")
Back to Journals » Journal of Inflammation Research » Volume 7
Epigallocatechin-3-gallate suppresses proinflammatory cytokines and chemokines induced by Toll-like receptor 9 agonists in prostate cancer cells
Authors Mukherjee S, Siddiqui M, Dayal S, Ayoub Y, Malathi K
Received 13 March 2014
Accepted for publication 19 April 2014
Published 17 June 2014 Volume 2014:7 Pages 89—101
DOI https://doi.org/10.2147/JIR.S61365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sushovita Mukherjee, Mohammad Adnan Siddiqui, Shubham Dayal, Yasmine Zakaria Ayoub, Krishnamurthy Malathi
Department of Biological Sciences, University of Toledo, Toledo, OH, USA
Abstract: Chronic inflammation of the prostate contributes to the increased risk of prostate cancer. Microbial pathogens in the prostate cause inflammation that leads to prostatitis and proliferative inflammatory atrophy frequently associated with the development of prostate cancer. Bacterial lipopolysaccharides and DNA mediate immune responses by engaging Toll-like receptor (TLR) 4 and 9, respectively. Synthetic oligodeoxynucleotides containing CpG motifs (CpG-ODN) mimic bacterial DNA and signal through TLR9 to initiate innate immune responses. Here, we show that stimulation of DU145, PC3, or LnCap prostate cancer cells by the TLR9 agonists, CpG-ODN, induces mRNA expression of IL-6, IL-8, CXCL1, IP-10, CCL5, and TGFβ. In addition, activity of matrix metalloproteinase (MMP)-9 and -2 and cell migration increased on CpG-ODN treatment. Induction of cytokines and chemokines was mediated by NF-ΚB activation and translocation to the nucleus. Treatment with epigallocatechin-3-gallate (EGCG), the major constituent of green tea, prior to CpG-ODN stimulation, inhibits cytokine and chemokine gene induction, activity of MMP-9 and -2, and cell migration. EGCG treatment sequesters the p65 subunit of transcription factor NF-ΚB in the cytoplasm and inhibits transcriptional activity of the NF-ΚB-driven promoter in response to CpG-ODN. Our results suggest that the ability of the TLR9 agonists, CpG-ODN, to induce cytokines, chemokines, and MMP activity, as well as suppression by EGCG are independent of the androgen receptor and p53 status of the cells. EGCG may provide protective effects against inflammation in the prostate and benefit prostate cancer treatment.
Keywords: CpG-ODN, EGCG, inflammation, NF-ΚB
Introduction
Prostate cancer is the second leading cause of death among men in the US. Age, hormonal, environmental, and genetic factors contribute to the pathogenesis of the disease. Several epidemiological studies, including meta-analyses, suggest an important role for intraprostatic inflammation in the pathogenesis of prostate cancer.1–6 Several case-controlled studies have reported a statistically significant association of prostate cancer with sexually transmitted infections.7,8 In studies of men with advanced and hormone refractory prostate cancer, high plasma concentrations of inflammatory cytokines like interleukin-6 (IL-6), IL-8, IL-1, tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) have been reported.5,9,10 Microorganisms that cause prostatitis are believed to promote chronic inflammation by inflicting cellular damage, cellular hyperproliferation, and increased production of cytokines.11 While cytokines are required to limit tissue damage by replacing damaged cells, angiogenesis, and tissue repair, uncontrolled response promotes the progression from benign prostatitis to intraepithelial neoplasia (PIN) and cancer.12,13 Therefore, preventing or reducing inflammation would serve as an attractive mechanism in the chemoprevention of prostate cancer.
Epigallocatechin-3-gallate (EGCG), a major polyphenolic constituent of green tea, has chemopreventive and chemotherapeutic effects in many tumor models.14–16 EGCG selectively induces apoptosis and alters the expression of cell cycle proteins and inhibits cell growth in cancer cells compared to normal cells.17–20 Increases in the expression of p21, p27, p18, and p16, and a corresponding decrease in cyclin D1, D2, and cyclin E as well as CDK 2, 4, and 6 were observed in cells treated with EGCG.21–24 In prostate cancer cells, EGCG treatment reduced the expression of antiapoptotic proteins like phospho-PI3K and its substrate AKT, thereby limiting cell growth.25,26 In addition, EGCG reduces nuclear localization of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which has significant roles in cell survival and induction of cytokines.27,28 Proteolysis of the extracellular matrix components and induction of neovasculature by angiogenesis are required for tumor development. EGCG inhibits the production and activation of vascular endothelial growth factor (VEGF) in breast and colon cancer, and leukemia cells.29–31 Proteolytic activity of matrix metalloproteinases (MMPs) in prostate cancer cells is inhibited in response to EGCG treatment.32
Toll-like receptors (TLRs) are evolutionarily conserved transmembrane proteins that recognize conserved molecular motifs specific for microbial components. The mammalian TLR family consists of eleven members, each able to detect a distinct pathogen-derived ligand to induce signaling pathways involved in innate immune responses.33,34 TLR signaling pathways utilize TIR-domain-containing adaptors like MyD88 (myeloid differentiation primary response 88), TIRAP/MAL (toll-interleukin 1 receptor [TIR] domain containing adaptor protein/MyD88 adapter-like), TRIF (TIR-domain-containing adapter-inducing IFNβ), and TRAM (translocating chain-associated membrane protein), and differentially use transcription factors like NF-κB or interferon regulatory transcription factor (IRFs) to fine-tune specific responses.35 Bacterial components such as Gram-negative lipopolysaccharide (LPS) are recognized by TLR4, whereas bacterial DNA stimulates innate immune responses by engaging TLR9.36 It has been shown that viral or bacterial DNA contain unmethylated CpG dinucleotide motifs (CpG motifs) that have potent immunostimulatory activity.37–41 There are two types of CpG DNA that are structurally distinct: type A has greater ability to induce interferon α (IFNα) from plasmacytoid dendritic cells, and type B, which is more conventional and induces inflammatory cytokines. Both types recognize and bind TLR9 to exert antimicrobial responses.33 On binding a ligand, TLR9 associates with the adaptor, MyD88, and recruits IRAK and TRAF6 to activate the IκB kinase complex (IKK). Phosphorylation of IκB by the IKK complex causes nuclear translocation of NF-κB, which induces expression of inflammatory cytokines.36 Synthetic phosphorothioate-stabilized oligonucleotides containing the CpG motifs (CpG-ODN) mimic the immunostimulatory activity of microbial DNA and are recognized by TLR9.42,43 Expression of TLR9 is not limited to immune cells. Normal epithelial and tumor cells express TLR9, and stimulation with ligands induces production of pro-inflammatory cytokines.44–46 TLR9 agonists have been shown to promote migration of breast cancer cells, astrocytes, and prostate cancer cells.47–49
Persistent infection in the prostate contributes microbial components, which can induce production of cytokines initiating inflammation. Bacterial or viral unmethylated CpG-DNA released by dying or proliferating pathogens is recognized by TLR9 to initiate immune responses, which in turn can alter the tumor microenvironment.50 In this study, we compared the role of the TLR9 agonists, CpG-ODN, in cytokine induction in DU145, PC3, and LnCap prostate cancer cells, and examined the effect of EGCG in suppressing cytokine induction. Our results show that stimulation of prostate cancer cells with CpG-ODN induces messenger ribonucleic acid (mRNA) expression of cytokines and chemokines. In addition, the activity of MMP-9 and -2 are increased over basal levels of expression on CpG-ODN treatment, which correlates with increased migration. The induction of cytokine gene transcription is dependent on NF-κB activation and translocation to the nucleus. EGCG treatment inhibits both cytokine gene induction and activity of MMP-9 and -2 by inhibiting translocation of NF-kB to the nucleus and transcriptional activity of the NF-kB-driven promoter in response to TLR9 agonists. Our results have compared cytokine gene induction and MMP activity in three prostate cancer cell lines. DU145 and PC3 are androgen-independent cell lines and LnCap expresses androgen receptor (AR) and is androgen-dependent. In addition, DU145 cells express mutant p53, PC3 cells lack p53 expression, and LnCap cells express functional p53 protein. Our results suggest that the ability of CpG-ODN to induce cytokines, chemokines, and MMPs, and the suppression by EGCG, is independent of the AR and p53 status of the cells, which is an important property of chemoprevention for prostate cancer treatment.
Materials and methods
Chemicals, reagents, and antibodies
Chemicals, unless indicated otherwise, were obtained from Sigma-Aldrich Co, (St Louis, MO, USA). Antibodies to NF-κB p65 (C-20) used for immunoblotting and immunofluorescence and IκBα were from Santa Cruz Biotechnology Inc., (Dallas, TX, USA). Antibody to β-actin was from Sigma-Aldrich Co, and Histone H2A was from EMD Millipore (Billerica, MA, USA). Anti-mouse immunoglobulin G (IgG) and anti-rabbit IgG horseradish peroxidase (HRP) linked secondary antibodies were from Cell Signaling, Inc., (Danvers, MA, USA) and enhanced chemiluminescence reagents were from GE Healthcare Bio-Sciences Corp (Piscataway, NJ, USA). EGCG was purchased from Cayman Chemical Co, (Ann Arbor, MI, USA). Phosphorothioate-modified, human-specific, class B CpG-ODN (5′-TCGTCGTTTTGTCGTTTTGTCGTT-3′) was from InvivoGen Corporation (San Diego, CA, USA) and dissolved in endotoxin-free sterile water as per the manufacturer’s instructions and used at the indicated concentrations. TRIzol reagent, Alexa Fluor® 488 Donkey Anti-Rabbit IgG (H + L) antibody, and Lipofectamine 2000 were from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). The luciferase construct containing five copies of NF-kB response elements linked to firefly luciferase complementary DNA (cDNA) (pNF-kB-Luc) was obtained from Stratagene (Agilent Technologies, Santa Clara, CA, USA). Dual luciferase reporter assay reagents and CellTiter 96® AQueous One Solution Cell Proliferation were from Promega Corporation (Fitchburg, WI, USA).
Cell culture and treatment of cells
The human prostate cancer cell lines DU145, PC3 (kindly provided by Aimin Zhou, Cleveland State University, OH, USA), and LnCap (kindly provided by William Taylor, University of Toledo, OH, USA) were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with streptomycin (100 μg/mL), penicillin (100 units/mL), 2 mmol/L glutamine, and 10% fetal bovine serum (FBS; Invitrogen). Cells were maintained in 95% air, 5% CO2 at 37°C. EGCG was dissolved in dimethyl sulfoxide (DMSO) at 20 mg/mL, and aliquoted and stored at −20°C. Cells were treated at the indicated dose and time in complete RPMI medium. In some experiments, cells were treated with Bay11-7082 (10 μM; Santa Cruz Biotechnology) for 1 hour at 37°C followed by transfection with CpG-ODN and analyzed. Cells that were used as controls (mock) were incubated with vehicle only.
Cell viability assays
The viability of cells was determined using the CellTiter 96 AQueous Cell Proliferation Assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [MTT] assay; Promega). Briefly, 2×104 cells were seeded in a 96-well culture plate and treated with varying concentrations of EGCG or CpG-ODN (1 μM). At indicated times after treatment, MTT reagent was added to each well and absorbance was measured at 490 nm with a plate reader (Spectra Max 340; Molecular Devices, LLC, Sunnyvale, CA, USA). Cell viability was normalized to untreated cells. Experiments were performed in triplicate and standard deviation (SD) was calculated.
RNA isolation and quantitative reverse transcription polymerase chain reaction
RNA was isolated using TRIzol reagent as per the manufacturer’s instructions and used for cDNA synthesis using random decamers and a RETROscript cDNA synthesis kit (Life Technologies; Thermo Fisher Scientific, Waltham, MA, USA). Expression of cytokines and chemokines was determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using SYBR Green PCR Master Mix (Bio-Rad Laboratories Inc., Hercules, CA, USA) using the gene-specific primers listed below and normalized to GAPDH expression. The primers used are: IL-8F 5′ ACC ACA CTG CGC CAA CAC AGA AAT 3′, IL-8R 5′ AAA CTT CTC CAC AAC CCT CTG CAC 3′, CXCL1 F 5′ TCC AAA GTG TGA ACG TGA AGT CCC 3′, CXCL1 R 5′ CAA GCT TTC CGC CCA TTC TTG AGT 3′, TGFβ F 5′ CGA GAG GAG CGA CGA AGA GT 3′, TGFβ R 5′ AGG GCG GCA TGT CTA TTT TG 3′, CCL5F 5′ TTT CTA CAC CAG TGG CAA GTG CTC 3′, CCL5 R 5′ TCT TCT CTG GGT TGG CAC ACA CTT 3′, IL-6F 5′ TGT GAA AGC AGC AAA GAG GCA CTG 3′, IL-6R 5′ CAC CAG GCA AGT CTC CTC ATT GAA 3′, IP-10F 5′ ACC GTA CGC TGT ACC TGC AT 3′, IP-10R 5′ TCT TGA TGG CCT TCG ATT CT 3′, GAPDH 5′ TCG ACA GTC AGC CGC ATC TTC TTT 3′, and GAPDH R 5′ ACC AAA TCC GTT GAC TCC GAC CTT 3′.
Cell lysates and immunoblotting
Cells were lysed in buffer and separated into nuclear and cytoplasmic extracts using a nuclear/cytosol fractionation kit (MBL International Corporation, Woburn, MA, USA) using the protocol provided by the manufacturer. Protein concentrations in the supernatants were determined using bovine serum albumin as a standard (Bio-Rad protein assay kit). The protein in the cell lysates were separated in 10% or 15% polyacrylamide/sodium dodecyl sulfate (SDS) gels and transferred to a nitrocellulose membrane (Bio-Rad). Membranes were probed with different primary antibodies according to the manufacturer’s protocols. The membranes were washed with Tris-buffered saline with 1% Tween 20 and incubated with goat anti-mouse or goat anti-rabbit antibody tagged with HRP for 1 hour. Immunoreactive bands were detected by enhanced chemiluminescence (GE Healthcare Bio-Sciences Corp).
Luciferase reporter assays for NF-kB
DU145 or PC3 cells (2×105 cells per well) were cotransfected with 1.0 μg of pNF-kB-Luc plasmid along with 0.1 μg of pRL-TK (Renilla luciferase vector) using lipofectamine 2000 reagent according to the protocol provided by the manufacturers. The IL-8 promoter fused to luciferase (IL-8 WT-Luc) and constructs with mutations in NF-κB (IL-8mNF-κB) or AP-1 binding sites (IL-8mAP-1; kindly provided by Dr Gary Wu, University of Pennsylvania, PA, USA) were cotransfected with the pRL-TK plasmid. After 24 hours, some samples were transfected with CpG-ODN (1 μM) using lipofectamine 2000. EGCG (40 μg/mL) was added to some experiments prior to CpG-ODN transfection. Cells were lysed in lysis buffer after 16–18 hours and luciferase activity was determined and normalized to Renilla luciferase activity using a dual luciferase assay kit (Promega).
NF-κB nuclear translocation by immunofluorescence assay
DU145 cells were grown on glass coverslips and mock treated or treated with CpG-ODN (1 μM) or TNFα (10 ng/mL; R&D Systems, Inc., Minneapolis, MN, USA) for 1 hour with or without pretreatment with EGCG for 24 hours. Cells were rinsed with phosphate-buffered saline (PBS) and permeabilized with 0.1% Triton-X100 for 15 minutes. After 1 hour of incubation with NF-κB p65 subunit antibody (1:250), cells were washed once with 0.01% Tween 20 followed by two washes with PBS. Cells were incubated for 1 hour with Alexa Fluor 488 donkey anti-rabbit secondary antibody (1:400). Cells were washed with 0.01% Tween 20 and PBS, mounted in VECTASHIELD® with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs, Burlingame, CA, USA). Cells were imaged with a Leica CS SP5 multi-photon laser scanning confocal microscope (Leica Microsystems, Weitzler, Germany).
MMP-9 and -2 gelatin zymography
Activity of MMP-9 and -2 was determined in culture supernatants by gelatin zymography. DU145, PC3, or LnCaP cells (2×105 cells) were treated as indicated with CpG-ODN (1 μM) with or without EGCG (40 μg/mL) pretreatment. Culture supernatant was electrophoresed in 8% SDS-polyacrylamide gel electrophoresis (PAGE) containing 0.1% gelatin (Sigma-Aldrich Co). Gels were soaked in 2.5% Triton-X100 for 2 hours, followed by incubation in digestion buffer (10 mM CaCl2, 50 mM Tris, pH 7.4, 1% Triton-X100) for 20 hours at 37°C. The gels were then stained with Coomassie Brilliant Blue R-250 (0.2% in 40% methanol, 10% acetic acid) and destained in 20% methanol, 10% acetic acid solution. Clear bands representing MMP-9 and -2 activity were imaged.
Cell migration assay
Transwell cell migration assays were performed using a modified Boyden chamber (Corning Inc., Corning, NY, USA) containing a fibronectin-coated polycarbonate membrane filter (6.5 mm diameter, 8 μm pore size). DU145 or PC3 cells (2×105) were pretreated or not with EGCG (40 μg/mL) for 24 hours followed by transfection with CpG-ODN (1 μM). Cells were plated in the upper chamber and the lower chamber contained culture medium with 10% FBS. Cells were incubated for 24 hours at 37°C in 5% CO2. Nonmigrated cells were scraped from the upper surface of the membrane with a cotton swab, and migrated cells remaining on the bottom surface were trypsinized and counted with a hemocytometer.
Statistical analysis
All values are presented as mean ± SD. Student’s t-tests were used for determining statistical significance between groups. P<0.05 was considered significant.
Results
Effect of CpG-ODN and EGCG on viability of prostate cancer cells
CpG-ODN, like bacterial DNA, has substantial immunostimulatory effects and serves as TLR9 agonists. The prostate cancer cells DU145, PC3, and LnCap cells have been shown to express TLR9 and its adaptor, MyD88. To determine if CpG-ODN affects the viability of prostate cancer cells, DU145, PC3, and LnCap cells were transfected with varying concentrations of CpG-ODN (1 μM or 10 μM) or 1 μM of CpG-ODN for 8 or 24 hours using lipofectamine 2000. Cell viability was determined by MTT colorimetric assays (Figure 1A and B). A high concentration (10 μM) of CpG-ODN transfection induced significant cell death in all three cell lines. LnCap cells showed more resistance compared to DU145 and PC3 cells, whereas treatment with 1 μM of CpG-ODN had very little effect on viability; 85%–91% of the cells were viable after 24 hours of treatment. All experiments were therefore performed with 1 μM CpG-ODN, which induced less cytopathicity.
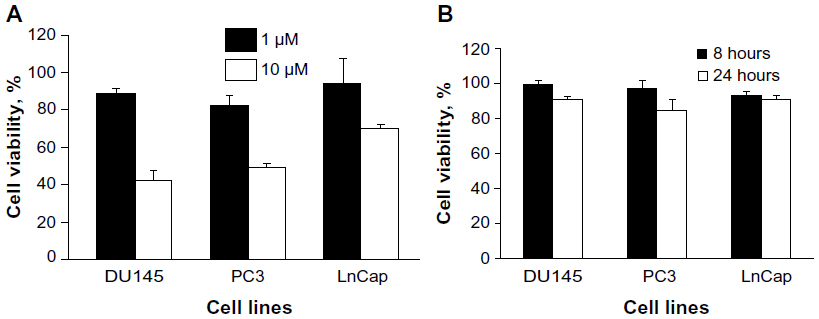 | Figure 1 Effect of CpG-ODN on cell viability in DU145, PC3, and LnCap cells. |
EGCG inhibits the growth of several types of cancer cells, including prostate cancer cells. To determine the effect of EGCG on DU145, PC3, or LnCap cells, we treated cells with 20–100 μg/mL EGCG for 24 hours. Cell viability was determined by MTT colorimetric assays (Figure 2). At 40 μg/mL, 79% of DU145 cells and 85% of PC3 or LnCap cells were viable. A high concentration of EGCG (100 μg/mL) reduced cell viability of DU145 cells to 51%, compared to 61% of PC3 and 76% of LnCap cells. These results indicate that EGCG inhibits cell growth of the prostate cancer cell lines tested in a dose-dependent manner.
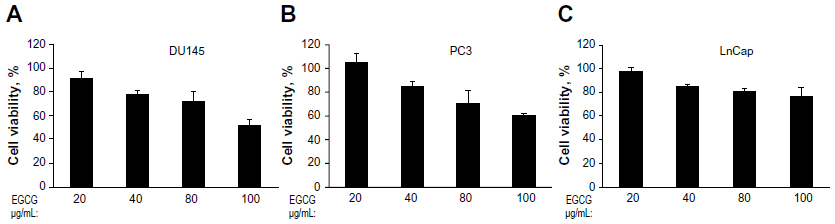 | Figure 2 Dose-dependent effect of EGCG on cell viability on DU145, PC3, and LnCap cells. |
CpG-ODN induces expression of proinflammatory cytokines and chemokines
To determine if CpG-ODN can induce proinflammatory cytokines and chemokines in DU145, PC3, and LnCap cells, CpG-ODN (1 μM) was transfected for 6 or 24 hours. Levels of mRNAs for IL-6, IL-8, CXCL1, IP-10, CCL5, and TGFβ were determined by qRT-PCR and normalized to GAPDH levels. Induction of IL-6 and IL-8 followed similar kinetics in all the cell lines (Figure 3). CXCL1 levels dropped significantly 24 hours postinduction in LnCap cells compared to DU145 and PC3 cells. In PC3 cells, the kinetics of induction of IP-10, CCL5, and TGFβ were significantly different compared those in DU145 and LnCap cells. These results suggest that CpG-ODN induced proinflammatory cytokines and chemokines in prostate cancer cells, albeit with different kinetics involving a mechanism that is independent of expression of AR and p53 status of the cells.
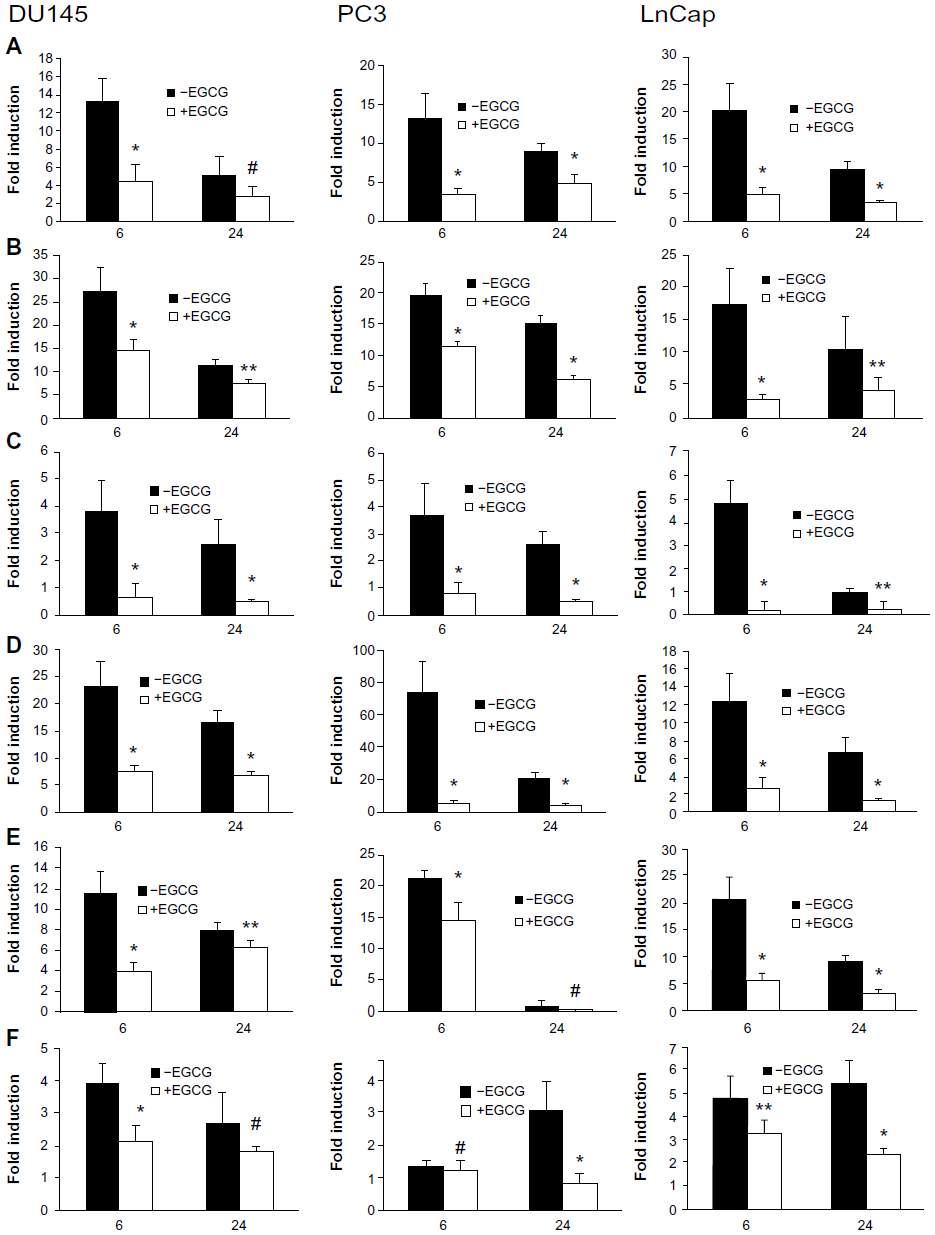 | Figure 3 CpG-ODN stimulation induces the expression of proinflammatory cytokines and chemokines in prostate cancer cells. |
EGCG suppresses proinflammatory cytokines induced by CpG-ODN
EGCG modulates multiple signal transduction pathways, providing growth inhibitory effects. To investigate if EGCG regulates CpG-ODN-induced gene expression, prostate cancer cells were pretreated with EGCG (40 μg/mL) for 24 hours followed by transfection with CpG-ODN (1 μM) for 6 or 24 hours. Expression of mRNA for cytokines and chemokines was determined by RNA isolation and qRT-PCR, and compared to cytokine gene induction without EGCG treatment (Figure 3). Our results show that EGCG potently inhibits gene induction of most cytokines in response to CpG-ODN in all three prostate cancer cell lines. However, suppression of IL-8 and TGFβ induction in DU145 cells and CCL5 in PC3 was not significant at 24 hours. Similarly, no significant suppression of TGFβ in PC3 cells was observed at 6 hours.
Effect of EGCG on CpG-ODN- induced NF-κB activation
TLR9 agonists signal through the adaptor protein MyD88 and activate transcription factor NF-κB to induce target gene expression. Other studies have shown that CpG-ODN activates NF-κB signaling pathways in several cancer cell lines.49,51 To determine the role of NF-κB in gene induction of chemokines and cytokines by CpG-ODN, DU145 or PC3 cells were pretreated with a pharmacological inhibitor of NF-κB and Bay11-7083 (10 μM), and a level of mRNAs as determined by RNA isolation and qRT-PCR (Figure 4). Inhibiting the activity of NF-κB potently reduced mRNA levels induced by CpG-ODN suggesting requirement of NF-κB transcriptional activity. We further evaluated the role of NF-κB more specifically in the induction of IL-8 by CpG-ODN in promoter luciferase assays. The activity of the -135bp promoter containing NF-κB and AP-1 transcription factor binding sites fused to luciferase cDNA was determined in response to CpG-ODN transfection. To determine the contribution of NF-κB or AP-1 to gene induction, promoter constructs with point mutations abolishing the binding of mutant NF-κB (mNF-κB) or mutant AP-1 (mAP-1) were tested in luciferase assays (Figure 5). Loss of the NF-κB binding site reduced induction of IL-8 in both DU145 and PC3 cells, whereas the AP-1 site mutation did not significantly affect IL-8 gene induction by CpG-ODN.
 | Figure 4 Effect of inhibition of NF-κB on CpG-ODN-induced expression of chemokines and cytokines. |
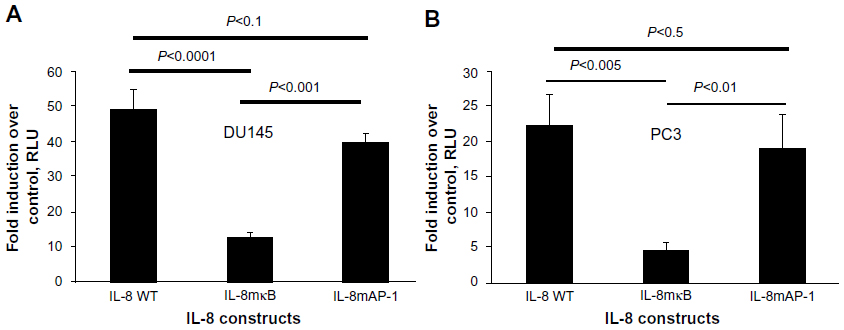 | Figure 5 Induction of the IL-8 promoter by CpG-ODN requires NF-κB transcriptional activity. |
To further determine if CpG-ODN stimulated NF-κB transcriptional activity, we cotransfected DU145 or PC3 cells with promoter constructs containing five copies of NF-κB binding sites fused to luciferase cDNA along with Renilla luciferase vector for normalization. We observed an increase in NF-κB-dependent luciferase activity following transfection with CpG-ODN (Figure 6). Further, pretreatment with EGCG for 24 hours prior to CpG-ODN transfection resulted in a decrease in NF-κB-dependent transcriptional activity; the suppression was greater in DU145 cells compared to PC3 cells.
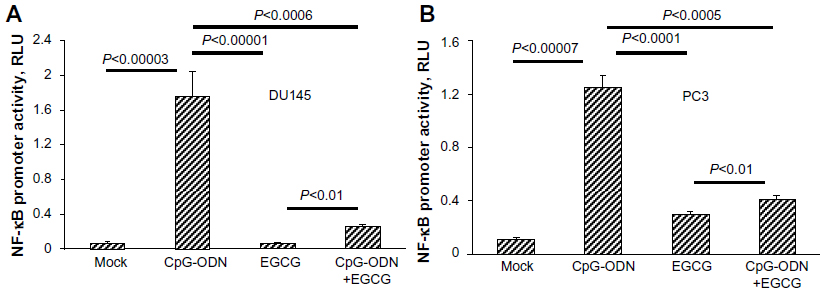 | Figure 6 EGCG inhibits CpG-ODN-induced NF-κB transcriptional activation. |
CpG-ODN-induced nuclear translocation of NF-κB
NF-κB is made of p65 and p50 subunits that are sequestered in the cytoplasm in an inactive form through interaction with an inhibitory subunit, IκBα. Phosphorylation of IκBα by the upstream kinases, IKKs, leads to rapid ubiquitination and degradation of IκBα by the 26S proteasome system, which allows NF-κB to translocate to the nucleus to activate expression of target genes.52–54 Our results along with others demonstrate that EGCG inhibits activation of NF-κB and prevents degradation of IκBα.55 As shown in Figure 7, in mock-treated DU145 cells, NF-κB is predominantly cytoplasmic. Transfection with CpG-ODN for 1 hour resulted in translocation of NF-κB to the nucleus, similar to treatment with TNFα. Pretreatment with EGCG inhibited CpG-ODN-induced translocation of NF-κB, which accumulates in the cytoplasm. We further evaluated the translocation of NF-κB in nuclear and cytosolic extracts of cells treated with CpG-ODN with or without EGCG pretreatment. On immunoblots, we observed less cytosolic NF-κBp65 protein in CpG-ODN-treated cells and more in the nuclear fraction (Figure 8). Cytosolic levels of IκBα are low as would be expected in cells where phosphorylation by IKK would lead to degradation by the proteosomal system (Figure 8). However, increase in cytosolic accumulation of the p65 subunit was observed in cytosolic lysates of cells pretreated with EGCG prior to CpG-ODN transfection. EGCG has been shown to inhibit degradation of IκBα in many cells, including both normal and tumor cells, by inhibiting phosphorylation of IκBα.16 We observed inhibition of IκBα degradation in cells pretreated with EGCG prior to CpG-ODN as well as EGCG alone. The accumulation of the p65 subunit was more significant in EGCG treatment alone. Our results show that EGCG suppresses the induction of cytokines and chemokines induced by CpG-ODN by inhibiting the transcriptional activity of NF-κB.
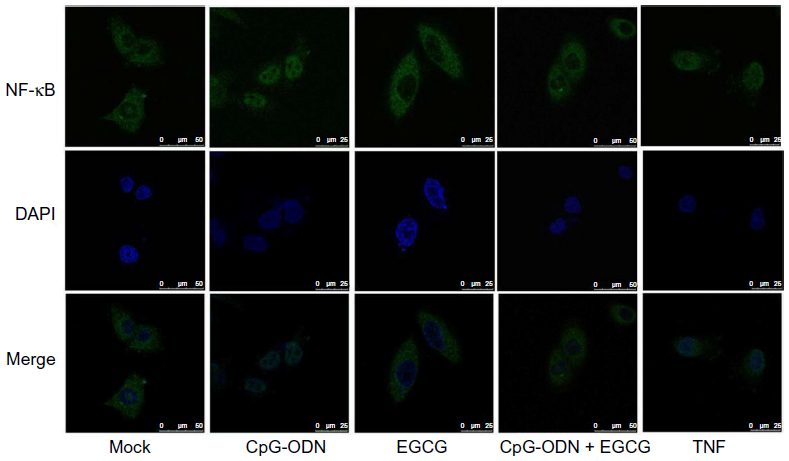 | Figure 7 EGCG inhibits CpG-ODN-induced nuclear translocation of NF-κB. |
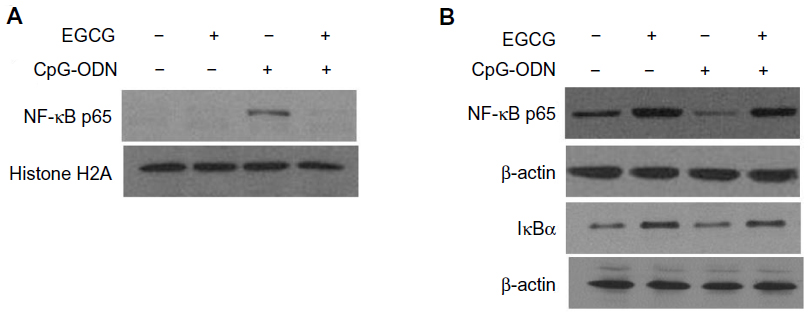 | Figure 8 EGCG sequesters NF-κB p65 subunit in the cytosol. |
CpG-ODN induces MMP activity and cell migration
TLR9 agonists have been shown to promote invasion of prostate cancer cells and MMP-13 activity in breast cancer cells. MMPs degrade the extracellular matrix and promote cell invasion. To test if CpG-ODN regulates MMP activity, in addition to inducing proinflammatory cytokines, culture supernatants of prostate cancer cells transfected with CpG-ODN with or without EGCG pretreatment was assessed by gelatin zymography. Both PC3 and DU145 cells produce significant amounts of MMP-9 and MMP-2, compared to LnCap cells. Treatment with CpG-ODN induced MMP-9 and -2 activity 2.5-fold each in DU145 and LnCap cells and 3.5-fold in PC3 cells (Figure 9). In bladder cancer cells and pancreatic cancer cells, EGCG inhibits the activity of MMP-9 by inhibiting NF-κB activity.56–58 Our results show that pretreatment with EGCG inhibited the induction of MMP-9 and -2 by CpG-ODN, but did not alter the basal level of MMP-9 and -2 activities in all three prostate cancer cell lines.
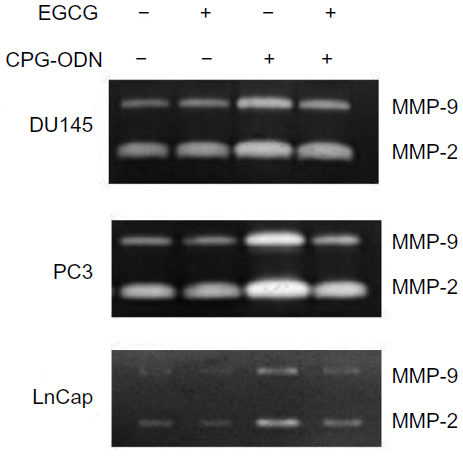 | Figure 9 EGCG inhibits CpG-ODN-induced activity of MMP-9 and -2. |
MMP activity causes remodeling of cell–matrix and cell–cell interactions and promotes cell migration. To determine if the upregulation of MMP-9 and -2 activity by CpG-ODN translates into increased migration, DU145 or PC3 cells were pretreated or not with EGCG followed by CpG-ODN transfection. Substantially higher numbers of cells treated with CpG-ODN migrated across the membrane, which correlates with an increase in MMP-9 and -2 activity (Figure 10). Cells pretreated with EGCG showed significant inhibition of migration and MMP-9 and -2 activity. EGCG suppressed both MMP-9 and -2 activity and migration of cells induced by CpG-ODN.
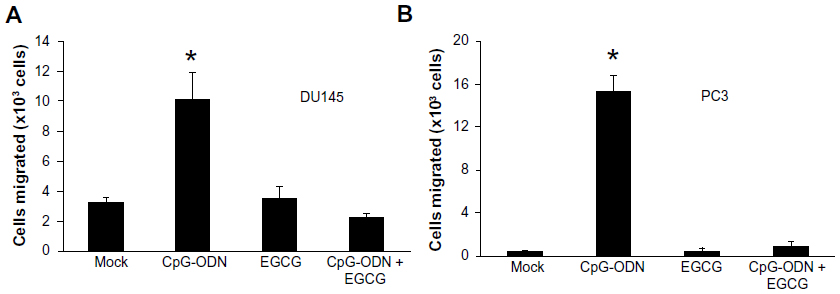 | Figure 10 EGCG inhibits CpG-ODN-induced migration of DU145 and PC3 cells. |
Discussion
Inflammation caused by microbial pathogens, host genetics, and the environment contribute to the development of prostate cancer. Persistent inflammation causes increased proliferation to replace damaged tissue accompanied by elaborating production of cytokines, which alters the prostatic microenvironment and promotes tumorigenesis. Host innate immune response allows recognition of invading pathogens by TLRs to activate inflammatory and antimicrobial responses. The inflammatory response is transmitted by the TLR–MyD88 pathway, leading to activation of NF-κB and AP-1 responsive genes. Bacterial DNA, contributed by infection of the prostate, is a potent immunostimulator and produces inflammatory cytokines by engaging TLR9 and its adaptor, MyD88. Synthetic CpG-ODN mimics bacterial DNA and has been shown to induce TLR9-dependent cytokines in many cell lines. In this study, we have evaluated the role of the TLR9 agonist, CpG-ODN, in inducing proinflammatory cytokines and MMPs in DU145, PC3, and LnCap prostate cancer cells. Further, we determined the role of EGCG, a major constituent of green tea, in the suppression of cytokines and MMPs and its antiproliferative effects in prostate cancer cells. Our results presented here, demonstrate that CpG-ODN induces mRNA expression of IL-6, IL-8, CXCL1, IP-10, CCL5, and TGFβ with variable kinetics in all three prostate cancer cell lines. Induction of the mRNAs involved the activity of NF-κB, since the pharmacological inhibitor Bay11-7082 significantly decreased mRNA levels. Analysis of the induction of the IL-8 promoter in response to CpG-ODN using constructs with mutations in the NF-κB or AP-1 binding site shows the requirement of the NF-kB binding site. Activity of MMP-9 and -2 was induced by CpG-ODN 2.5- to 3.5-fold over the basal levels expressed in these cells, which correlates with increased migration. EGCG significantly suppressed cytokine gene induction, activity of MMP-9 and -2, and cell migration by inhibiting the transcriptional activity and nuclear translocation of NF-κB, as well as inhibiting the degradation of IκBα. Our results suggest that TLR9 agonists induce inflammatory cytokines and MMPs independent of the expression of AR or p53, but involving transcriptional activity of NF-κB.
TLR9 is expressed in all types of immune cells and a variety of cancer cells. Several studies have shown that in addition to protective roles during pathogen infection, TLR9 engagement promotes cell survival and tumor progression by facilitating NF-κB activation. Stimulation of neutrophil granulocytes, B cells, and macrophages induced production of proinflammatory cytokines, such as IL-6, IL-8, TNFα, IL-12, MIP1α, MIP1β, IgM, IgG, and type II IFN, respectively.39,59,60 Expression in RAW264.7 macrophages of CpG-DNA-induced cytokines was significantly decreased in cells with knockdown of TLR9.61 In various cancer cells, CpG-ODN promotes the release of cytokines and chemokines through the TLR9 pathway. For instance, lung cancer cells stimulated with CpG-ODN produced elevated levels of IL-1, IL-8, and MMP-2, which correlated with increased metastasis.45 In murine astrocytes and microglia, CpG-ODN induces expression of adhesion molecules, MMP-9, and inflammatory cytokines involving activation of NF-κB and Jun N-terminal kinase (JNK) pathways.49 In PC3 cells, CpG-ODN can induce IL-8 and TGFβ1 release through the TLR9 pathway and promotes cellular invasion.46,48 Consistent with these studies, our results show that CpG-ODN induces IL-6, IL-8, CXCL1, IP-10, CCL5, and TGFβ genes. IL-8 is produced constitutively by prostate cancer cells and its expression correlates with tumorigenicity and metastasis.62,63 Following CpG-ODN treatment, a 17- to 26-fold increase is observed in induced levels of IL-8, and IL-8 promoter activity is reduced when the NF-κB binding site is mutated. Increased plasma IL-6 levels in metastatic and castration-resistant prostate cancer correlate inversely with tumor survival and chemoresistance.64,65 In all the cell lines, levels of IL-6 increased sharply at 6 hours and decreased by 24 hours. Induction of CXCL1 in prostate cancer cells increases migration and invasion and correlates with high-grade malignancy of tumors.66,67 CpG-ODN induced CXCL1 levels 3- to 4-fold in our experiments. Proinflammatory chemokine IP-10 was induced more significantly in DU145 and PC3 cells compared to LnCap cells. CCL5 and its receptor, CCR5, are expressed in all prostate cancer cells and increase in CCL5 stimulates proliferation and invasion in vitro.68 TGFβ1 regulates cell proliferation, apoptosis, and invasion. In our studies, the levels of TGFβ1 increased at 6 hours in DU145 and LnCap cells, and at 24 hours in PC3 cells. MMPs degrade the extracellular matrix and promote tumor invasion and metastasis. Expression of MMP-9 is regulated by NF-κB, and the MMP-9 promoter has binding sites for NF-κB.69 We observed increased activity of MMP-9 and -2 in the supernatants of DU145, PC3, and LnCap cells following stimulation with CpG-ODN. Accordingly, migration increased 3.5-fold in DU145 and 10-fold in PC3 cells, which is reduced in cells pretreated with EGCG. LnCap cells have very low levels of MMP, which correlates with reduced migration and invasion in vitro. In all, our results show that expression of cytokines, chemokines, and MMPs is induced significantly in prostate cancer cells in response to CpG-ODN. It is conceivable that infection in the prostate may promote the release of cytokines, chemokines, and MMPs with roles in propagating inflammation and promoting invasion that could lead to progression of prostate cancer.
EGCG has anti-inflammatory and antitumorigenic effects in several cell types in cell culture and animal models. These effects are exerted by inducing apoptosis, cell cycle arrest, decreasing the expression of antiapoptotic proteins such as PI3K and AKT, suppressing prosurvival transcription factors such as NF-κB, and inhibiting the activity of proteins that promote invasion. Treatment of prostate cancer cells decreases cell viability with increasing doses of EGCG. Studies in LnCap cells showed that increase in cellular p53 levels accompanied a decrease in viability.27 We did not observe significant differences in cell viability between DU145, PC3, and LnCap cells, which differ in the expression of p53. Since CpG-ODN stimulates cytokines involving NF-κB, and EGCG inhibits NF-κB translocation, we tested to see if EGCG suppresses NF-κB target genes following CpG-ODN stimulation. We show that pretreatment with EGCG prior to CpG-ODN stimulation inhibits the induction of cytokines and MMP-9 and -2 activities. NF-κB is an antiapoptotic protein that also increases cell proliferation and is constitutively expressed in many cancers, including prostate cancer. We demonstrate that EGCG decreases NF-κB transcriptional activity in DU145 and PC3 cells, which can be explained by inhibition of nuclear translocation of the p65 subunit of NF-κB. In addition, IκBα degradation by ubiquitination is inhibited by EGCG. We observed an increase in cytosolic IκBα in our experiments, possibly due to inhibition of degradation. It is also likely that EGCG decreases levels of antiapoptotic proteins such as bcl2 and increases proapoptotic proteins such as BAX to increase apoptosis as shown in LnCap cells.27,70 These effects are predicted to involve p53. In our studies, the induction of proinflammatory cytokines and MMPs are independent of the expression of AR and p53, since DU145 and PC3 cells lack AR and are mutant, or lack p53 compared to LnCap cells, which express AR and p53. EGCG has broad antitumorigenic roles specifically in cancer cells and anti-inflammatory roles independent of p53, which is frequently mutated in cancers. These attributes suggest that EGCG can be used for the chemoprevention of tumors, more specifically, prostate tumors, which are promoted by inflammation.
Conclusion
Like bacterial DNA, TLR9 agonists like CpG-ODN induce proinflammatory cytokines in DU145, PC3, and LnCap prostate cancer cells. The mRNA for cytokines and chemokines are induced in all three cell lines but with variable kinetics. Activity of matrix MMP-9 and -2, which degrade the extracellular matrix to promote cell invasion, increased in cells stimulated with CpG-ODN. The cytokine transcriptional response and MMP activity induced by CpG-ODN was mediated by a transcription factor, NF-κB. EGCG pretreatment suppressed cytokine gene induction and MMP-9 and -2 activities. The inhibitory effect of EGCG was mediated by inhibiting the translocation of the p65 subunit of NF-κB to the nucleus and transcriptional activity. These studies demonstrate that TLR9 agonists induce proinflammatory cytokines and MMPs, and that EGCG suppresses inflammatory cytokines independent of the expression of AR and p53 status of the cells. EGCG may provide protection against inflammation, which contributes to the development of prostate cancer.
Acknowledgments
This work was supported by the National Institutes of Health Grants AI089518 (KM) and startup funds from the University of Toledo. YA was supported by FYSRE, Sullivan Grant, and USRCAP fellowships for undergraduate research from the University of Toledo.
Disclosure
The authors report no conflicts of interest in this work.
References
Dennis LK, Dawson DV. Meta-analysis of measures of sexual activity and prostate cancer. Epidemiology. 2002;13(1):72–79. | |
Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology. 2002;60(1):78–83. | |
Roberts RO, Bergstralh EJ, Bass SE, Lieber MM, Jacobsen SJ. Prostatitis as a risk factor for prostate cancer. Epidemiology. 2004;15(1):93–99. | |
Mishina T, Watanabe H, Araki H, Nakao M. Epidemiological study of prostatic cancer by matched-pair analysis. Prostate. 1985;6(4):423–436. | |
Platz EA, De Marzo AM. Epidemiology of inflammation and prostate cancer. J Urol. 2004;171(2 Pt 2):S36–S40. | |
Platz EA, De Marzo AM, Giovannucci E. Prostate cancer association studies: pitfalls and solutions to cancer misclassification in the PSA era. J Cell Biochem. 2004;91(3):553–571. | |
Patel DA, Bock CH, Schwartz K, Wenzlaff AS, Demers RY, Severson RK. Sexually transmitted diseases and other urogenital conditions as risk factors for prostate cancer: a case – control study in Wayne County, Michigan. Cancer Causes Control. 2005;16(3):263–273. | |
Fernández L, Galán Y, Jiménez R, et al. Sexual behaviour, history of sexually transmitted diseases, and the risk of prostate cancer: a case-control study in Cuba. Int J Epidemiol. 2005;34(1):193–197. | |
Maggio M, Blackford A, Taub D, et al. Circulating inflammatory cytokine expression in men with prostate cancer undergoing androgen deprivation therapy. J Androl. 2006;27(6):725–728. | |
Wise GJ, Marella VK, Talluri G, Shirazian D. Cytokine variations in patients with hormone treated prostate cancer. J Urol. 2000; 164(3 Pt 1):722–725. | |
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420(6917):860–867. | |
De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155(6):1985–1992. | |
Putzi MJ, De Marzo AM. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology. 2000;56(5):828–832. | |
Khan N, Mukhtar H. Modulation of signaling pathways in prostate cancer by green tea polyphenols. Biochem Pharmacol. 2013;85(5):667–672. | |
Katiyar SK, Perez A, Mukhtar H. Green tea polyphenol treatment to human skin prevents formation of ultraviolet light B-induced pyrimidine dimers in DNA. Clin Cancer Res. 2000;6(10):3864–3869. | |
Ahmad N, Gupta S, Mukhtar H. Green tea polyphenol epigallocatechin-3- gallate differentially modulates nuclear factor kappaB in cancer cells versus normal cells. Arch Biochem Biophys. 2000;376(2):338–346. | |
Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst. 1997;89(24):1881–1886. | |
Chen ZP, Schell JB, Ho CT, Chen KY. Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett. 1998;129(2):173–179. | |
Otsuka T, Ogo T, Eto T, Asano Y, Suganuma M, Niho Y. Growth inhibition of leukemic cells by (-)-epigallocatechin gallate, the main constituent of green tea. Life Sci. 1998;63(16):1397–1403. | |
Yang GY, Liao J, Kim K, Yurkow EJ, Yang CS. Inhibition of growth and induction of apoptosis in human cancer cell lines by tea polyphenols. Carcinogenesis. 1998;19(4):611–616. | |
Gupta S, Ahmad N, Nieminen AL, Mukhtar H. Growth inhibition, cell-cycle dysregulation, and induction of apoptosis by green tea constituent (-)-epigallocatechin-3-gallate in androgen-sensitive and androgen-insensitive human prostate carcinoma cells. Toxicol Appl Pharmacol. 2000;164(1):82–90. | |
Gupta S, Hastak K, Ahmad N, Lewin JS, Mukhtar H. Inhibition of prostate carcinogenesis in TRAMP mice by oral infusion of green tea polyphenols. Proc Natl Acad Sci U S A. 2001;98(18):10350–10355. | |
Gupta S, Hussain T, Mukhtar H. Molecular pathway for (-)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch Biochem Biophys. 2003;410(1):177–185. | |
Agarwal R. Cell signaling and regulators of cell cycle as molecular targets for prostate cancer prevention by dietary agents. Biochem Pharmacol. 2000;60(8):1051–1059. | |
Siddiqui IA, Adhami VM, Afaq F, Ahmad N, Mukhtar H. Modulation of phosphatidylinositol-3-kinase/protein kinase B- and mitogen-activated protein kinase-pathways by tea polyphenols in human prostate cancer cells. J Cell Biochem. 2004;91(2):232–242. | |
Adhami VM, Siddiqui IA, Ahmad N, Gupta S, Mukhtar H. Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer Res. 2004;64(23):8715–8722. | |
Hastak K, Gupta S, Ahmad N, Agarwal MK, Agarwal ML, Mukhtar H. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene. 2003;22(31):4851–4859. | |
Shimizu M, Deguchi A, Lim JT, Moriwaki H, Kopelovich L, Weinstein IB. (-)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin Cancer Res. 2005;11(7):2735–2746. | |
Jung YD, Kim MS, Shin BA, et al. EGCG, a major component of green tea, inhibits tumour growth by inhibiting VEGF induction in human colon carcinoma cells. Br J Cancer. 2001;84(6):844–850. | |
Sartippour MR, Shao ZM, Heber D, et al. Green tea inhibits vascular endothelial growth factor (VEGF) induction in human breast cancer cells. J Nutr. 2002;132(8):2307–2311. | |
Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104(3):788–794. | |
Vayalil PK, Katiyar SK. Treatment of epigallocatechin-3-gallate inhibits matrix metalloproteinases-2 and -9 via inhibition of activation of mitogen-activated protein kinases, c-jun and NF-kappaB in human prostate carcinoma DU-145 cells. Prostate. 2004;59(1):33–42. | |
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17(1):1–14. | |
Takeda K, Akira S. Roles of Toll-like receptors in innate immune responses. Genes Cells. 2001;6(9):733–742. | |
Akira S. Mammalian Toll-like receptors. Curr Opin Immunol. 2003; 15(1):5–11. | |
Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. | |
Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. | |
Jakob T, Walker PS, Krieg AM, Udey MC, Vogel JC. Activation of cutaneous dendritic cells by CpG-containing oligodeoxynucleotides: a role for dendritic cells in the augmentation of Th1 responses by immunostimulatory DNA. J Immunol. 1998;161(6):3042–3049. | |
Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A. 1996;93(7):2879–2883. | |
Yi AK, Klinman DM, Martin TL, Matson S, Krieg AM. Rapid immune activation by CpG motifs in bacterial DNA. Systemic induction of IL-6 transcription through an antioxidant-sensitive pathway. J Immunol. 1996;157(12):5394–5402. | |
Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(6522):546–549. | |
Hanagata N. Structure-dependent immunostimulatory effect of CpG oligodeoxynucleotides and their delivery system. Int J Nanomedicine. 2012;7:2181–2195. | |
Jurk M, Vollmer J. Therapeutic applications of synthetic CpG oligodeoxynucleotides as TLR9 agonists for immune modulation. BioDrugs. 2007;21(6):387–401. | |
Schmausser B, Andrulis M, Endrich S, Muller-Hermelink HK, Eck M. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: an implication for interaction with Helicobacter pylori. Int J Med Microbiol. 2005;295(3):179–185. | |
Ren T, Wen ZK, Liu ZM, Liang YJ, Guo ZL, Xu L. Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell: functional active role of TLR9 on tumor metastasis. Cancer Biol Ther. 2007;6(11):1704–1709. | |
Di JM, Pang J, Pu XY, et al. Toll-like receptor 9 agonists promote IL-8 and TGF-beta1 production via activation of nuclear factor kappaB in PC-3 cells. Cancer Genet Cytogenet. 2009;192(2):60–67. | |
Merrell MA, Ilvesaro JM, Lehtonen N, et al. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res. 2006;4(7):437–447. | |
Ilvesaro JM, Merrell MA, Swain TM, et al. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate. 2007;67(7):774–781. | |
Lee S, Hong J, Choi SY, et al. CpG oligodeoxynucleotides induce expression of proinflammatory cytokines and chemokines in astrocytes: the role of c-Jun N-terminal kinase in CpG ODN-mediated NF-kappaB activation. J Neuroimmunol. 2004;153(1–2):50–63. | |
Kundu SD, Lee C, Billips BK, et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate. 2008;68(2):223–229. | |
Di JM, Pang J, Sun QP, et al. Toll-like receptor 9 agonists up-regulates the expression of cyclooxygenase-2 via activation of NF-kappaB in prostate cancer cells. Mol Biol Rep. 2010;37(4):1849–1855. | |
Lin A, Karin M. NF-kappaB in cancer: a marked target. Semin Cancer Biol. 2003;13(2):107–114. | |
Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. | |
Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. | |
Gupta S, Hastak K, Afaq F, Ahmad N, Mukhtar H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene. 2004;23(14):2507–2522. | |
Qin J, Wang Y, Bai Y, et al. Epigallocatechin-3-gallate inhibits bladder cancer cell invasion via suppression of NF-kappaB–mediated matrix metalloproteinase-9 expression. Mol Med Rep. 2012;6(5):1040–1044. | |
Rieger-Christ KM, Hanley R, Lodowsky C, et al. The green tea compound, (-)-epigallocatechin-3-gallate downregulates N-cadherin and suppresses migration of bladder carcinoma cells. J Cell Biochem. 2007;102(2):377–388. | |
Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front Biosci. 2008;13:440–452. | |
Bohle B, Jahn-Schmid B, Maurer D, Kraft D, Ebner C. Oligodeoxynucleotides containing CpG motifs induce IL-12, IL-18 and IFN-gamma production in cells from allergic individuals and inhibit IgE synthesis in vitro. Eur J Immunol. 1999;29(7):2344–2353. | |
Akhtar M, Watson JL, Nazli A, McKay DM. Bacterial DNA evokes epithelial IL-8 production by a MAPK-dependent, NF-kappaB-independent pathway. FASEB J. 2003;17(10):1319–1321. | |
Qiao B, Li B, Yang X, et al. Specific siRNA downregulated TLR9 and altered cytokine expression pattern in macrophage after CpG DNA stimulation. Cell Mol Immunol. 2005;2(2):130–135. | |
Inoue K, Slaton JW, Eve BY, et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res. 2000;6(5):2104–2119. | |
Moore BB, Arenberg DA, Stoy K, et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am J Pathol. 1999;154(5):1503–1512. | |
Codony-Servat J, Marin-Aguilera M, Visa L, et al. Nuclear factor-kappa B and interleukin-6 related docetaxel resistance in castration-resistant prostate cancer. Prostate. 2013;73(5):512–521. | |
Malinowska K, Neuwirt H, Cavarretta IT, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer. 2009;16(1):155–169. | |
Vindrieux D, Escobar P, Lazennec G. Emerging roles of chemokines in prostate cancer. Endocr Relat Cancer. 2009;16(3):663–673. | |
Miyake M, Lawton A, Goodison S, Urquidi V, Rosser CJ. Chemokine (C-X-C motif) ligand 1 (CXCL1) protein expression is increased in high-grade prostate cancer. Pathol Res Pract. 2014;210(2):74–78. | |
Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate. 2006;66(2):124–134. | |
Yang J, Wei D, Liu J. Repressions of MMP-9 expression and NF-kappa B localization are involved in inhibition of lung carcinoma 95-D cell invasion by (-)-epigallocatechin-3-gallate. Biomed Pharmacother. 2005;59(3):98–103. | |
Hastak K, Agarwal MK, Mukhtar H, Agarwal ML. Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J. 2005;19(7):789–791. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.