier WA, Matute-Bello G, Hansbro PM, Knight DA. Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin Immunol (orlando, Fla). 2014;151(1):1–15. doi:10.1016/j.clim.2013.12.003
11. Moheimani F, Roth HM, Cross J, et al. Disruption of beta-catenin/CBP signaling inhibits human airway epithelial-mesenchymal transition and repair. Int J Biochem Cell Biol. 2015;68:59–69. doi:10.1016/j.biocel.2015.08.014
12. Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(8):726–733. doi:10.1513/pats.200605-126SF
13. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169. doi:10.1165/rcmb.2017-0200TR
14. Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease. smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11(Suppl 5):S252–S258. doi:10.1513/AnnalsATS.201402-049AW
15. Shaykhiev R, Otaki F, Bonsu P, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68(5):877–892. doi:10.1007/s00018-010-0500-x
16. Sohal SS. Airway basal cell reprogramming and EMT: potential key to understanding early COPD. Am J Respir Crit Care Med. 2018;197:1644–1645. doi:10.1164/rccm.201712-2450LE
17. Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev Respir Med. 2014;8(5):547–559. doi:10.1586/17476348.2014.948853
18. Sohal SS. Endothelial to mesenchymal transition (EndMT): an active process in Chronic Obstructive Pulmonary Disease (COPD)? Respir Res. 2016;17(1):20. doi:10.1186/s12931-016-0337-4
19. Königshoff M. Lung cancer in pulmonary fibrosis: tales of epithelial cell plasticity. Respiration. 2011;81(5):353–358. doi:10.1159/000326299
20. Kumar JM, Chris W, Suji EM, et al. Epithelial–mesenchymal transition, a spectrum of states: role in lung development, homeostasis, and disease. Develop Dyn. 2018;247(3):346–358. doi:10.1002/dvdy.24541
21. Gurzu S, Turdean S, Kovecsi A, Contac AO, Jung I. Epithelial-mesenchymal, mesenchymal-epithelial, and endothelial-mesenchymal transitions in malignant tumors: an update. World J Clin Cases. 2015;3:393. doi:10.12998/wjcc.v3.i5.393
22. Sohal SS, Walters EH. Advanced non-small-cell lung cancer. N Engl J Med. 2017;377(20):1998–1999. doi:10.1056/NEJMc1712794
23. Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. doi:10.1136/thoraxjnl-2012-201761
24. Sohal SS, Reid D, Soltani A, et al. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12. doi:10.1186/1465-9921-12-130
25. Sohal SS, Walters EH. Epithelial mesenchymal transition (EMT) in small airways of COPD patients. Thorax. 2013;68:783–784. doi:10.1136/thoraxjnl-2013-203373
26. Mahmood MQ, Sohal SS, Shukla SD, et al. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction. Int J Chron Obstruct Pulmon Dis. 2015;10:1515–1524.
27. Sohal SS. Chronic obstructive pulmonary disease (COPD) and lung cancer: epithelial mesenchymal transition (EMT), the missing link? EBioMedicine. 2015;2:1578–1579. doi:10.1016/j.ebiom.2015.10.016
28. Sohal S, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–938. doi:10.1111/j.1440-1843.2010.01808.x
29. Schneider M, Hansen JL, Sheikh SP. S100A4: a common mediator of epithelial-mesenchymal transition, fibrosis and regeneration in diseases?. J Mol Med. 2008;86:507–522. doi:10.1007/s00109-007-0301-3
30. Sohal SS, Soltani A, Weston S, Wood-Baker R, Walters H. Intermediate filament vimentin and potential role in epithelial mesenchymal transition (EMT). Vimentin Concepts Mol Mech. 2013.
31. Sohal SS, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15:930–938. doi:10.1111/j.1440-1843.2010.01808.x
32. Soltani A, Walters EH, Reid DW, et al. Inhaled corticosteroid normalizes some but not all airway vascular remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2359–2367. doi:10.2147/COPD.S113176
33. Sohal SS, Reid D, Soltani A, Ward C, Weston S, Muller HK. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15:930–938.
34. Soltani A, Reid DW, Sohal SS, et al. Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: a cross-sectional study. Respir Res. 2010;11(1):105. doi:10.1186/1465-9921-11-62
35. Sohal SS, Soltani A, Reid D, et al. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study. Int J Chron Obstruct Pulmon Dis. 2014;9. doi:10.2147/COPD.S60179
36. Sohal SS, Mahmood QM, Walters HE. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer. Clin Transl Med. 2014;3. doi:10.1186/s40169-014-0033-2
37. Raymakers AJ, McCormick N, Marra CA, Fitzgerald JM, Sin D, Lynd LD. Do inhaled corticosteroids protect against lung cancer in patients with COPD? A systematic review. Respirology. 2017;22(1):61–70. doi:10.1111/resp.12919
38. Milara J, Peiro T, Serrano A, et al. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28(2):138–148. doi:10.1016/j.pupt.2014.02.001
39. Gohy ST, Hupin C, Fregimilicka C, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015. doi:10.1183/09031936.00135814
40. Wang Q, Wang Y, Zhang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013;14(1):67. doi:10.1186/1465-9921-14-19
41. Froidure A, Ladjemi MZ, Pilette C. Interleukin-1α: a key player for epithelial-to-mesenchymal signalling in COPD? Eur Respir J. 2016;48(2):301–304. doi:10.1183/13993003.01180-2016
42. Osei ET, Noordhoek JA, Hackett TL, et al. Interleukin-1alpha drives the dysfunctional cross-talk of the airway epithelium and lung fibroblasts in COPD. Eur Respir J. 2016;48(2):359–369. doi:10.1183/13993003.01911-2015
43. Soltani A, Sohal S, Reid D, Weston S, Wood-Baker R, Walters E. Vessel-associated transforming growth factor-Beta1 (TGF-beta1) is increased in the bronchial reticular basement membrane in COPD and normal smokers. PLoS One. 2012;7(6):e39736. doi:10.1371/journal.pone.0039736
44. Mahmood MQ, Reid D, Ward C, et al. Transforming growth factor (TGF) beta1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology. 2017;22(1):133–140. doi:10.1111/resp.12882
45. Springer J, Scholz FR, Peiser C, Groneberg DA, Fischer A. SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smoke. Biol Chem. 2004;385(7):649–653. doi:10.1515/BC.2004.080
46. Zandvoort A, Postma DS, Jonker MR, et al. Altered expression of the Smad signalling pathway: implications for COPD pathogenesis. Eur Respir J. 2006;28(3):533–541. doi:10.1183/09031936.06.00078405
47. Verhamme FM, Bracke KR, Joos GF, Brusselle GG. Transforming growth factor-β superfamily in obstructive lung diseases. more suspects than TGF-β alone. Am J Respir Cell Mol Biol. 2015;52(6):653–662. doi:10.1165/rcmb.2014-0282RT
48. Mahmood MQ, Walters EH, Shukla SD, et al. β-catenin, twist and snail: transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci Rep. 2017;7(1):10832. doi:10.1038/s41598-017-11375-x
49. Zou W, Zou Y, Zhao Z, Li B, Ran P. Nicotine-induced epithelial-mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304(4):L199–L209. doi:10.1152/ajplung.00094.2012
50. Kneidinger N, AÖ Y, Callegari J, et al. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med. 2011;183(6):723–733. doi:10.1164/rccm.200910-1560OC
51. Skronska-Wasek W, Mutze K, Baarsma HA, et al. Reduced frizzled receptor 4 expression prevents WNT/β-catenin–driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196(2):172–185. doi:10.1164/rccm.201605-0904OC
52. Baarsma HA, Königshoff M. ‘WNT-er is coming’: WNT signalling in chronic lung diseases. Thorax. 2017;72(8):746–759. doi:10.1136/thoraxjnl-2016-209753
53. Heijink IH, de Bruin HG, Dennebos R, et al. Cigarette smoke-induced epithelial expression of WNT-5B: implications for COPD. Eur Respir J. 2016;48(2):504–515. doi:10.1183/13993003.01541-2015
54. DiRenzo DM, Chaudhary MA, Shi X, et al. A crosstalk between TGF-β/Smad3 and Wnt/β-catenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal. 2016;28(5):498–505. doi:10.1007/s12032-017-0900-y
55. Zhou B, Liu Y, Kahn M, et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem. 2012;287(10):7026–7038. doi:10.1074/jbc.M111.276311
56. Jiang B, Guan Y, Shen H-J, et al. Akt/PKB signaling regulates cigarette smoke-induced pulmonary epithelial-mesenchymal transition. Lung Cancer. 2018;122:44–53. doi:10.1016/j.lungcan.2018.05.019
57. Lai T, Li Y, Chen M, et al. Heparin-binding epidermal growth factor contributes to COPD disease severity by modulating airway fibrosis and pulmonary epithelial-mesenchymal transition. Lab Invest. 2018;98(9):1159–1169. doi:10.1038/s41374-018-0049-0
58. Eapen MS, Sharma P, Thompson IE, et al. Heparin-binding epidermal growth factor (HB-EGF) drives EMT in patients with COPD: implications for disease pathogenesis and novel therapies. Lab Invest. 2019;99(2):150–157. doi:10.1038/s41374-018-0146-0
59. Sun J, Gu X, Wu N, Zhang P, Liu Y, Jiang S. Human antigen R enhances the epithelial-mesenchymal transition via regulation of ZEB-1 in the human airway epithelium. Respir Res. 2018;19. doi:10.1186/s12931-018-0805-0
60. Zhou Y, Chang R, Ji W, et al. Loss of scribble promotes snail translation through translocation of HuR and enhances cancer drug resistance. J Biol Chem. 2016;291(1):291–302. doi:10.1074/jbc.M115.693853
61. Gu XM, Wang XG, Sun J, Wang N, Jiang SJ. The role of HuR in mediating snail expression in human small airway epithelium induced by cigarette smoke extract. Zhonghua Jie He He Hu Xi Za Zhi. 2017;40(7):515–519. doi:10.3760/cma.j.issn.1001-0939.2017.07.008
62. Kim I, Hur J, Jeong S. HuR represses Wnt/β-catenin-mediated transcriptional activity by promoting cytoplasmic localization of β-catenin. Biochem Biophys Res Commun. 2015;457(1):65–70. doi:10.1016/j.bbrc.2014.12.052
63. Sohal SS, Hansbro PM, Walters EH. Epithelial mesenchymal transition in chronic obstructive pulmonary disease, a precursor for epithelial cancers: understanding and translation to early therapy. Ann Am Thorac Soc. 2017;14(9):1491–1492. doi:10.1513/AnnalsATS.201705-387LE
64. Sohal SS. Chronic Obstructive Pulmonary Disease (COPD) and lung cancer: epithelial mesenchymal transition (EMT), the missing link? EBioMedicine. 2015;2(11):1578–1579. doi:10.1016/j.ebiom.2015.10.016
65. Mahmood MQ, Ward C, Muller HK, Sohal SS, Walters EH. Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): a mutual association with airway disease. Med Oncol. 2017;34(3):45.
66. Sohal SS, Eapen MS, Ward C, Walters EH. Epithelial mesenchymal transition (EMT): a necessary new therapeutic target in COPD? Am J Respir Crit Care Med. 2017. doi:10.1164/rccm.201704-0771LE
67. Andersson-Sjöland A, Nihlberg K, Eriksson L, Bjermer L, Westergren-Thorsson G. Fibrocytes and the tissue niche in lung repair. Respir Res. 2011;12(1):76. doi:10.1186/1465-9921-12-122
68. Sohal SS. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig. 2017;55(2):104–113.
69. Eapen MS, Myers S, Lu W, Tanghe C, Sharma P, Sohal SS. sE-cadherin and sVE-cadherin indicate active epithelial/endothelial to mesenchymal transition (EMT and EndoMT) in smokers and COPD: implications for new biomarkers and therapeutics. Biomarkers. 2018;23:1–5.
70. Burgess JK, Mauad T, Tjin G, Karlsson JC, Westergren‐Thorsson G. The extracellular matrix – the under‐recognized element in lung disease? J Pathol. 2016;240(4):397–409.
71. Baarsma HA, Spanjer AIR, Haitsma G, et al. Activation of WNT/β-catenin signaling in pulmonary fibroblasts by TGF-β1 is increased in chronic obstructive pulmonary disease. PLoS One. 2011;6(9):e25450.
72. Akhmetshina A, Palumbo K, Dees C, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3:735.
73. Al-Habeeb FF, Nair PK, Azuelos I, Baglole C. The role of HuR in myofibroblast differentiation: implication for pulmonary fibrosis. A68 molecular determinants of remodeling in lung fibrosis. Am Thorac Soc Int Conf Abstr. 2018;197:A2205–A.
74. Eapen MS, Sohal SS. Understanding novel mechanisms of microbial pathogenesis in chronic lung disease: implications for new therapeutic targets. Clin Sci (Lond). 2018;132(3):375–379.
75. Sohal SS. Inhaled corticosteroids and increased microbial load in COPD: potential role of epithelial adhesion molecules. Eur Respir J. 2018;51(2):1702257.
76. Sohal SS, Eapen MS, Ward C, Walters EH. Airway inflammation and inhaled corticosteroids in COPD. Eur Respir J. 2017;49(6):1700289.
77. Eapen MS, McAlinden K, Tan D, et al. Profiling cellular and inflammatory changes in the airway wall of mild to moderate COPD. Respirology. 2017;22(6):1125–1132.
78. Eapen MS, Hansbro PM, McAlinden K, et al. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. 2017;7(1):13392.
79. Zhu L, Fu X, Chen X, Han X, Dong P. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol Int. 2017;41(9):960–968.
80. Yu CC, Chien CT, Chang TC. M2 macrophage polarization modulates epithelial-mesenchymal transition in cisplatin-induced tubulointerstitial fibrosis. BioMedicine. 2016;6(1):5.
81. Braga TT, Agudelo JSH, Camara NOS. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol. 2015;6:602.
82. Eapen MS, Sharma P, Moodley YP, Hansbro PM, Sohal SS. Dysfunctional immunity and microbial adhesion molecules in smoking-induced pneumonia. Am J Respir Crit Care Med. 2018;199:250–251.
83. Jubrail J, Kurian N, Niedergang F. Macrophage phagocytosis cracking the defect code in COPD. Biomed J. 2017;40(6):305–312.
84. McKendry RT, Spalluto CM, Burke H, et al. Dysregulation of antiviral function of CD8(+) T cells in the chronic obstructive pulmonary disease lung. role of the PD-1-PD-L1 axis. Am J Respir Crit Care Med. 2016;193(6):642–651.
85. Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest. 2012;122(1):48–61.
86. Katsanou V, Papadaki O, Milatos S, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. 2005;19(6):777–789.
87. Eapen MS, Hansbro PM, Larsson-Callerfelt AK, et al. Chronic obstructive pulmonary disease and lung cancer: underlying pathophysiology and new therapeutic modalities. Drugs. 2018;78(16):1717–1740.
88. Sohal SS, Walters EH. Essential need for rethink of COPD airway pathology: implications for new drug approaches for prevention of lung cancer as well as small airway fibrosis. Int J Chron Obstruct Pulmon Dis. 2017;12:2677–2679.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.