Back to Journals » Journal of Inflammation Research » Volume 12
Epigenetics of chronic inflammatory diseases
Authors Stylianou E
Received 31 July 2018
Accepted for publication 17 September 2018
Published 20 December 2018 Volume 2019:12 Pages 1—14
DOI https://doi.org/10.2147/JIR.S129027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Eleni Stylianou
Consultant Biomedical Scientist and Bioinformaticist, North Royalton, OH, USA
Abstract: Chronic, noncommunicable, and inflammation-associated diseases remain the largest cause of morbidity and mortality globally and within the United States. This is mainly due to our limited understanding of the molecular mechanisms that underlie these complex pathologies. The available evidence indicates that studies of epigenetics (traditionally defined as the heritable changes to gene expression that are independent of changes to DNA) are significantly advancing our knowledge of these inflammatory conditions. This review will focus on epigenetic studies of three diseases, that are among the most burdensome globally: cardiovascular disease, the number one cause of deaths worldwide, type 2 diabetes and, Alzheimer’s disease. The current status of epigenetic research, including the ability to predict disease risk, and key pathophysiological defects are discussed. The significance of defining the contribution of epigenetic defects to nonresolving inflammation and aging, each associated with these diseases, is highlighted, as these are likely to provide new insights into inflammatory disease pathogenesis.
Keywords: epigenetics, nonresolving inflammation, inflammatory diseases, atherosclerosis, type 2 diabetes, Alzheimer’s disease
Introduction
Our fascination with inflammation is centuries old, yet the most recent figures indicate that inflammation-associated diseases remain the most common health problem worldwide and within the United States (http://www.who.int/chp/about/integrated_cd/en/ and https://www.cdc.gov/chronicdisease/index.htm). These chronic, noncommunicable, and complex pathologies include atherosclerosis, metabolic diseases such as type 2 diabetes (T2DM), and neurodegenerative disorders. Our knowledge of the molecular mechanisms that dysregulate a physiological, beneficial, inflammatory response, and render it pathological, remains limited.
Definition of the epigenetic changes that regulate genes associated with chronic inflammatory diseases is advancing both our ability to predict disease risk and our understanding of the underlying pathophysiological defects. Traditionally, epigenetics is defined as heritable changes to gene expression that are independent of changes to the DNA sequence.1 Detailed discussion of the exceptions to this, eg, the dependence of DNA methylation on allele-specific single nucleotide polymorphisms (SNPs), is beyond the scope of this review but will be addressed briefly.2
This review will focus on explaining the current status of epigenetic research in three chronic disorders that are among the most burdensome worldwide. First: atherosclerosis, the leading cause of cardiovascular disease and deaths globally; second: T2DM, a rapidly emerging pandemic; and third: Alzheimer’s disease (AD), responsible for the international exponential growth of neurodegenerative disorders. Although reviews on epigenetics exist for each of the aforementioned diseases, this review provides an update on the major types of epigenetic modifications of all three pathologies together. This will allow comparisons to be made and will address the contribution that epigenetic mechanisms of nonresolving inflammation and aging could bring to our understanding of these diseases. Areas where further research is needed will be highlighted.
Physiological and pathological inflammation
Physiological inflammation is an integral part of innate immunity and functions as a protective combatant of infection, tissue stress, and injury with the aim of promoting tissue repair and restoring homeostasis.3,4 In chronic inflammatory disease, nonresolving or pathological inflammation comprises persistent inflammatory events that damage tissue and compromise function at the cellular or tissue level (Figure 1). The nature of the inflammatory trigger and the microenvironment in which the inflammation takes place determine the recognition mechanism and the type of pathways and genes that are activated. The available evidence suggests that epigenetic changes determine the specificity and outcome of these physiological and pathological inflammatory pathways.5
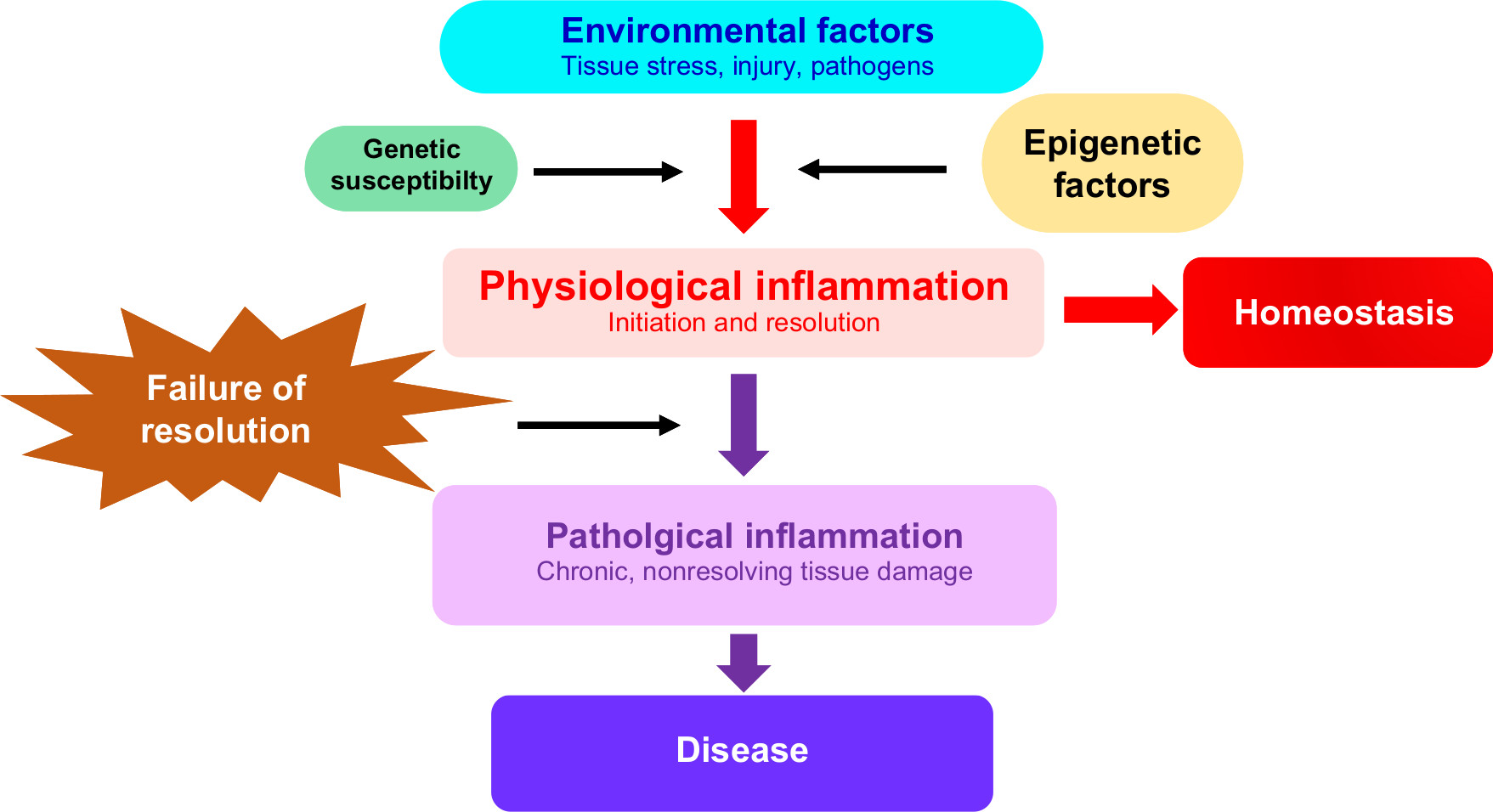 | Figure 1 Epigenetic factors are mediators of inflammation and chronic inflammatory disease. Notes: Environmental triggers of inflammation initiate a series of regulated molecular events that are mediated by epigenetic, genetic, as well as other factors (such as the microbiome which is not discussed here). This leads to the initiation and resolution of physiological inflammation and the restoration of homeostasis. Epigenetic modifications also contribute to failure of the inflammatory response to resolve, leading to tissue damage and chronic inflammatory disease. |
A detailed description of the highly intricate and context-dependent processes of physiological and pathological inflammation is beyond the scope of this article. The reader is referred to excellent reviews elsewhere on this topic.3–8 The salient features of the inflammatory response will be summarized in order to identify the key stages at which epigenetic mechanisms have a regulatory role. I will begin by discussing the initiation and resolution of inflammation.
Initiation
The initiation of inflammation involves an intricate network of molecular and cellular pathways and interactions. Many of these, from the outset, contribute to both initiation and resolution of the inflammatory response.7 Interestingly, many of the classically regarded activators of inflammation simultaneously drive anti-inflammatory and proresolution responses at both the transcriptional and posttranscriptional levels.6 Both the latter levels of regulation are mediated by epigenetic changes.
Triggers of inflammation can be pathogens, trauma, pain, or tissue damage.3,4,8 Pattern recognition receptors (PRRs) can be extracellular or intracellular and function to recognize and bind pathogen-associated molecular patterns (PAMPs), and/or the products of cell necrosis termed damage-associated molecular patterns (DAMPs). Examples of PRRs include Toll-like receptors and NOD-like receptors. The engagement of these receptors by PAMPs, DAMPs, or inflammatory cytokines results in multiple signaling pathways that culminate in the activation of gene transcription.8 Epigenetic modifiers (proteins that alter chromatin structure), transcription factors, and other proteins form transcription complexes that regulate the expression of inflammatory genes. Inflammatory mediators induce the extravasation of neutrophils, and recruit additional immune cells (eg, by chemokine attraction of monocyte-derived macrophages) followed by the activation of adaptive immune cells and residential stromal cells. The inflammatory response is perpetuated largely through cytokine-mediated feed-forward loops by both immune and resident cells to remove the initiating stimulus, activate tissue repair, and restore homeostasis.
Resolution
The resolution phase of inflammation, initiated at the outset of the inflammatory response, is designed to remove pathogens, cell debris, and inhibit proinflammatory cytokine production. Proresolution factors, including interleukin-10, transforming growth factor-β, annexin A1, lipoxins, resolvins, protectins, and maresins7,9,10 are produced or activated by inflammatory and/or other cells involved in the initial phase. In addition, neutrophils undergo apoptosis and are cleared by efferocytosis (nonphlogistic phagocytosis). Inflammatory macrophages exit from the site of injury and switch to a proresolving phenotype. Tregs are recruited with the purpose of restoring the vasculature, architecture, and function of target tissues. Proresolving mediators actively resolve inflammation whereas anti-inflammatory mediators inhibit the length and amplification of the inflammatory response.10 Defects in pathways of resolution appear to be key to the establishment of chronic inflammation and disease (Figure 1).
Epigenetics, aging, and chronic inflammatory disease
Despite intensive investigations of the pathways of inflammation at the cellular and molecular level, the epigenetic mechanisms that contribute to the nonresolution of inflammation and to the transition between protective and pathological states remain elusive. Accumulating evidence suggests that epigenetic changes have a fundamental role in regulating pathophysiological cellular processes including expression of protein and RNA coding genes, embryogenesis, cell differentiation, DNA replication, and repair.11 Epigenetic modifications can be stochastic (determined during development by inherited genetic variants) or environmentally induced.12 They are also reversible and transmissible between cell generations.
A variety of epigenetic defects have been associated with aging and age-related diseases.13,14 These include global loss of genomic DNA methylation and changes in methylation at specific CpG sites. Aging has also been associated with increased levels of specific histone modifications due to the dysregulated expression of chromatin-modifying enzymes, DNA methylation of noncoding RNAs (ncRNAs), and specific microRNA (miRNA) activity.13,14 A major challenge is to identify the epigenetic changes that are causal or drivers of dysregulated gene expression, genomic instability, aging, and disease.
The number, variety and complexity of epigenetic marks (chemical/structural modifications of DNA, RNA, and the histone protein components of chromatin) associated with chronic inflammatory diseases is expanding continuously.15,16 Candidate gene approaches or genome-wide analysis of a particular type of epigenetic modification at a given point in time, or condition, ie, epigenomic profiling, has identified key genomic regions that may be functionally important in pathological gene expression. Epigenomic studies have also helped to define epigenetic modifications associated with increased risk, which can act as biomarkers of early pathology and which can help stratify disease subtypes. In the following section, I will summarize the key types of epigenetic modification and explain briefly what is currently known about their functional significance.
DNA modifications
DNA methylation on the cytosines of CpG dinucleotides was originally described as being inversely correlated with transcription; however, more recent studies have shown that this is not always the case.17 DNA methylation outside of CpG islands may have other functions. For example within gene bodies, methylated DNA has been linked with enhanced gene transcription.
Currently, six different types of modified DNA bases have been identified.16 Four of these are modifications of position C5 on cytosine: C5-methylcytosine (5mC), C5-hydroxymethylcytosine (5hmC), C5-formylcytosine (5fc), and C5-carboxyl cytosine (5caC). 5hmC, 5fc, and 5caC are formed by stepwise oxidation and demethylation of 5mC by the Ten-eleven translocation family (TET) enzymes. A further two modifications comprise 1) the chemical addition of a methyl group to N3 of cytosine to form N3-methylcytosine and 2) the addition of a methyl group by a currently unknown mechanism to N6 on adenine to form N6-methyladenine. Currently, DNA methylation has been linked to the regulation of a number of pathophysiological processes or states including imprinting, genome instability, and chromosomal translocations. The role of DNA methylation in aging is an active area of investigation as it accurately reflects the chronological and biological age of cells and tissues.13 It is evident that DNA methylation may not only be a promising and reliable biomarker for early diagnosis and classification of disease subtypes, but has the potential to provide new insights into aging and chronic disease pathogenesis.
Histone modifications
Currently, 12 distinct types of histone modifications have been identified: acetylation, methylation, phosphorylation, butyrylation, formylation, sumoylation, propionylation, citrullination, ubiquitylation, crotonylation, proline isomerization, and ADP ribosylation.11 These are all enzymatically catalyzed. To date, 130 sites of modification have been identified within the N terminal tails of the four core histones (H2A, H2B, H3, and H4) and in 30 histone variants. Increased acetylation and trimethylation of histone H3, lysine 4 (H3K4), H3K36, and H3K79 are associated with accessible, actively transcribed euchromatin, whereas low acetylation and increased levels of methylated H3K9, H3K27, and H4K20 are linked with transcriptionally inactive heterochromatin. Gene expression requires crosstalk between the binding of key transcription factors and specific levels/types of histone modifications or marks.15,16 The permutations that are possible with respect to combinations of histone marks and their interactions with other proteins in the transcription machinery are staggering. Only a fraction of these have been studied in development, normal physiology, or chronic pathologies.
Non-coding RNAs (ncRNAs)
A number of ncRNAs function as structural, functional, and regulatory molecules with essential roles in cellular homeostasis.18,19 Hence, defects in the transcription, maturation, and function of these may be causal factors in numerous pathologies. The number and types of identified ncRNAs are expanding rapidly and much remains to be discovered about the regulation of their expression as well as their physiological and pathophysiological activities. The major categories of ncRNAs are as follows:20 1) long noncoding RNAs (lncRNAs) (>200 nt), 2) mid-size RNAs (20–300 nt) and 3) short ncRNAs (<200 nt), which include miRNAs (21–23 nt) and short interfering RNAs (20–30 nt). ncRNAs generally function as silencers and also mediators of site-specific transcriptional and posttranscriptional processes including nuclear organization, RNA processing, and transposon suppression.18 A single miRNA can directly repress the expression of hundreds of proteins, and a protein-coding gene can be modulated by more than one miRNA.
The lncRNAs field is in its infancy.18 Currently, lncRNAs are classified as natural antisense (those RNAs transcribed from the opposite DNA strand of other RNA transcripts) and long intergenic RNAs (transcribed from intergenic regions). These lncRNAs are thought to modulate gene expression through interactions with chromatin modifiers as well as miRNAs, RNA binding proteins, and mRNAs to form regulatory complexes.21
Chromatin architecture and epigenetic modifiers
The chromatin packaging and topology of high-order chromatin structures are important determinants of gene expression. Chromatin undergoes active reorganization of its architecture to regulate transcription and thus a variety of physiological and pathological processes. A compact chromatin structure (heterochromatin) represses gene expression by preventing access of transcription proteins to regulatory elements. In contrast, an open chromatin conformation (eucromatin) allows proteins to bind to DNA regions required for gene activation. An example of research in this area is the recently devised 4D Nucleome project, which aims to link genetic and epigenomic features with their interacting enhancer and promoter regions in 3D space (https://www.4dnucleome.org/). Posttranslationally modified nonhistone protein complexes containing epigenetic modifiers modulate chromatin structure and the accessibility to chromatin of the transcription machinery.
Research is ongoing to determine the mechanism by which the many epigenetic factors, including modifications of DNA, RNA, histone, and nonhistone proteins, and epigenetic modifiers interact with chromatin to affect disease phenotypes.15,16 Elegant techniques involving genetic engineering of modifications or epigenetic modifiers and targeting of chromatin enzymes to manipulate modifications in situ (epigenetic editing) are exciting developments. Such approaches will permit the contribution of specific epigenetic changes to transcriptional states to be determined. Epigenetic editing could also provide a hierarchical order of epigenetic modifications that would distinguish between causal/driver and consequential or passenger ‘epimutations’. A great deal of research remains to be done to identify the epigenetic changes relevant to specific pathophysiological conditions.
Epigenetics and risk factors for chronic disease
Chronic nonautoimmune diseases such as cardiovascular disease, T2DM, and Alzheimer’s disease share preventable biological risk factors such as unhealthy diet, physical inactivity, and tobacco use. It is compelling that aging is also associated with the development of each of these diseases. Importantly, both aging and lifestyle factors impact epigenetic changes and the regulation of pathological gene expression.13 Select studies of epigenetic changes in atherosclerosis, (the focus of most research on cardiovascular disease), T2DM and Alzheimer’s disease will be described in the following sections.
Atherosclerosis
Molecular and clinical features
Atherosclerosis is responsible for the majority of morbidity and mortality worldwide.22,23 Altered laminar flow leads to arterial endothelial dysfunction, and oxidized low-density lipoprotein (OxLDL), the major carrier of cholesterol, activates an endothelial cell inflammatory response. Monocyte-derived macrophages ingest the OxLDL and form foam cells. Continued activation of endothelial cells results in the further accumulation of inflammatory cells including foam cells and T cells. Defective resolution of inflammation, eg, defective efferocytosis by macrophages,24 leads to focal necrosis and vascular smooth muscle cell (VSMC) proliferation which thickens the arterial wall. Ultimately, a “fibrolipid plaque” is formed consisting of a necrotic core rich in lipids and capped by VSMCs and a collagen-containing matrix. The arterial lumen decreases in size, hampering blood flow when the plaque becomes too large, and the artery can no longer dilate to compensate (remodeling). Impaired, aging-associated vascular repair, failure in resolution of vascular inflammation, together with rupture of the plaque and thrombosis, result in myocardial infarction.25
Epigenetics of atherosclerosis
Genome-wide association studies (GWAS) have revealed that genetic variants associated with cardiovascular disease are insufficient to explain disease susceptibility.26 This “missing heritability” component has directed interest toward epigenetics. Risk factors for atherosclerosis such as diet and physical activity, smoking,27 hypertension, hyperlipidemia, psychological stress, and diabetes all reprogram epigenetic modifications and modulate gene expression during inflammation.22 Defining the epigenetic mechanisms involved is likely to make a major contribution to the development of new diagnostics and treatments for atherosclerosis and cardiovascular disease.
DNA methylation
The data on the involvement of DNA methylation in atherosclerosis are conflicting. A range of observations including no change in global DNA methylation, DNA hypermethylation, and hypomethylation of cytosines within CpG dinucleotides have all been associated with atherosclerosis.28 Candidate gene approaches and epigenome-wide association studies (EWAS) have reported links between atherosclerosis and differentially methylated DNA at specific gene loci (eg, LINE, ALU, CETP, LPL, FOXP3, ESR1, and MCT3). This is significant as the location of DNA methylation within the genome is a determinant of its mode of action (see earlier section on DNA modifications, in epigenetics, aging, and chronic inflammatory disease). A correlation between atherosclerotic lesion severity and DNA hypermethylation has also been observed.29 Consistent differences in methylated genes with important roles in the atherosclerosis inflammatory response and in obesity and lipid metabolism have been found in multiple EWAS studies.28 Disparities in the outcome of DNA methylome studies are most likely due to methodological differences, eg, sample size, the type of methylome profiling, and the data analysis pipeline. In addition, the majority of DNA methylation studies have been performed on peripheral blood and not on purified T-cell subsets, smooth muscle cells, or endothelial cells. This is significant as each of the latter will have cell type–specific changes in DNA methylation.
Pathway analysis of differentially methylated genes associated with atherosclerosis has consistently identified enrichment of the RhoA signaling pathway across a number of studies. RhoA is a mediator of OxLDL-induced endothelial cell proliferation30 and a key contributor to plaque growth and inflammation in atherosclerosis. The impact on DNA methylation of environmental factors relevant to atherosclerosis susceptibility and pathogenesis has been investigated. One example is the reported link between socioeconomic status and DNA hypermethylation at select stress and inflammatory gene loci.31 Another is the association between disturbed blood flow and upregulation of DNA methyltransferases and hypermethylation of mechanosensitive genes.32
TET methylcytosine dioxygenase 2 (TET2), a DNA demethylase, has been reported to suppress the phenotypic transformation of VSMC and to inhibit inflammation in endothelial cells and macrophages during atherosclerosis.33 Interestingly, a loss of function mutation in TET2 is associated with accelerated atherosclerosis in mice. These intriguing observations indicate that the relative balance between DNA methylation and demethylation merits further research to determine its functional significance in atherosclerosis in vivo.
Histone modifications
Distinct roles for a few histone modifications and epigenetic modifiers in atherosclerosis have been suggested by a number of studies in various types of human endothelial cells in vitro that are either established cell lines or isolated from nonatherosclerotic individuals.34 One recent study on VSMCs and macrophages within human carotid plaques showed a decrease in methylated histone H3 lysine 27 (H3K27) and H3K9 methylation and an increase in methylated H3K4 (a marker of active transcription).35 In the same study, increased histone acetylation at H3K9 and H3K27 were also observed in association with advanced atherosclerosis.35 Experiments following exposure of primary human monocytes to OxLDL showed that increased trimethylation of H3K4 on the promoters of IL-6, IL-18, TNF-a, MCP-1, MMP2, MMP9, CD36, and scavenger receptor-A correlated with increased expression of these genes, all of which have important roles in arterial inflammation.36 These observations need to be corroborated using genome-wide analyses of histone modifications in patients with atherosclerosis compared with controls to determine the location and function of such changes in regulating genes in the disease. At the time of writing, no studies on global profiling of histone modifications in atherosclerosis have been published.
Epigenetic studies in mouse models have reported correlations between histone modifications, epigenetic modifiers, and atherosclerosis.34 Data from histone deacetylase 3 (HDAC3) knockout mice showed increased deposition of collagen in atherosclerotic lesions, stabilization of plaques and a decreased inflammatory phenotype, suggesting that HDAC3 promotes arterial inflammation.37 In ApoE-/- mice, the expression of the histone methylase EZH2 was induced in response to high levels of homocysteine, an independent risk factor for atherosclerosis. Also, high levels of H3K27 trimethylation correlated with the size of atherosclerotic lesions. This change in histone methylation is the opposite to observations in human atherosclerotic plaques, the reasons for which are currently unclear. In other studies, the offspring from ApoE-/- mothers fed a postnatal high cholesterol diet showed differences in histone trimethylation and gene expression in endothelial cells and VSMCs compared with wild-type mothers.38 Validation of methylated histones in individuals with hyperhomocysteinemia or hypercholesterolemia would determine the contribution of epigenetic modifications to pathological changes in the human vasculature and would provide evidence that vascular pathology is mediated by diet induced epigenetic changes.
MicroRNAs
Multiple studies have revealed the central involvement of miRNAs in monocytes, endothelial cells, and VSMCs in atherosclerotic disease highlighting the potential of these RNAs to be used as new, effective therapeutic targets for atherosclerosis.39 Pertinent examples of miRNA function include associations between aberrant expression of miRNAs and 1) atherosclerosis susceptibility (miR 33a and mir-124–3p40) and 2) disturbed arterial flow and endothelial cell dysfunction (miR-92a, miR-126, miR-146, and miR-18).39 miRNAs have also been identified as having a protective function. miR-10a and mir-23b reduce the endothelial cell inflammatory response and block cell cycle progression, respectively, and miR-126-5p promotes the proliferation of endothelial cells and prevents formation of atherosclerotic lesions.41 Other miRNAs, such as miR-33a/b, have been shown to both promote and protect against foam cell formation and atherogenesis.40,42 Studies are continuing to emerge in cultured cells to determine the mechanistic function of miRNAs in atherosclerosis.
Experimental data on patients with hyperhomocysteinemia showed a significant correlation of specific miRNAs with homocysteine or lipid parameters that are of potential diagnostic and predictive value for atherosclerosis in such individuals.43 The precise functions of several miRNAs have demonstrated therapeutic potential, and a variety of miRNA-based tools are under investigation and in development.39 Profiling of the miRNAome in large cohorts of atherosclerosis patients is needed as this would help identify miRNAs that are robust biomarkers and that merit further functional characterization.
Long noncoding RNAs
lncRNAs encoding loci have been associated with several types of cardiovascular disease and are upregulated in patients with atherosclerosis.39 The lncRNAs H19 chromosome 11p15.5 is one such example. Intriguingly, this locus encodes an imprinted gene that is downregulated postnatally, but its expression is induced following vascular injury and within atherosclerotic plaques. Functional validation of its role in atherosclerosis is merited. Of further interest are the lncRNAs that are hypoxia induced in endothelial cells, eg, linc00323-003 and MIR503HG, both of which may control cell proliferation through inhibition of transcription factor GATA-binding protein 2. The lncRNAs SENCR (smooth muscle and endothelial cell-enriched migration/differentiation associated lncRNAs) stabilize the VSMC contractile phenotype and inhibit VSMC migration44 indicating that modulating the activity of these ncRNAs has therapeutic potential. Clearly, both miRNAs and lncRNAs appear to have fundamental cell type–specific roles in cardiovascular physiology.
The selectivity and specificity of ncRNAs in the pathogenesis of atherosclerosis is a dynamic area of research. The development of lncRNAs- and miRNA inhibitors and mimics is a further active area of investigation.39
Integrative omics studies
Integrative genome-wide analyses of atherosclerosis are emerging. One recent example is integration of the epigenome with the transcriptome of purified blood monocytes.45 Expression of the epigenetic modifier, AT-rich interactive domain-containing protein 5 (which demethylates dimethylated H3K9), promotes chronic inflammation and atherosclerosis. These events are inversely correlated with DNA methylation of the AIRD5B enhancer and provide a novel pathway by which inflammation is dysregulated during atherosclerosis. Another study has integrated GWAS loci, epigenomic data, and transcriptomic profiles of human coronary artery smooth muscle cells to reveal new links between coronary artery disease SNPs, open chromatin, acetylated H3K27 (a marker of active enhancers), and gene expression.46 This has revealed new regulatory loci and the basis for defining the link between risk and epigenetic mechanisms of gene regulation relevant to atherosclerosis.
The fascinating observation that allele-specific DNA methylation may be disrupted by SNPs indicates that in specific contexts, epigenetic changes may be dependent on genetic variation.12 In atherosclerosis, methylated DNA in and around SNPs indicates a link with disease susceptibility and provides the potential for these DNA methylated sites to serve as novel disease biomarkers.47
Type 2 diabetes
Molecular and clinical features
T2DM is characterized by hyperglycemia and occurs due to defective insulin secretion and increased resistance to insulin by dysfunctional β cells in the islets of Langerhans of the pancreas.48 Islets maintain glucose homeostasis by detecting and secreting insulin in response to changes in the concentration of blood glucose and by regulating multiple pathways including insulin and glucagon secretion. Other key processes include absorption of sugar from the intestine, the synthesis of glucose by the liver, and glucose uptake by adipose, muscle, and other tissues. The insufficient metabolism of glucose and low-level chronic inflammation of adipose tissue are associated with aging and are central to T2DM pathophysiology. Fascinating evidence suggests that obesity both activates T2DM-associated inflammation and induces a phenotypic switch in adipose tissue macrophages resulting in a failure of inflammation to resolve and the development of obesity-associated insulin resistance.49 T2DM is also characterized by life-threatening complications, which can reduce life expectancy, particularly cardiovascular disease.50,51
Epigenetics of T2DM
Considerable research has been performed on the genetic etiology and pathogenesis of T2DM.51 GWAS studies have identified hundreds of T2DM-specific SNPs that increase the probability of developing the disease and that are associated with T2DM-related levels of insulin, proinsulin, and glucose. However, these variants have modest odds ratios and explain only a minor proportion of disease heritability.52 Efforts to identify less common SNPs by the GOT2D and T2D-GENES consortia have concluded that low-frequency variants do not have a significant role in conferring disease risk.
Many environmental and lifestyle risk factors such as diet, lack of physical activity, smoking, stress, BMI, waist circumference as well as aging have been identified for T2DM and are shared by cardiovascular disease and obesity.51 Both maternal malnutrition and overnutrition have been associated with altered epigenetic modifications and increase susceptibility to the development of T2D in offspring53.
Significant advances in understanding T2DM pathophysiology have been achieved by investigating the role of epigenetic changes in regulating candidate genes and signaling pathways.54 Among the challenges in analyzing the T2DM epigenome is obtaining sufficient numbers of purified homogeneous cells to identify cell type–specific epigenetic changes. This is one reason why pancreatic islets have been intensively studied even though these are composed of heterogeneous cell types.54–57 Single-cell epigenome analyses, especially of constituent islet cell populations, are emerging, but current challenges include donor variability in the proportion of islet subpopulations and the isolation and analysis of rarer islet cell types.
DNA methylation
Genome-wide DNA methylome studies have been performed in blood and human pancreatic islets from patients with T2DM, including on monozygotic twins discordant for the disease as well as several integrative omics studies.58 Examples of the latter include demonstration of important roles for DNA methylation in association with specific histone modifications in altering gene expression in T2DM.59 In human pancreatic isletsfrom T2DM patients differentially methylated sites are associated with altered expression of genes involved in dysregulated glucagon and insulin secretion.55 Fascinating evidence from a study comparing prediabetic, nondiabetic, and T2DM patients indicates that DNA methylation varies according to glucose tolerance. In addition, micro-RNA genes were hypermethylated suggesting that DNA methylation is an important epigenetic modification that regulates the expression of miRNAs in T2DM pathogenesis.60
The impact of a variety of environmental factors on DNA methylation in T2DM has also been studied. One recent example of this has reported that exposure of human islets from T2DM patients to high glucose (19 mM) alters both gene expression and DNA methylation in five genes (GLRA1, RASD1, VAC14, SLCO5A1, and CHRNA5) involved in insulin secretion.61 Another has revealed that DNA methylation markers may be as good or even better at predicting the risk of developing T2DM than traditional risk factors such as BMI, obesity, or abnormal fasting glucose.62 Further intriguing studies in human pancreatic islets in vivo have shown that specific DNA methylation changes and associated alterations in gene expression in peripheral blood reflect aging and are linked with insulin secretion in T2DM.63 The numerous investigations of the relationship between DNA methylation and genetic variation have shown that approximately half of T2DM-associated SNPs are linked with changes in methylated DNA and provide the basis for investigating a potential causal role for DNA methylation in disease development.64
Histone modifications and chromatin architecture
Global profiling of histone modifications in pancreatic islets has shown that the presence of H3 mono/di/tri-methylated on lysine 4 was higher in promoters containing a CpG island. Furthermore, distinct combinations of methylated H3 proteins were associated with different promoters.65 Highly expressed genes, such as insulin and glucagon, were marked by very low levels of H3K4me3 indicating that other epigenetic modifications may regulate these genes. In addition, global profiling of histone methylation (on H3, lysines 4 and 79), CTCF transcription factor binding, and DNAse I hypersensitive sites (indicative of active/open chromatin) in human pancreatic islets identified regulatory elements enriched for T2DM noncoding variants.57 Further evidence for genetic-epigenetic interactions comes from sophisticated studies showing that islet-specific gene activation occurs through binding of transcription factors to clusters of enhancers within three-dimensional chromatin domains.56 T2DM SNPs are located in these enhancer clusters that disrupt both transcription factor binding and enhancer activity revealing a novel regulatory mechanism in T2DM pathophysiology.
Mouse studies have provided experimental evidence using in vitro fertilization that offspring of parents fed a high fat diet are more likely to develop diabetes and obesity.66 Modifications to chromatin during prenatal and early postnatal development appear to be altered by local changes in nutrient availability and metabolite concentration.67 In addition, remodeled chromatin, specific transcription factors, and histone modifications in mice fed a high fat diet were also found in the offspring.68
Research on chromatin enzyme cofactors, eg, acetyl CoA for histone acetylases, S adenosyl methionine for histone methylases, and NAD+ for Sirtuin deacetylases has revealed direct links between chromatin modifications and both glucose and lipid metabolic pathways relevant to T2DM. These have been extensively reviewed elsewhere.53
MicroRNAs
miRNAs have fundamental roles in the differentiation, proliferation, and function of pancreatic cells; the development of insulin resistance, defective glucose and lipid metabolism, adipocytokine signaling, and the onset of T2DM.69 To determine the mechanisms involved, the network of interactions between miRNAs and their target mRNAs has been investigated using high-throughput sequencing, microarrays, and bioinformatics tools. Integration of multiple datasets from different omics has been performed for adipose tissue of insulin-sensitive and insulin-resistant individuals.70 Differential expression of 17 miRNAs was identified. Of these, 16 were downregulated and their corresponding target genes upregulated in the insulin-resistant individuals compared with the insulin-sensitive group. This study provides new miRNA–mRNA networks linked with insulin resistance. In addition, evidence from GWAS and bioinformatics analyses indicates that there are SNPs located within miRNA binding sites that impact the function of genes involved in T2DM pathophysiology.71
Circulating miRNAs found in various types of body fluids such as serum, plasma, and urine,69 are promising, predictive biomarkers for T2DM due to their stability, noninvasiveness, and unique expression patterns. Comparison of profiles of plasma miRNAs in normal individuals vs T2DM-susceptible and T2DM patients revealed that only miR-126 was significantly reduced in patients with T2DM or in those at risk of the disease, identifying it as a potential biomarker of disease susceptibility.69 Similar studies of patients with prediabetes and newly diagnosed T2DM demonstrated that plasma miR-1249, miR-320b, and miR-572 levels could also act as early diagnostic biomarkers. Identification of miRNA candidate biomarkers has led to clinical trials that are now underway to validate their efficacy.
Long noncoding RNAs
lncRNAs are involved in diverse aspects of cell metabolism and are associated with T2DM and its complications.72 These RNAs are highly specific and have been shown to regulate the development and function of pancreatic islets. Numerous research labs have profiled lncRNA expression in mouse and human pancreatic islets as well as β cells.73,74 Tissue-specific and dysregulated lncRNAs often mapping close to islet-specific chromatin domains and associated genes or close to T2DM risk loci have been discovered. Integral functions for lncRNAs in β-cell differentiation and maturation have also been reported.74 Such observations suggest that lncRNAs have critical regulatory roles in insulin resistance, diabetes, and its associated complications, though the mechanisms involved are not understood. Important functional roles for lncRNAs in pancreatic β cells in regulating glucose metabolism and adipocyte differentiation have also been suggested.72,74–76 lncRNAs are regarded as prospective, robust biomarkers of T2D due to their 1) specificity, 2) expression patterns in tissues, and 3) stability in diverse body fluids that reflect disease progression.
Integrative omics
More recently, integrative GWAS, epigenome, and transcriptome studies have revealed links between T2D SNPs, epigenetic changes, and alterations in gene expression in multiple tissues. For example, in islets, SNPs enriched in islet-specific regulatory regions, factors that bind to these regions, and islet-specific enhancers have been identified.54,56,58,77 A recent EWAS has shown that BMI, a measure of adiposity, is associated with changes in DNA methylation but the latter was a consequence and not a cause of adiposity. However, in the same study, dysregulated DNA methylation was found to predict the development of T2DM.62 Further research in purified cell types from diabetic patients to independently replicate and functionally validate changes in histone modifications as a result of hyperglycemia, insulin resistance, and other changes associated with T2DM is needed. The curation of information on transcription factor binding, chromatin architecture, sequence motifs, enhancer clusters, etc, into a single online database, the Islet Regulome Browser (http://www.isletregulome.org/), will significantly facilitate research in this area.
Alzheimer’s disease
Molecular and clinical features
AD is a progressively destructive, irreversible, and mainly sporadic neurodegenerative disorder resulting in severe cognitive impairment. In the context of an unprecedented aging population, it is also a major contributor to ill health globally and is rapidly increasing in prevalence worldwide.78–80 The classic molecular hallmarks of AD-associated dementia are the accumulation of beta amyloid (Aβ) plaques and neurofibrillary tangles containing abnormally phosphorylated, acetylated, and misfolded tau proteins.81 Defects in nonresolving inflammation include a failure to clear these neurotoxic peptides.79 Dysregulated tau metabolism and the accumulation of extracellular Aβ plaques or aggregates are major contributors to neuroinflammation and disease progression.78–80 AD pathology involves multiple cell types and pathways and inflammatory mechanisms in the brain, eg, the binding of misfolded and aggregated proteins to glial cell surface receptors that initiates a pathological innate immune response. In addition, genes that regulate glial clearance of misfolded proteins and inflammatory reactions have been associated with increased risk for AD. Emerging evidence indicates that failure of resolution of inflammation in the brain may be a major contributor to AD pathology and targeting this could be of therapeutic benefit.82
Epigenetics of AD
GWAS has shown that ~20 common genetic variants explain disease susceptibility in around 30% of cases.83 This missing heritability, together with 1) the distinct vulnerability of different regions of the brain to AD, and 2)the age of onset, suggest that epigenetics has an important role in AD pathogenesis. In addition, epigenetic changes are known to play important roles in brain functionality and memory as well as in physiological aging.84
The epigenetics field in AD has transitioned from reductionist, candidate gene approaches to genome-wide profiling of disease-associated epigenetic changes.85 The major challenges in defining epigenetic mechanisms in AD are the involvement of multiple brain regions and cell types, the changes in DNA methylation observed in healthy aging, and the region-dependent changes in cellular content in the brain with progression of disease.84 For example, neuronal cells are lost and different types of glial cells increase in number (gliosis). Some analyses have accounted for neuron/glial cell ratios as covariates (independent variables) in bioinformatic analyses, but the isolation of sufficient numbers of purified brain cell populations for epigenetic studies remains a major challenge.
DNA methylation
Two major genome-wide epigenetic studies have revealed robust and reproducible changes in DNA methylation in four genes not previously associated with AD, namely, ANK1, CDH23, RHBDF2, and RPL13.86,87 Both studies employed the 450K Illumina platform for analyzing DNA methylation, which only represents 2% of the human genome’s CpGs and does not distinguish DNA methylation and DNA hydroxyl-methylation. Both studies found changes in the brain DNA methylome at the onset of AD, though whether these cause or are an early consequence of AD pathology remains to be determined. In another study, differential DNA methylation was found in six genes associated with AD risk (BIN1, CLU, ABCA7, MS4A6A, CD2AP, and APOE),88 indicating that epigenetics contributes to AD susceptibility. A recent DNA methylome study89 has shown that hypermethylated DNA regions overlapped with poised promoters marked with H3K27me3 and H3K4me3. Consistent regions of differentially methylated DNA were found between this and previous AD-associated DNA methylome studies.86,87 In addition, global increases in DNA methylation and DNMT1 expression in the peripheral blood of patients are associated with late-onset AD.90 These studies provide the basis for developing convenient methods of screening for risk of this neurodegenerative disorder.
Relatively new methods are now available that distinguish between 5mC and 5hmC. These have been successfully measured in different regions of the human brain.91 Within the last year, two studies have profiled genome-wide changes in 5-hydroxymethylcytosine in the brain of AD patients.92,93 A great deal remains to be discovered about the functional significance of this and other types of DNA methylation and their contribution to AD pathogenesis. One study reported differentially hydroxymethylated DNA in association with plaques and neurofibrillary tangles in AD.93 The other, using single base resolution mapping, showed significant alterations in hydroxymethylation in a variety of hippocampal genes in AD patients.92
DNA methylation has also been implicated in defective neural plasticity associated with declining cognitive function and aging.84 Whether it is possible to reverse impaired brain function in AD by targeting such epigenetic changes will depend on the outcome of future research.
Histone modifications
The number of studies globally profiling histone modifications in AD brains remains very limited. There are currently no published studies using high-throughput sequencing techniques such as ChIP-Seq or formaldehyde-assisted isolation of regulatory elements (FAIRE-seq).16 Moreover, as with many other epigenetic studies, methodological differences have a major impact on the interpretability and comparability of the data. Histone H3 and H4 acetylation in AD has been reported94 to correlate significantly with levels of gliosis as well as with tau and amyloid accumulation in postmortem AD brains. Cellular and in vivo models of AD are conflicting in showing both hyper- and hypoacetylation.95 Animal models of AD have reported hypoacetylation, which is reversible with HDAC inhibitors and improves cognition, but this has not been replicated in human studies. Fascinating studies have shown that HDAC2 blocks gene transcription associated with cognitive decline in AD and that this is potentially reversible by targeting the activity of HDAC2.96
MicroRNAs
Accumulating evidence indicates that miRNAs are major regulators of gene expression in AD and are also reliable candidate AD biomarkers due to their stability in biofluids.21 miRNAs specifically expressed in the temporal cortex of AD patients compared with control brains have been shown to be differentially DNA methylated indicating that epigenetic regulation of these ncRNAs may contribute to AD pathogenesis.97 In evaluating the activity and expression of different miRNAs in AD, it is important to account for the different methodologies used to quantitate miRNAs including qPCR, microarrays, and RNA seq. miRNAs in AD brains function by directly targeting, through sequence complementarity, mRNAs of proteins linked to AD pathology.21 Such proteins include amyloid precursor protein and other proteins involved in the Aβ processing such as BACE1 that regulate Aβ production.21,98 Other miRNAs can directly regulate tau expression and tau phosphorylation or the mRNAs of proteins involved in the neuronal innate immune response.99 An international collaboration between several groups analyzed data from three different brain areas – hippocampus, prefrontal cortex, and temporal gyrus – and produced differentially expressed miRNAs for each tissue.100 In addition, evidence for altered editing of miRNAs (by modification of adenosine to inosine) has been found in AD.101 Currently, the field lacks studies in which the observed differences in AD miRNAs have been independently validated and/or which correlate specific miRNA expression/function with clinical phenotype. More studies to determine which AD-associated miRNAs are robust biomarkers that are functionally and specifically relevant to disease pathogenesis are also needed. With such knowledge, miRNAs could provide improved, early detection of AD prior to irreversible neurological damage. In vivo testing of miRNA mimics, anti-miRNAs, and analogs of miRNA precursors has begun though the challenges of targeted delivery and off-target effects remain to be overcome.
Long noncoding RNA
RNA sequencing has identified lncRNAs that are intergenic and significantly altered in the brain and plasma in AD.102 Research on the functions of lncRNAs in AD is in its infancy, and with the exception of BACE1-AS lncRNA (which has been shown to stabilize BACE1 mRNA, increase BACE1 protein and Aβ formation), no clear pathological roles for the majority of these RNAs have yet been defined. Other even less studied types of RNA include circRNAs and Y RNAs that may also play important roles in the pathology of AD.21,103
Future research focusing on integrative omics studies, (eg, epigenome with GWAS, transcriptome, and clinical data) is likely to provide important insights into AD etiopathogenesis.
Conclusion and future perspectives
The field of epigenetics of chronic inflammatory diseases has undergone a major transition. The shift from GWAS studies to EWAS and integrative omics is dynamic and current. There is increasing recognition that GWAS studies have only explained a minor portion of the heritability of these disorders. Furthermore, while candidate gene approaches have generated important data, EWAS studies have provided comprehensive analyses of epigenetic modifications. Important insights into the content of epigenomes in atherosclerosis, T2DM, and AD have been gained, but the data are largely descriptive and have only profiled a minority of epigenetic changes. Definition of the links between epigenetic defects and nonresolving inflammation or aging is in its infancy and determining which epigenetic modifications are casual, predict risk, or explain chronic disease pathogenesis remains a major challenge. Integrative analyses of the epigenome, genome, transcriptome along with the microbiome and clinical data are needed to generate a more complete picture of chronic disease risk, to stratify disease subtypes and to define relevant pathological mechanisms. The microbiome has already significantly advanced our understanding of atherosclerosis and is likely to make further important contributions to understanding the pathophysiology of other aging-related diseases.
Universal standards for tissue preparation, minimum sample size, methods to address cellular heterogeneity, and bioinformatics analyses still need to be achieved and are vital to enable rigorous comparisons to be made between data from independent groups. Improvements in sequencing strategies and other experimental techniques for identifying disease-specific signatures at the single-cell level are also likely to have an impact on progress in this area. The use of reference epigenomes and predictive models is also vital to generate interactomes and pathways that are functionally and therapeutically relevant.16 Furthermore, the number of openly accessible databases is growing, eg, iMethyl (http://imethyl.iwate-megabank.org/) which integrates DNA-methylated CpG sites, SNPs, and transcriptome in inflammatory cells. However, additional computational tools are needed to facilitate progress in defining disease mechanisms. Such approaches, together with genome editing,16 could provide paradigm-shifting advances in defining the contribution of epigenetic defects to the etiopathogenesis of chronic inflammatory disease.
Disclosure
The author reports no conflicts of interest in this work.
References
Deans C, Maggert KA. What do you mean, “epigenetic”? Genetics. 2015;199(4):887–896. | ||
Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20(7):883–889. | ||
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. | ||
Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140(6):771–776. | ||
Perkins DJ, Patel MC, Blanco JC, Vogel SN. Epigenetic mechanisms governing innate inflammatory responses. J Interferon Cytokine Res. 2016;36(7):454–461. | ||
Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. 2016;7:160. | ||
Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6(12):1191–1197. | ||
Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3):a006049. | ||
Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339(6116):166–172. | ||
Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–327. | ||
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. | ||
Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1757–1764. | ||
Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16(10):593–610. | ||
Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2(7):e1600584. | ||
Stylianou E. Epigenetics: the fine-tuner in inflammatory bowel disease? Curr Opin Gastroenterol. 2013;29(4):370–377. | ||
Stricker SH, Köferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18(1):51–66. | ||
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. | ||
Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15(6):423–437. | ||
Wang S, Wu W, Claret FX. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics. 2017;12(3):187–197. | ||
Han Y, He X. Integrating epigenomics into the understanding of biomedical insight. Bioinform Biol Insights. 2016;10:267–289. | ||
Maoz R, Garfinkel BP, Soreq H. Alzheimer’s disease and ncRNAs. Adv Exp Med Biol. 2017;978:337–361. | ||
Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med. 2010;14(6A):1225–1240. | ||
Bentzon JF. Targeting inflammation in atherosclerosis. J Am Coll Cardiol. 2016;68(25):2794–2796. | ||
Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86. | ||
Kasikara C, Doran AC, Cai B, Tabas I. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. 2018;128(7):2713–2723. | ||
Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 2010;7(9):510–519. | ||
Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–447. | ||
Fernández-Sanlés A, Sayols-Baixeras S, Subirana I, Degano IR, Elosua R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis. 2017;263:325–333. | ||
Valencia-Morales MP, Zaina S, Heyn H, et al. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics. 2015;8:7. | ||
Zhang C, Adamos C, Oh MJ, et al. OxLDL induces endothelial cell proliferation via Rho/ROCK/Akt/p27kip1 signaling: opposite effects of oxLDL and cholesterol loading. Am J Physiol Cell Physiol. 2017;313(3):C340–C351. | ||
Smith JA, Zhao W, Wang X, et al. Neighborhood characteristics influence DNA methylation of genes involved in stress response and inflammation: The Multi-Ethnic Study of Atherosclerosis. Epigenetics. 2017;12(8):662–673. | ||
Dunn J, Thabet S, Jo H. Flow-dependent epigenetic DNA methylation in endothelial gene expression and atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(7):1562–1569. | ||
Liu Y, Peng W, Qu K, et al. TET2: a novel epigenetic regulator and potential intervention target for atherosclerosis. DNA Cell Biol. 2018;37(6):517–523. | ||
Jiang W, Agrawal DK, Boosani CS. Cell-specific histone modifications in atherosclerosis (Review). Mol Med Rep. 2018;18(2):1215–1224. | ||
Greißel A, Culmes M, Burgkart R, et al. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. 2016;25(2):79–86. | ||
Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014;34(8):1731–1738. | ||
Hoeksema MA, Gijbels MJ, van den Bossche J, et al. Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6(9):1124–1132. | ||
Alkemade FE, van Vliet P, Henneman P, et al. Prenatal exposure to apoE deficiency and postnatal hypercholesterolemia are associated with altered cell-specific lysine methyltransferase and histone methylation patterns in the vasculature. Am J Pathol. 2010;176(2):542–548. | ||
Lucas T, Bonauer A, Dimmeler S. RNA therapeutics in cardiovascular disease. Circ Res. 2018;123(2):205–220. | ||
Kim SH, Kim GJ, Umemura T, Lee SG, Cho KJ. Aberrant expression of plasma microRNA-33a in an atherosclerosis-risk group. Mol Biol Rep. 2017;44(1):79–88. | ||
Schober A, Nazari-Jahantigh M, Wei Y, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20(4):368–376. | ||
Price NL, Rotllan N, Canfrán-Duque A, et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. 2017;21(5):1317–1330. | ||
Liu K, Xuekelati S, Zhou K, et al. Expression profiles of six atherosclerosis-associated microRNAs that cluster in patients with hyperhomocysteinemia: a clinical study. DNA Cell Biol. 2018;37(3):189–198. | ||
Bell RD, Long X, Lin M, et al. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol. 2014;34(6):1249–1259. | ||
Liu Y, Reynolds LM, Ding J, et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat Commun. 2017;8(1):393. | ||
Miller CL, Pjanic M, Wang T, et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat Commun. 2016;7:12092. | ||
Zaina S. Unraveling the DNA methylome of atherosclerosis. Curr Opin Lipidol. 2014;25(2):148–153. | ||
Skyler JS, Bakris GL, Bonifacio E, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes. 2017;66(2):241–255. | ||
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. | ||
Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127(1):1–4. | ||
Kraniotou C, Karadima V, Bellos G, Tsangaris GT. Predictive biomarkers for type 2 of diabetes mellitus: bridging the gap between systems research and personalized medicine. J Proteomics. 2018;188:59–62. | ||
Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41–47. | ||
Keating ST, El-Osta A. Epigenetics and metabolism. Circ Res. 2015;116(4):715–736. | ||
Lawlor N, Khetan S, Ucar D, Stitzel ML. Genomics of islet (Dys)function and type 2 diabetes. Trends Genet. 2017;33(4):244–255. | ||
Dayeh T, Volkov P, Salö S, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160. | ||
Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46(2):136–143. | ||
Stitzel ML, Sethupathy P, Pearson DS, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12(5):443–455. | ||
Kwak SH, Park KS. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp Mol Med. 2016;48:e220. | ||
Volkov P, Bacos K, Ofori JK, et al. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes. 2017;66(4):1074–1085. | ||
Pheiffer C, Erasmus RT, Kengne AP, Matsha TE. Differential DNA methylation of microRNAs within promoters, intergenic and intragenic regions of type 2 diabetic, pre-diabetic and non-diabetic individuals. Clin Biochem. 2016;49(6):433–438. | ||
Hall E, Dekker Nitert M, Volkov P, et al. The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol Cell Endocrinol. 2018;472:57–67. | ||
Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541(7635):81–86. | ||
Bacos K, Gillberg L, Volkov P, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. 2016;7:11089. | ||
Elliott HR, Shihab HA, Lockett GA, et al. Role of DNA methylation in type 2 diabetes etiology: using genotype as a causal anchor. Diabetes. 2017;66(6):1713–1722. | ||
Bhandare R, Schug J, Le Lay J, et al. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20(4):428–433. | ||
Huypens P, Sass S, Wu M, et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genet. 2016;48(5):497–499. | ||
Schroeder JW, Conneely KN, Cubells JC, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. 2011;6(12):1498–1504. | ||
Leung A, Trac C, du J, Natarajan R, Schones DE. Persistent chromatin modifications induced by high fat diet. J Biol Chem. 2016;291(20):10446–10455. | ||
Feng J, Xing W, Xie L. Regulatory roles of microRNAs in diabetes. Int J Mol Sci. 2016;17(10):1729. | ||
Kirby TJ, Walton RG, Finlin B, et al. Integrative mRNA-microRNA analyses reveal novel interactions related to insulin sensitivity in human adipose tissue. Physiol Genomics. 2016;48(2):145–153. | ||
Cheng M, Liu X, Yang M, Han L, Xu A, Huang Q. Computational analyses of type 2 diabetes-associated loci identified by genome-wide association studies. J Diabetes. 2017;9(4):362–377. | ||
Goyal N, Kesharwani D, Datta M. Lnc-ing non-coding RNAs with metabolism and diabetes: roles of lncRNAs. Cell Mol Life Sci. 2018;75(10):1827–1837. | ||
Ku GM, Kim H, Vaughn IW, et al. Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome. Mol Endocrinol. 2012;26(10):1783–1792. | ||
Moran I, Akerman I, van de Bunt M, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–448. | ||
Akerman I, Tu Z, Beucher A, et al. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017;25(2):400–411. | ||
Fadista J, Vikman P, Laakso EO, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A. 2014;111(38):13924–13929. | ||
Parker SC, Stitzel ML, Taylor DL, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110(44):17921–17926. | ||
Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184(1–2):69–91. | ||
Johansson JU, Woodling NS, Shi J, Andreasson KI. Inflammatory cyclooxygenase activity and PGE2 signaling in models of Alzheimer’s disease. Curr Immunol Rev. 2015;11(2):125–131. | ||
Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612–623. | ||
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | ||
Fraga VG, Carvalho MDG, Caramelli P, de Sousa LP, Gomes KB. Resolution of inflammation, n-3 fatty acid supplementation and Alzheimer disease: a narrative review. J Neuroimmunol. 2017;310:111–119. | ||
Ridge PG, Mukherjee S, Crane PK, Kauwe JS, Alzheimer’s Disease Genetics Consortium. Alzheimer’s disease: analyzing the missing heritability. PLoS One. 2013;8:e79771. | ||
Qazi TJ, Quan Z, Mir A, Qing H. Epigenetics in Alzheimer’s disease: perspective of DNA methylation. Mol Neurobiol. 2018;55(2):1026–1044. | ||
Lord J, Cruchaga C. The epigenetic landscape of Alzheimer’s disease. Nat Neurosci. 2014;17(9):1138–1140. | ||
Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17(9):1164–1170. | ||
de Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17(9):1156–1163. | ||
Chibnik LB, Yu L, Eaton ML, et al. Alzheimer’s loci: epigenetic associations and interaction with genetic factors. Ann Clin Transl Neurol. 2015;2(6):636–647. | ||
Watson CT, Roussos P, Garg P, et al. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016;8(1):5. | ||
di Francesco A, Arosio B, Falconi A, et al. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav Immun. 2015;45:139–144. | ||
Lunnon K, Hannon E, Smith RG, et al. Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol. 2016;17:27. | ||
Ellison EM, Bradley-Whitman MA, Lovell MA. Single-base resolution mapping of 5-hydroxymethylcytosine modifications in hippocampus of Alzheimer’s disease subjects. J Mol Neurosci. 2017;63(2):185–197. | ||
Zhao J, Zhu Y, Yang J, et al. A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer’s disease. Alzheimers Dement. 2017;13(6):674–688. | ||
Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol Dis. 2015;74:281–294. | ||
Narayan P, Dragunow M. Alzheimer’s disease and histone code alterations. Adv Exp Med Biol. 2017;978:321–336. | ||
Gräff J, Rei D, Guan JS, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–226. | ||
Villela D, Ramalho RF, Silva AR, et al. Differential DNA methylation of microRNA genes in temporal cortex from Alzheimer’s disease individuals. Neural Plast. 2016;2016:2584940. | ||
Hébert SS, Horré K, Nicolaï L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–6420. | ||
Denk J, Boelmans K, Siegismund C, Lassner D, Arlt S, Jahn H. MicroRNA profiling of CSF reveals potential biomarkers to detect Alzheimer’s disease. PLoS One. 2015;10(5):e0126423. | ||
Lau P, Bossers K, Janky R, et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med. 2013;5(10):1613–1634. | ||
Manassero G, Guglielmotto M, Zamfir R, et al. Beta-amyloid 1-42 monomers, but not oligomers, produce PHF-like conformation of Tau protein. Aging Cell. 2016;15(5):914–923. | ||
Magistri M, Velmeshev D, Makhmutova M, Faghihi MA. Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-β homeostasis, and deregulated expression of long noncoding RNAs. J Alzheimers Dis. 2015;48(3):647–665. | ||
Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.