")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Epigallocatechin-3-gallate attenuates apoptosis and autophagy in concanavalin A-induced hepatitis by inhibiting BNIP3
Authors Li S, Xia Y, Chen K , Li J, Liu T, Wang F, Lu J, Zhou Y, Guo C
Received 29 October 2015
Accepted for publication 11 December 2015
Published 15 February 2016 Volume 2016:10 Pages 631—647
DOI https://doi.org/10.2147/DDDT.S99420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Wei Duan
Sainan Li, Yujing Xia, Kan Chen, Jingjing Li, Tong Liu, Fan Wang, Jie Lu, Yingqun Zhou, Chuanyong Guo
Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Background: Epigallocatechin-3-gallate (EGCG) is the most effective compound in green tea, and possesses a wide range of beneficial effects, including anti-inflammatory, antioxidant, antiobesity, and anticancer effects. In this study, we investigated the protective effects of EGCG in concanavalin A (ConA)-induced hepatitis in mice and explored the possible mechanisms involved in these effects.
Methods: Balb/C mice were injected with ConA (25 mg/kg) to induce acute autoimmune hepatitis, and EGCG (10 or 30 mg/kg) was administered orally twice daily for 10 days before ConA injection. Serum liver enzymes, proinflammatory cytokines, and other marker proteins were determined 2, 8, and 24 hours after the ConA administration.
Results: BNIP3 mediated cell apoptosis and autophagy in ConA-induced hepatitis. EGCG decreased the immunoreaction and pathological damage by reducing inflammatory factors, such as TNF-α, IL-6, IFN-γ, and IL-1β. EGCG also exhibited an antiapoptotic and antiautophagic effect by inhibiting BNIP3 via the IL-6/JAKs/STAT3 pathway.
Conclusion: EGCG attenuated liver injury in ConA-induced hepatitis by downregulating IL-6/JAKs/STAT3/BNIP3-mediated apoptosis and autophagy.
Keywords: concanavalin A, hepatitis, EGCG, autophagy, apoptosis, BNIP3, STAT3, JAKs, IL-6
Introduction
Autoimmune hepatitis (AIH) is a chronic disease closely related to abnormal immune stimulation of liver cells, leading to liver cirrhosis, liver cancer, and even death.1 However, the only available treatments for AIH are immunosuppressants and liver transplantation, which is far from ideal.1–5 Therefore, determination of the pathogenesis of immune-related liver disease and identification of a safe and effective therapy are urgently needed.6–8 Of the animal models currently used, intravenous administration of the plant lectin concanavalin A (ConA) is widely used to induce acute hepatitis in mice and has unique pathogenic features and similarities in immune-mediated hepatitis to those seen in humans, such as in AIH, acute viral hepatitis, and hepatitis caused by drug toxicity.5 ConA-induced liver injury is mainly caused by abnormal immune response against liver cells and secretion of cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, interferon (IFN)-γ, and IL-1β, thus mediating liver damage.9–11 Of these cytokines, IL-6 is particularly significant due to its multiple functions in the immune system and liver injury.12,13
Apoptosis, as a major constituent of programmed cell death, is choreographed by a set of previously dormant proteases, the caspases,14 which can be triggered by both extrinsic (the “death receptors”) and intrinsic (the “mitochondria”) pathways.15–23 The Bcl-2 family, including the BCL-2 homology (BH) 3-only proteins, contributes the procedure.24–26 Bcl-2/E1B-19K interacting protein 3 (BNIP3) is one of the BH-3-only proteins. BNIP3 dissociates Beclin-1 from the Beclin-1/Bcl-2 complex by competing with Beclin-1 for binding with Bcl-2; therefore, when excess BNIP3 is present to bind Bcl-2, more Beclin-1 is free to induce autophagy.24,27–30 Autophagy has both prosurviving and prodeath functions by accumulating autophagosomes and lysosomes, leading to the degradation of mitochondria and endoplasmic reticulum. Recent evidence suggests that autophagy acts against liver protection.31–41 Beclin-1 is an essential autophagy gene, which physically interacts with Bcl-2 and forms the Beclin-1/Bcl-2 complex, and is regulated by the BH-3-only proteins.21,42 Thus, the BH-3-only proteins are involved in the regulation of both apoptosis and autophagy. In in vitro experiments using glioblastoma cells, the genetic or pharmaceutical inhibition of phosphorylated STAT3 abrogates ConA-induced BNIP3 expression, indicating that the increased expression of BNIP3 is regulated by STAT3 phosphorylation.43 Therefore, the IL-6/JAKs/STAT3 signaling pathway may be involved in the stimulation of BNIP3.43–49
Epigallocatechin-3-gallate (EGCG), the most abundant component in green tea, exhibits a wide range of biological and pharmacological properties, such as antioxidant, anticancer, anti-infection, and anti-inflammatory activities.50–56 Several animal models have been established to identify the properties and mechanisms of EGCG in liver damage, including ischemia/reperfusion liver injury, carbon tetrachloride (CCl4)-induced liver injury, and hepatitis B virus-induced liver injury.51–55 Recently, its function in improving the symptoms and pathological conditions in autoimmune diseases has attracted much attention. Previous studies have shown that EGCG exhibits hepatoprotective effects in ConA-induced AIH;57 Liu et al58 found that EGCG could suppress NF-κB-mediated inflammation. However, whether apoptosis and autophagy are involved in these actions and their possible mechanisms have not yet been fully elucidated. Several in vitro and in vivo studies on EGCG have shown that it inhibits the secretion of cytokines such as IL-6 in liver injury and acts as a protective factor in cancer via the JAKs/STAT3 pathway.57–64
In this study, we investigated the effect of BNIP3 on ConA-induced AIH and the possible involvement of the IL-6/JAKs/STAT3 pathway in this effect. We hypothesized that EGCG acts as a protective factor in ConA-induced AIH and identified the underlying mechanism of its functions.
Materials and methods
Reagents
Both EGCG and ConA were purchased from Sigma-Aldrich (St Louis, MO, USA). The alanine aminotransferase (ALT) and aspartate aminotransferase (AST) microplate test kits were purchased from Jiancheng Bioengineering Institute (Jiancheng Biotech, Nanjing, People’s Republic of China). Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α, IL-6, IFN-γ, and IL-1β were purchased from R&D Systems (Minneapolis, MN, USA). The antibodies used in this study, which included TNF-α, IL-6, IFN-γ, IL-1β, Bcl-2, Bax, Caspase-3, Caspase-9, Beclin-1, LC3I/II, LC3II, P62, JAK1, JAK2, STAT3, p-STAT3, and BNIP3, were obtained from Cell Signaling Technology (Danvers, MA, USA). The RNA polymerase chain reaction (PCR) kit was purchased from TaKaRa Biotechnology (Dalian, People’s Republic of China).
Animals
Male Balb/c mice (6–8 weeks old, 23±2 g) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, People’s Republic of China). The mice were housed in an environment maintained at a temperature of 24°C±2°C and 55% humidity under a 12:12-hour light:dark cycle, with free access to a chow diet and water. All animal experiments were performed based on the National Institutes of Health Guidelines and approved by the Animal Care and Use Committee of Shanghai Tongji University.
Hepatocyte isolation
Primary hepatocytes were isolated using a two-step perfusion method.65 Mice were executed and then laparotomized after soaking in 75% ethanol. The hepatic portal vein was perfused with 10 mL of prewarmed D-Hanks’ buffer for 10 minutes, followed by 5 mL of 0.02% type V collagenase solution. Liver tissues were cut into small pieces and placed in collagenase V solution in a shaking water bath for approximately 30 minutes. The cell suspensions were then filtered into a glass tube and centrifuged at 800× g for 5 minutes. Roswell Park Memorial Institute (RPMI)-1640 culture medium was added to the washed primary hepatocytes and then incubated at 37°C with 5% CO2. Isolated hepatocyte viability was determined with Trypan blue exclusion, which exceeded 95%.
Cell culture and cell proliferation analysis
The primary hepatocytes were cultured in RPMI-1640 culture medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Hyclone, Argentina, South America), 100 U/mL penicillin, and 100 g/mL streptomycin (Thermo Fisher Scientific) in a humidified incubator at 37°C with 5% CO2. The cells were plated in 96-well plates and were cultured with the indicated concentrations of EGCG only or treated with EGCG 24 hours before stimulation with ConA at a concentration of 30 μg/mL.32 Approximately 10 μL CCK-8 solution (Peptide Institute Inc., Osaka, Japan) was then added to each well. The plate was maintained in the incubator for 4 hours. The absorbance was measured at 450 nm using a microplate reader.
The primary hepatocytes were divided into four groups:
- Control group: treated with phosphate-buffered saline (PBS) only as vehicle;
- EGCG group: treated with EGCG diluted in PBS at a concentration of 20 μM;
- ConA group: treated with ConA dissolved in PBS solution at a concentration of 30 μg/mL;
- ConA + EGCG group: EGCG administered 24 hours before stimulation with ConA.
Preliminary study
PBS was used to dissolve EGCG. Seventy-two mice were randomly divided into four groups (n=18):
- Control group: no treatment;
- PBS group: PBS by gavage;
- Low EGCG group: 10 mg/kg EGCG by gavage twice daily for 10 days;
- High EGCG group: 30 mg/kg EGCG by gavage twice daily for 10 days.
Six mice from each group were randomly selected and sacrificed. All serum and liver tissue samples were collected and stored at −80°C.
Drug administration
Normal saline was used to dissolve ConA and the resulting solution was injected via the tail vein at a dose of 25 mg/kg to induce acute hepatic injury according to previous research.1,32 The EGCG dose (10 or 30 mg/kg) was orally administered to mice twice daily for 10 days before the induction of hepatitis.57,58 Ninety-six mice were randomly divided into four groups (n=24):
- Normal group: PBS by gavage;
- ConA group: ConA injected via tail vein following PBS by gavage;
- Low EGCG group: ConA injected via the tail vein with 10 mg/kg EGCG by gavage;
- High EGCG group: ConA injected via the tail vein with 30 mg/kg EGCG by gavage.
Eight mice from each group were randomly selected and sacrificed at the time points 2, 8, and 24 hours.
Biochemical analysis
Serum aminotransferase assay
After blood collection, serum was separated by centrifugation at 4,300× g for 10 minutes at room temperature. Serum AST and ALT were measured by an automated chemistry analyzer (Olympus AU1000, Olympus, Tokyo, Japan).
Serum cytokine measurement
ELISA kits were used to assess the serum levels of TNF-α, IL-6, IFN-γ, and IL-1β according to the manufacturer’s instructions.
Histopathology
A portion of liver tissue was incubated in 4% paraformaldehyde for at least 24 hours and then embedded in paraffin. Sections (3 μm thick) were cut for hematoxylin and eosin (H&E) staining, and the degree of inflammation and tissue damage was observed by light microscopy.
Immunohistochemistry
After heating in a baking oven at 60°C for 20 minutes, the prepared paraffin-embedded sections were dewaxed and rehydrated with xylene and various concentrations of alcohol. Antigens recovered in citrate buffer (pH 6.0) were then heated in a 95°C water bath for 20 minutes. Endogenous peroxidase activity was blocked by incubating in 3% hydrogen peroxide for 20 minutes at 37°C. Membranes were ruptured with 0.2% Triton at room temperature for 30 minutes and nonspecific binding sites were blocked with 5% bovine serum albumin at 37°C for 20 minutes, followed by incubation at room temperature for 10 minutes. The liver slices were then incubated overnight with the primary antibodies anti-LC3II (1:500), anti-Beclin-1 (1:500), anti-IL-6 (1:500), anti-JAK1 (1:1,000), anti-JAK2 (1:1,000), anti-p-STAT3 (1:1,000), and anti-BNIP3 (1:500). On the second day, the slices were incubated with secondary antibody (goat anti-rabbit; Epitomics, Burlingame, CA, USA) for 30 minutes at room temperature and were analyzed with a diaminobenzidine kit. The slices were then counterstained with hematoxylin, dehydrated with graded ethanol and xylene, and mounted with Entellan. The slices were then observed by light microscopy. Color development was filmed with a digital camera (Olympus) connected to a microscope (Leica, Wetzlar, Germany). The integrated optical densities (IODs) of the different indicators were calculated using Image-Pro Plus software 6.0 (Media Cybernetics, Silver Spring, MD, USA).
Western blotting analysis
Liver tissues were removed from storage at −80°C and lysed with radioimmunoprecipitation assay lysis buffer and protease inhibitors. Protein concentrations were detected by the bicinchoninic acid protein assay (Kaiji Biology, Nanjing, People’s Republic of China). Samples were then boiled with 5× sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample-loading buffer. The treated samples were separated by SDS-PAGE according to standard protocols and then transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk (dissolved in PBS) for 1 hour and the blots were then incubated overnight at 4°C with the following primary antibodies: β-actin (1:1,000), TNF-α (1:500), IL-6 (1:500), IFN-γ (1:500), IL-1β (1:500), Bcl-2 (1:500), Bax (1:500), Caspase-3 (1:500), Caspase-9 (1:500), Beclin-1 (1:500), LC3I/II (1:500), P62 (1:500), JAK1 (1:1,000), JAK2 (1:1,000), STAT3 (1:1,000), p-STAT3 (1:1,000), and BNIP3 (1:500). β-Actin was used as an internal reference for cytoplasmic proteins. All PVDF membranes were washed with PBST (PBS containing 0.1% Tween), then incubated with a secondary goat anti-mouse or anti-rabbit antibody (1:2,000) for 1 hour at 37°C. Finally, the membranes were washed with PBST three times for 10 minutes each time and the proteins were detected with the Odyssey® two-color infrared laser imaging system (fluorescence detection).
Reverse transcription (RT)-PCR and quantitative real-time (qRT)–PCR
Total RNA was extracted from liver tissues by TRIzol reagent (Thermo Fisher Scientific), transcribed into cDNA using the reverse transcription kit (TaKaRa Biotechnology), and then was detected and analyzed by SYBR Green qRT-PCR using a 7900HT fast real-time PCR system (ABI, Foster City, CA, USA) according to the SYBR Premix EX Taq instructions (TaKaRa Biotechnology). The primers used in the PCR are listed in Table 1.
 | Table 1 Primer sequences used for polymerase chain reactions |
Transmission electron microscopy (TEM)
A portion of liver tissue was placed in 2% glutaraldehyde buffer and the cells were observed by TEM (JEM 1230, JEOL, Tokyo, Japan).
TUNEL assay
Apoptosis of liver tissues was assessed using the TUNEL assay. Paraffin-embedded sections (5 μm) were cut and deparaffinized, followed by digestion with 20 μg/mL proteinase K (Sigma-Aldrich) for 15 minutes at room temperature. The slides were washed four times, then incubated with 2% hydrogen peroxide in PBS for 5 minutes at room temperature, washed twice, and then immersed in Terminal deoxynucleotidyl Transferase-containing buffer for 15 minutes. An antidigoxigenin antibody fragment carried a conjugated reporter enzyme (peroxidase) to the reaction sites, and then localized peroxidase generated an intense signal from the chromogenic substrate diaminobenzidine. The counterstain was methyl green.
Detection of apoptosis by flow cytometry
Primary hepatocytes were cultured in six-well plates. Cells in the PBS, EGCG, ConA, and ConA + EGCG groups were collected after 24 hours. The cells were centrifuged, washed twice with PBS, and mixed in 100 μL of 1× binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). After culturing for 15 minutes at room temperature in Annexin-V/PI (BD Biosciences, San Jose, CA, USA) double-staining liquid, the cells were examined by flow cytometry (Cytomics FC500; Beckman Coulter, Fullerton, CA, USA). The percentage of apoptotic cells was calculated using ModFitLT software (Verity Software House Inc, Topsham, ME, USA).
Statistical analysis
Data are expressed as mean ± standard deviation. ELISA and real-time PCR data were analyzed by one-way analysis of variance. The serum levels of ALT and AST, necrotic or edematous areas on histopathology, and Western blot were analyzed by Student’s t-test. In all comparisons, P<0.05 was considered statistically significant. All statistical analyses were calculated with GraphPad Prism version 6.0 for Windows (GraphPad Software, Inc., San Diego, CA, USA).
Results
EGCG does not affect liver function or the inflammatory response
To test whether the drug solvent affected liver function, we analyzed the levels of liver enzymes and cytokine release in the presence of PBS and EGCG. As shown in Figure 1A, the levels of serum ALT and AST in the four groups were similar, as were the serum levels of TNF-α, IL-6, IFN-γ, and IL-1β (Figure 1B). In addition, H&E staining showed no obvious necrosis (Figure 1C). Therefore, these results illustrated that the drug solvent had no effect on liver function or the inflammatory response. In addition, in the in vitro experiment, treatment with EGCG only showed no obvious difference compared with the PBS group (Figure 2A–D), indicating that EGCG showed no potential hepatocyte toxicity even at the highest dose (30 μM) in our experiment.
 | Figure 1 Effects of PBS and EGCG on the liver function and pathology of healthy mice. |
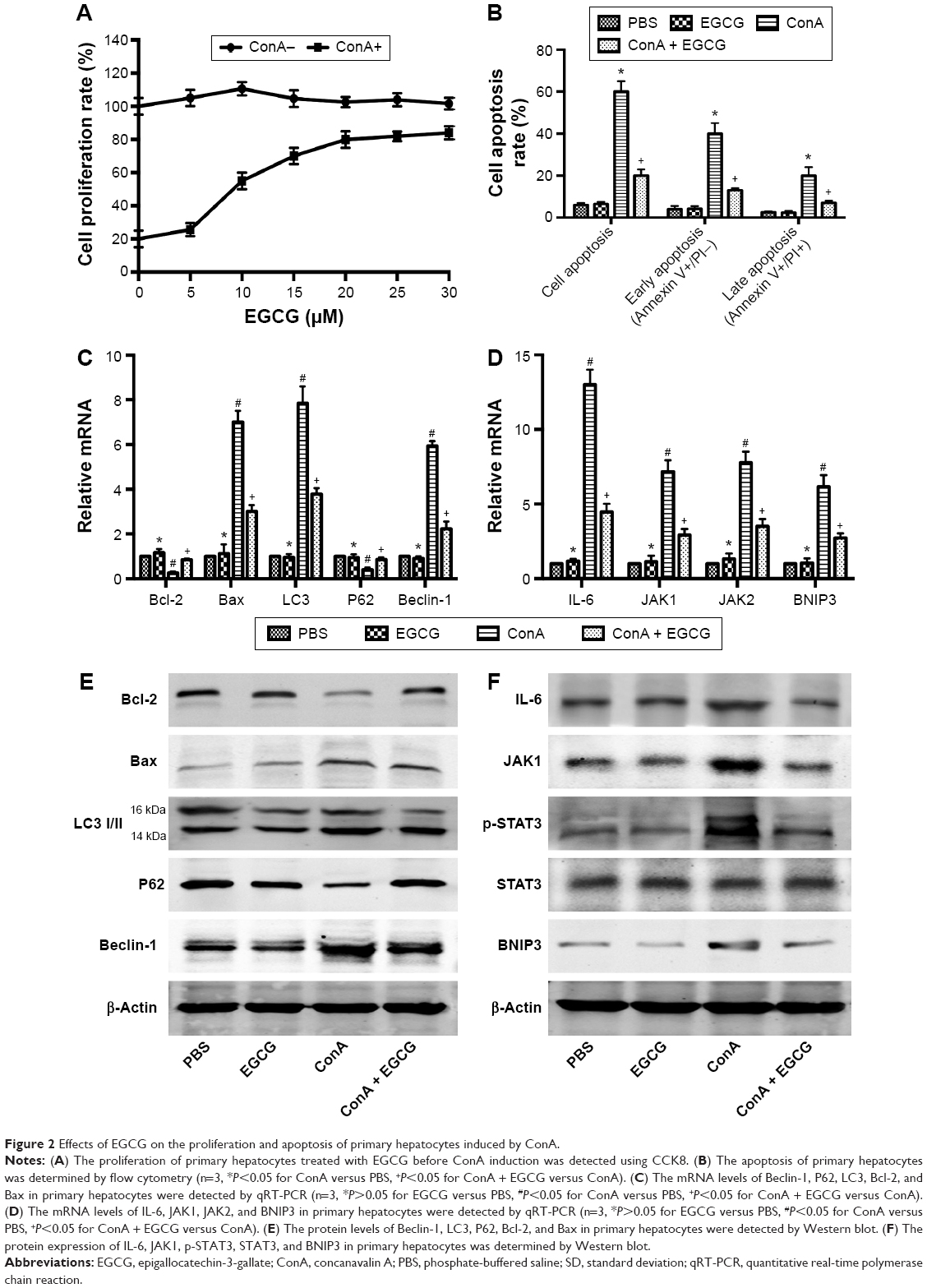 | Figure 2 Effects of EGCG on the proliferation and apoptosis of primary hepatocytes induced by ConA. |
EGCG pretreatment attenuates ConA-induced hepatitis
To determine the effects of EGCG pretreatment on ConA-induced liver injury, the levels of ALT and AST in serum in each group were measured. We found that the levels of ALT and AST clearly increased following ConA injection at all time points, and reached a peak at 8 hours (Figure 3A). However, the elevation in ALT and AST was reversed by EGCG pretreatment. The same results were obtained in the immunohistochemical study (Figure 3B). Massive areas of necrosis were observed in the ConA-treated group. The severity of necrosis increased with time, with the greatest area of nuclear fragmentation seen at 24 hours. In contrast, the EGCG pretreatment groups showed minor liver damage, indicating that EGCG pretreatment significantly reduced liver necrosis. The EGCG 30 mg/kg dose resulted in a greater improvement in liver tissue. These results showed that EGCG pretreatment attenuated ConA-induced hepatitis in mice.
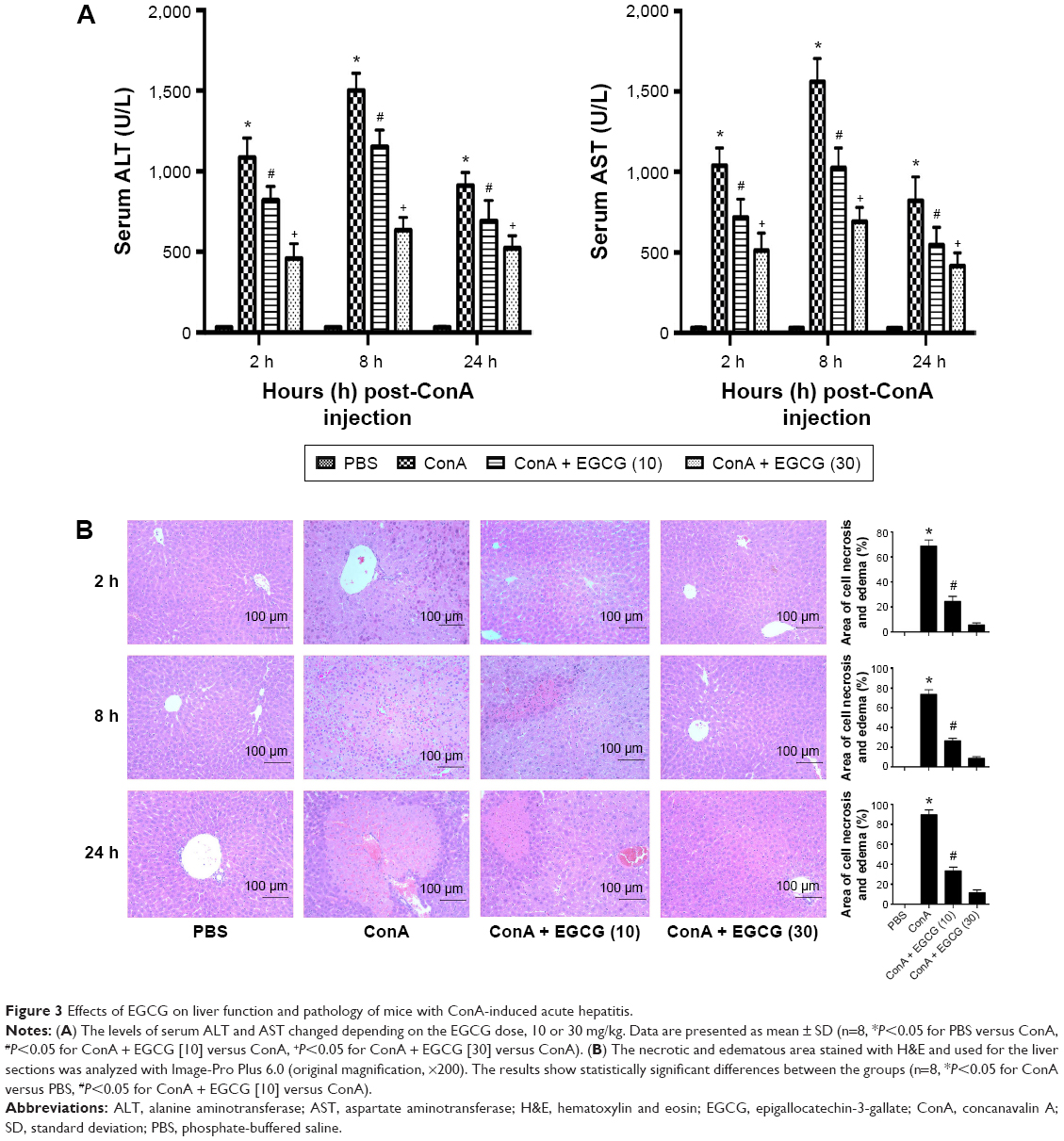 | Figure 3 Effects of EGCG on liver function and pathology of mice with ConA-induced acute hepatitis. |
EGCG pretreatment inhibits the production of TNF-α, IL-6, IFN-γ, and IL-1β in ConA-induced hepatitis
It has been found that the progression of liver injury is associated with a series of proinflammatory cytokines. Therefore, the serum levels of TNF-α, IL-6, IFN-γ, and IL-1β were examined and determined by ELISA (Figure 4A). As expected, the production of TNF-α, IL-6, IFN-γ, and IL-1β was substantially increased after ConA injection compared with the normal group and were prevented by EGCG pretreatment, especially at 8 hours. To confirm these observations, the mRNA expression of TNF-α, IL-6, IFN-γ, and IL-1β was determined by qRT-PCR (Figure 4B). The results showed that mRNA expression of these cytokines was significantly increased in the ConA-treated group, while EGCG pretreatment reduced the expression at all time points. The protein levels of TNF-α, IL-6, IFN-γ, and IL-1β were then assayed by Western blot (Figure 4C). The results showed that the expression of these cytokines decreased in the EGCG pretreatment group compared with the ConA-treated group, reached a peak at 8 hours, and was lower in the high-dose group compared with the low-dose group, similar to the result for mRNA expression. In addition, the expression of IL-6 and TNF-α assessed by immunohistochemistry was significantly increased in the ConA-treated group compared with that in the normal group at 8 hours (Figure 4D). Furthermore, EGCG pretreatment significantly attenuated the expression compared with the ConA-treated group. Thus, these results confirmed that EGCG pretreatment inhibited the production of proinflammatory cytokines, such as TNF-α, IL-6, IFN-γ, and IL-1β in ConA-induced acute hepatitis.
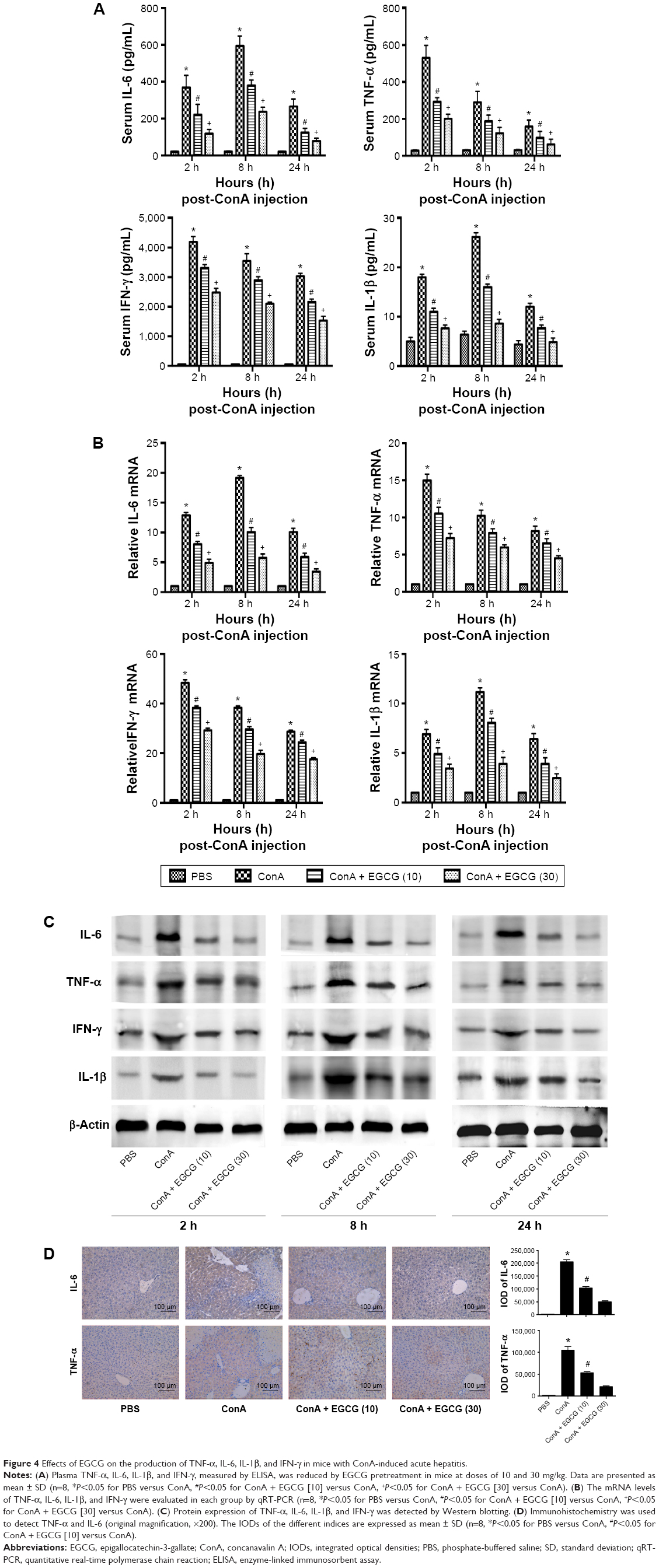 | Figure 4 Effects of EGCG on the production of TNF-α, IL-6, IL-1β, and IFN-γ in mice with ConA-induced acute hepatitis. |
EGCG pretreatment downregulated hepatocyte apoptosis and autophagy in ConA-induced hepatitis
Bcl-2, commonly known as the antiapoptosis protein, together with the proapoptosis proteins Bax, Caspase-3, and Caspase-9 were evaluated by qRT-PCR and Western blot (Figure 5A and B). As expected, with increased drug dose, EGCG pretreatment significantly increased the expression of Bcl-2 and reduced the expression of Bax, Caspase-3, and Caspase-9 compared with the ConA-treated group, with the most obvious change at 8 hours. TUNEL assay also showed clear improvement of apoptosis in EGCG-pretreated compared with ConA-treated mice (Figure 5D). Microtubule-associated protein 1 light chain 3, also known as LC3, is an important marker of autophagy; Beclin-1 plays a vital role in autophagy; and P62 is a ubiquitin binding protein involved in autophagy and can be degraded by the autophagosomes.66,67 Therefore, we determined the expression of LC3 and Beclin-1 in liver tissues using qRT-PCR and Western blot, and both showed a statistically significant increase in the ConA-treated group (P<0.01), while P62 showed the opposite effect, which was reversed by EGCG pretreatment (Figure 5A and B). Analysis of the immunohistochemical changes in mouse livers confirmed these results in both apoptosis and autophagy (Figure 5C). In addition, the formation of autophagosomes is a pivotal process in autophagy. Hence, electron microscopy was used to observe the ultrastructure of hepatic cells (Figure 5E). The ConA-treated group showed an obvious increase in lysosomes, autophagosomes as well as degraded mitochondria and endoplasmic reticulum, while the increase in these structures was prevented by EGCG pretreatment. In addition, all the antiapoptotic and antiautophagic effects of EGCG pretreatment correlated with dosage at all time points (Figure 5A–E). Taken together, these results demonstrate that EGCG pretreatment downregulated hepatocyte apoptosis and autophagy in ConA-induced hepatitis.
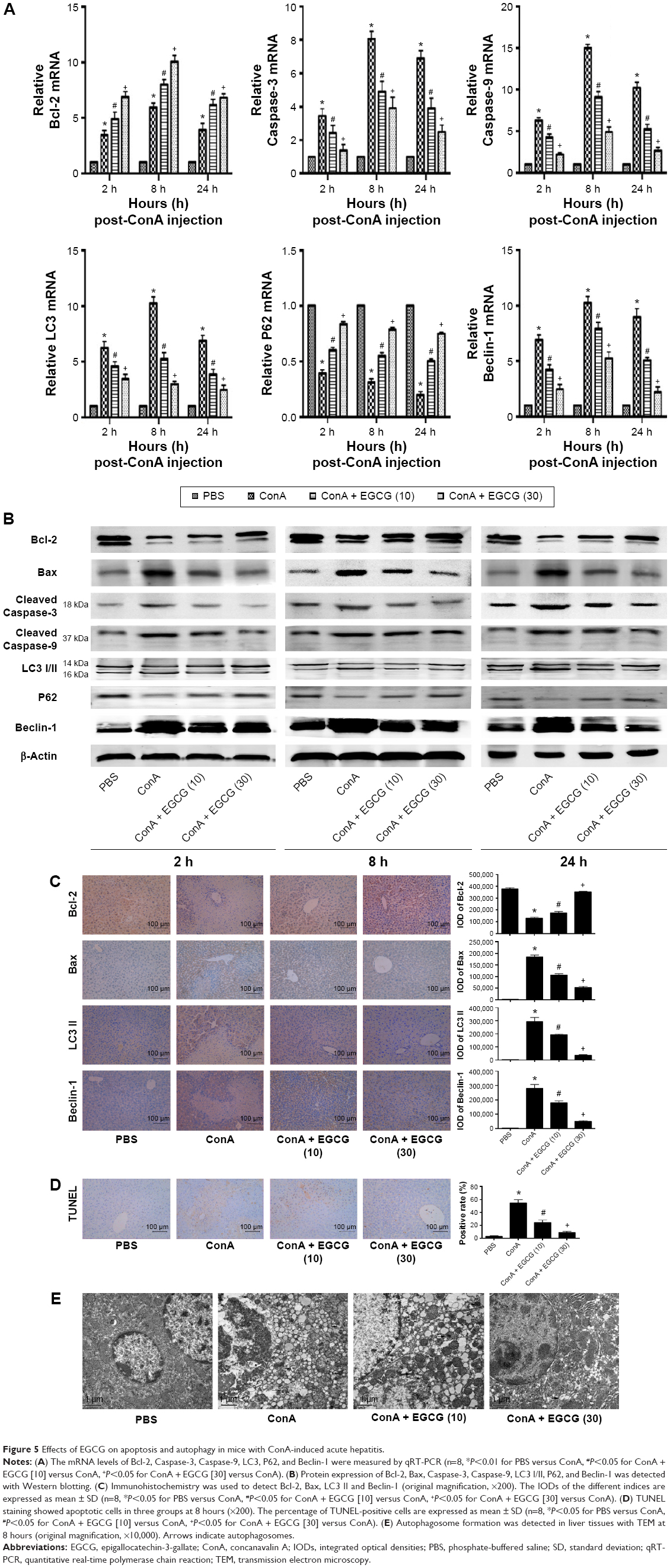 | Figure 5 Effects of EGCG on apoptosis and autophagy in mice with ConA-induced acute hepatitis. |
EGCG pretreatment reduced the expression of BNIP3 by blocking the IL-6/JAKs/STAT3 signal pathway in ConA-induced hepatitis
BNIP3 has been proven to be crucial in apoptosis and autophagy. To verify the possible mechanism of EGCG, we measured the content of BNIP3 in plasma and liver tissue using qRT-PCR and Western blot. As seen in Figure 6A and B, ConA promoted the expression of BNIP3 at both the mRNA and protein levels at all time points, while EGCG dose-dependently attenuated this effect. Immunohistochemical staining also supported the finding that EGCG impacted ConA-induced hepatitis by reducing the expression of BNIP3 (Figure 6C).
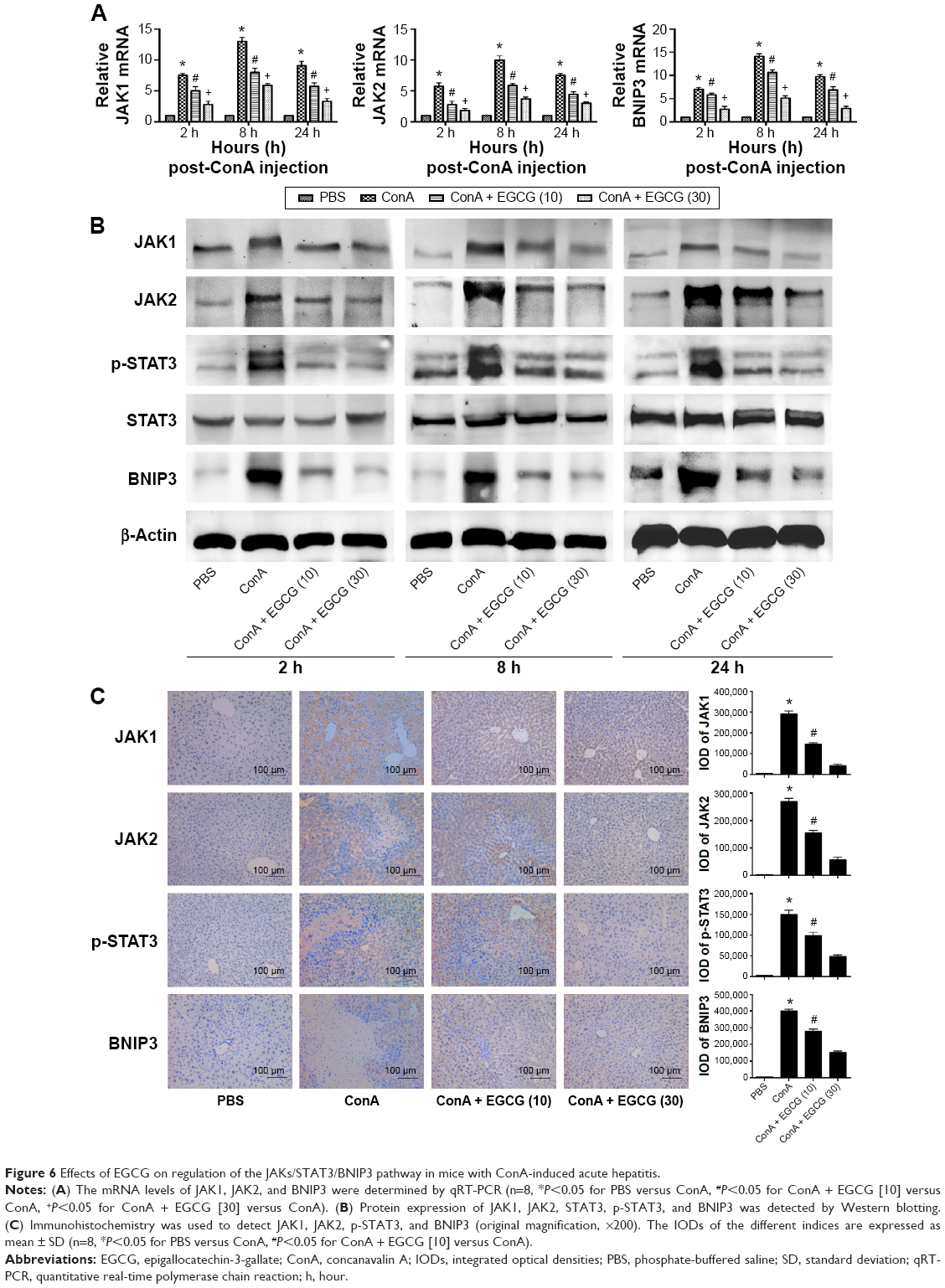 | Figure 6 Effects of EGCG on regulation of the JAKs/STAT3/BNIP3 pathway in mice with ConA-induced acute hepatitis. |
However, there was insufficient evidence to indicate that EGCG directly interacts with BNIP3. Therefore, we attempted to determine the way in which EGCG regulates BNIP3. Previous studies have shown that BNIP3 is regulated by several transcriptional factors; the IL-6/JAKs/STAT3 signal pathway is a significant factor.43–46,68–70 Therefore, the concentrations of IL-6, JAK1, JAK2, and p-STAT3 in plasma and liver tissue were evaluated. As seen in Figure 6B and C, ConA activated the phosphorylation of STAT3, and EGCG weakened this effect at all time points. The expression of JAK1 and JAK2 was consistent with the dose-dependent changes in BNIP3 and p-STAT3, and with IL-6 (Figure 4A–D), while the level of total-STAT3 remained unchanged. These findings indicated that EGCG downregulated the IL-6/JAKs/STAT3 signal pathway, especially the phosphorylation of STAT3. These results showed that EGCG pretreatment reduced the expression of BNIP3 by blocking the IL-6/JAKs/STAT3 signal pathway.
EGCG promoted the proliferation of primary hepatocytes induced by ConA and inhibited apoptosis via the JAKs/STAT3/BNIP3 pathway in vitro
Cell proliferation was analyzed using CCK8. Our results showed that the proliferation rate of primary hepatocytes treated with increasing concentrations (0–30 μM) of EGCG before ConA administration was dose-dependently promoted (Figure 2A). Approximately 20 μM was chosen as an effective dose of EGCG for subsequent experiments. The results of flow cytometry, qRT-PCR, and Western blotting showed that apoptosis was elevated after the administration of ConA, while pretreatment with EGCG attenuated the effect (Figure 2B, C, and E), indicating that EGCG protected the primary hepatocytes from ConA-induced hepatocyte damage. The protein and mRNA expression of IL-6, JAK1, JAK2, p-STAT3, STAT3, and BNIP3 was then detected with qRT-PCR and Western blot. The results showed increased expression of IL-6, JAK1, JAK2, p-STAT3, and BNIP3 in the ConA group, which was reversed with EGCG pretreatment (Figure 2D and F), demonstrating that the protective effect of EGCG in ConA-induced hepatocyte damage was partly through regulation of the IL-6/JAKs/STAT3/BNIP3 pathway.
Discussion
The liver is the largest antitoxic and anti-inflammation organ due to its metabolic functions. The compound EGCG is also well known for its antitoxic and anti-inflammation properties, similar to the liver. Thus, we hypothesized that EGCG may attenuate the pathological changes in liver injury and improve the symptoms. The specific pathogenic mechanisms of AIH, similar to other autoimmune diseases, have not yet been identified and there is no effective cure. Hence, in this study, we used the ConA model of AIH, with or without EGCG pretreatment, to determine the possible pathogenic mechanisms of AIH and to evaluate whether EGCG has a protective effect.
Both ALT and AST are transaminases that are released into the blood after hepatocyte death. As shown in Figure 3, the serum levels of ALT and AST were markedly elevated in the ConA-treated group and reached a peak at 8 hours, indicating increased cell death. Staining with H&E confirmed these results, and more necrotic regions were observed at 8 and 24 hours after ConA injection. The transaminase levels and the necrotic regions were reduced by EGCG pretreatment.
Activation of inflammatory cells is initiated during AIH, exhibiting direct cytotoxicity or releasing proinflammatory cytokines, which mediate liver damage.71,72 A previous study has demonstrated that TNF-α plays an important role in the early stages of inflammation.69 Research has shown that IL-6 is another vital mediator during the development of autoimmune diseases.47 As shown in Figure 4, PCR, Western blot, and immunohistochemistry revealed that the expression levels of TNF-α, IL-6, IFN-γ, and IL-1β in liver tissue increased sharply after ConA injection, especially at 8 hours, which was in accordance with previous studies.72 However, EGCG reversed this effect, in a dose-dependent manner. IL-6 was particularly reduced in the EGCG pretreatment groups, and was considered to be the specific initial mechanism in the protective effect of EGCG in liver damage.
Following the binding of IL-6 to its receptor, the JAK kinases, especially JAK1 and JAK2, contribute to phosphorylation of the IL-6 receptor complex, while STAT3 transiently binds to generate the latter. STAT3 is subsequently phosphorylated by the JAKs and dissociates to dimerize and translocate to the nucleus. Phosphorylated-STAT3, as a transcription factor, increases the expression of several target proteins, such as BNIP3.43,47,48 The increased expression of BNIP3 interacts with more Beclin-1/Bcl-2 complexes by binding to Bcl-2, resulting in free Beclin-1. The latter subsequently induces autophagy, while the former, the BNIP3/Bcl-2 complex, reduces the antiapoptotic effects of Bcl-2.27–30,73 In this study, we determined whether ConA functioned through BNIP3, and if the IL-6/JAKs/STAT3 pathway participated in the process. As seen in Figures 2 and 6, following ConA administration, the RNA and protein levels of JAK1, JAK2, p-STAT3, and BNIP3 increased immediately, while the level of total STAT3 remained unchanged, confirming our hypothesis. Previous studies have shown that EGCG can block the phosphorylation of STAT3 in hepatoma, chronic lymphocytic leukemia (CLL), and autoimmune arthritis.59–64 However, the mechanism of action of EGCG in immune-induced hepatitis remains unclear. In this study, we investigated whether EGCG blocked the JAKs/STAT3/BNIP3 pathway in ConA-induced AIH. As seen in Figures 2 and 6, EGCG pretreatment sharply abolished the elevation of JAK1, JAK2, p-STAT3, and BNIP3 at all time points, in a dose-dependent manner. These findings indicate that the JAKs/STAT3/BNIP3 signal pathway may be the mechanism involved in ConA-induced AIH, and EGCG functions via this pathway.
Following the binding of BNIP3 to Bcl-2 and the presence of excess Beclin-1, the BNIP3/Bcl-2 complex inhibits the antiapoptotic effects of Bcl-2, resulting in the initiation of apoptosis by Caspase-9 and Caspase-3,74 as shown in Figure 7. At the same time, the free Beclin-1 enhanced the induction of autophagy. The cytoplasmic marker, LC3-I, is converted into LC3-II during the formation of autophagosomes.75 The autophagy regulating effect of EGCG is under debate. Zhou et al76 demonstrated that EGCG stimulates autophagy in HepG2 cells and in mice on a high-fat diet,76 while further evidence has proved that EGCG exhibited an antiautophagic effect in Hep3B cells, retinal pigment epithelial cells, skeletal muscle cells, etc.77–82 Interestingly, in our mouse model of AIH with EGCG pretreatment, the change in gene and protein expression levels of Bcl-2, Caspase-9, and Caspase-3, as well as Beclin-1, P62, and LC3-II, suggested that as expected, EGCG had a protective effect in liver injury by inhibiting apoptosis and autophagy.
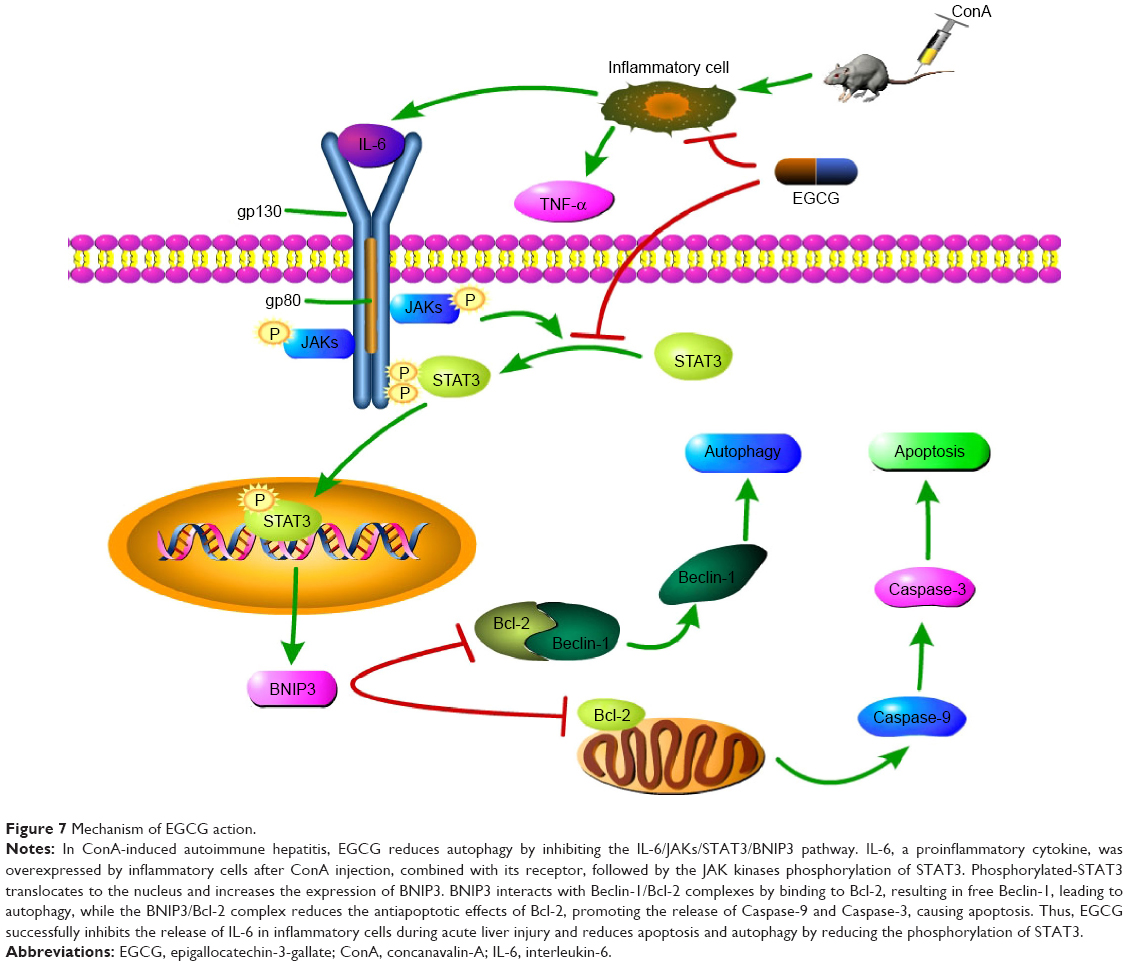 | Figure 7 Mechanism of EGCG action. |
In addition, as shown in Figures 1 and 2, treatment with EGCG alone did not affect normal liver function or hepatocytes even at the highest dose administered. A previous study proved that EGCG administered at 50 mg/kg/d for 16 weeks showed no liver toxicity.83 Thus, EGCG may be an ideal candidate for use as a therapeutic agent in AIH.
However, the mechanisms involved in ConA-induced hepatitis are complex, as is the function of EGCG; therefore, further research is required.
Conclusion
First, our study demonstrated that in ConA-induced AIH, the IL-6/JAKs/STAT3/BNIP3 signal pathway mediated cell apoptosis and autophagy. Second, we confirmed that EGCG suppressed liver injury caused by ConA in two ways: 1) EGCG reduced the immunoreaction and pathological damage by inhibiting inflammatory factors such as TNF-α, IL-6, IFN-γ, and IL-1β; and 2) EGCG downregulated the IL-6/JAKs/STAT3/BNIP3 signal pathway, which increased the antiapoptotic effect of Bcl-2 and blocked the proautophagic effect of Beclin-1, therefore reducing liver damage. Overall, these findings suggest that EGCG may be a promising potential therapeutic agent for AIH.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant numbers 81270515 and 81500466).
Disclosure
The authors report no conflicts of interest in this work.
References
Li J, Xia Y, Liu T, et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS One. 2015;10:e0120440. | ||
Hudspeth K, Pontarini E, Tentorio P, et al. The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun. 2013;46:55–65. | ||
Stein-Streilein J, Sonoda KH, Faunce D, Zhang-Hoover J. Regulation of adaptive immune responses by innate cells expressing NK markers and antigen-transporting macrophages. J Leukoc Biol. 2000;67:488–494. | ||
Tian Z, Gershwin ME, Zhang C. Regulatory NK cells in autoimmune disease. J Autoimmun. 2012;39:206–215. | ||
Wang HX, Liu M, Weng SY, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol. 2012;18:119–125. | ||
Zhang H, Yang J, Zhu R, et al. Combination therapy of ursodeoxycholic acid and budesonide for PBC-AIH overlap syndrome: a meta-analysis. Drug Des Devel Ther. 2015;9:567–574. | ||
Yin Q, Li J, Xia Y, et al. Systematic review and meta-analysis: bezafibrate in patients with primary biliary cirrhosis. Drug Des Devel Ther. 2015;9:5407–5419. | ||
Zhang Y, Li S, He L, et al. Combination therapy of fenofibrate and ursodeoxycholic acid in patients with primary biliary cirrhosis who respond incompletely to UDCA monotherapy: a meta-analysis. Drug Des Devel Ther. 2015;9:2757–2766. | ||
Heymann F, Hamesch K, Weiskirchen R, Tacke F. The concanavalin A model of acute hepatitis in mice. Lab Anim. 2015;49:12–20. | ||
Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. | ||
Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012;3:56. | ||
El Kasmi KC, Anderson AL, Devereaux MW, et al. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology. 2012;55:1518–1528. | ||
Nemeth E, Baird AW, O’Farrelly C. Microanatomy of the liver immune system. Semin Immunopathol. 2009;31:333–343. | ||
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. | ||
Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. | ||
Jacotot E, Costantini P, Laboureau E, et al. Mitochondrial membrane permeabilization during the apoptotic process. Ann N Y Acad Sci. 1999;887:18–30. | ||
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. | ||
Bantel H, Schulze-Osthoff K. Mechanisms of cell death in acute liver failure. Front Physiol. 2012;3:79. | ||
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. | ||
Czaja AJ. Targeting apoptosis in autoimmune hepatitis. Dig Dis Sci. 2014;59:2890–2904. | ||
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. | ||
Wang K. Molecular mechanisms of liver injury: apoptosis or necrosis. Exp Toxicol Pathol. 2014;66:351–356. | ||
Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015;14:1631–1642. | ||
Burton TR, Gibson SB. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009;16:515–523. | ||
Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008;27(Suppl 1):S2–S19. | ||
Vasagiri N, Kutala VK. Structure, function, and epigenetic regulation of BNIP3: a pathophysiological relevance. Mol Biol Rep. 2014;41:7705–7714. | ||
Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–S127. | ||
Delgado M, Tesfaigzi Y. BH3-only proteins, Bmf and Bim, in autophagy. Cell Cycle. 2013;12:3453–3454. | ||
Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. | ||
Ray R, Chen G, Vande Velde C, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. | ||
Cheng P, Wang F, Chen K, et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediators Inflamm. 2014;2014:935251. | ||
Chen K, Li J, Wang J, et al. 15-Deoxy- gamma 12,14-prostaglandin J2 reduces liver impairment in a model of ConA-induced acute hepatic inflammation by activation of PPAR gamma and reduction in NF-kappa B activity. PPAR Res. 2014;2014:215631. | ||
Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013;2013:461536. | ||
Li J, Wang F, Xia Y, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs. 2015;13:3368–3387. | ||
Wang C, Chen K, Xia Y, et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9:e108855. | ||
Shen M, Chen K, Lu J, et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-beta1 expression and autophagy. Mediators Inflamm. 2014;2014:954502. | ||
Zhou Y, Chen K, He L, et al. The protective effect of resveratrol on concanavalin-A-induced acute hepatic injury in mice. Gastroenterol Res Pract. 2015;2015:506390. | ||
Zhou Y, Dai W, Lin C, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013;2013:706156. | ||
Cheng P, Chen K, Xia Y, et al. Hydrogen sulfide, a potential novel drug, attenuates concanavalin A-induced hepatitis. Drug Des Devel Ther. 2014;8:1277–1286. | ||
Wang C, Xia Y, Zheng Y. Protective effects of N-acetylcysteine in concanavalin A-induced hepatitis in mice. Mediators Inflamm. 2015;2015:189785. | ||
Mao Y, Wang J, Yu F, et al. Ghrelin reduces liver impairment in a model of concanavalin A-induced acute hepatitis in mice. Drug Des Devel Ther. 2015;9:5385–5396. | ||
Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. | ||
Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014;26:917–924. | ||
Pietrocola F, Izzo V, Niso-Santano M, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310–322. | ||
You L, Wang Z, Li H, et al. The role of STAT3 in autophagy. Autophagy. 2015;11:729–739. | ||
Yurkova N, Shaw J, Blackie K, et al. The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circ Res. 2008;102:472–479. | ||
Horn F, Henze C, Heidrich K. Interleukin-6 signal transduction and lymphocyte function. Immunobiology. 2000;202:151–167. | ||
Lutticken C, Wegenka UM, Yuan J, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. | ||
Wang SW, Sun YM. The IL-6/JAK/STAT3 pathway: potential therapeutic strategies in treating colorectal cancer (Review). Int J Oncol. 2014;44:1032–1040. | ||
Byun JK, Yoon BY, Jhun JY, et al. Epigallocatechin-3-gallate ameliorates both obesity and autoinflammatory arthritis aggravated by obesity by altering the balance among CD4+ T-cell subsets. Immunol Lett. 2014;157:51–59. | ||
Fiorini RN, Donovan JL, Rodwell D, et al. Short-term administration of (−)-epigallocatechin gallate reduces hepatic steatosis and protects against warm hepatic ischemia/reperfusion injury in steatotic mice. Liver Transpl. 2005;11:298–308. | ||
Jamal MH, Ali H, Dashti A, et al. Effect of epigallocatechin gallate on uncoupling protein 2 in acute liver injury. Int J Clin Exp Pathol. 2015;8:649–654. | ||
Pang JY, Zhao KJ, Wang JB, Ma ZJ, Xiao XH. Green tea polyphenol, epigallocatechin-3-gallate, possesses the antiviral activity necessary to fight against the hepatitis B virus replication in vitro. J Zhejiang Univ Sci B. 2014;15:533–539. | ||
Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82:1807–1821. | ||
Tipoe GL, Leung TM, Liong EC, et al. Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology. 2010;273:45–52. | ||
Wu D, Wang J, Pae M, Meydani SN. Green tea EGCG, T cells, and T cell-mediated autoimmune diseases. Mol Aspects Med. 2012;33:107–118. | ||
Wang Y, Mei Y, Feng D, Xu L. (−)-Epigallocatechin-3-gallate protects mice from concanavalin A-induced hepatitis through suppressing immune-mediated liver injury. Clin Exp Immunol. 2006;145:485–492. | ||
Liu D, Zhang X, Jiang L, Guo Y, Zheng C. Epigallocatechin-3-gallate (EGCG) attenuates concanavalin A-induced hepatic injury in mice. Acta Histochem. 2014;116:654–662. | ||
Lee YK, Bone ND, Strege AK, et al. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104:788–794. | ||
Lee YK, Shanafelt TD, Bone ND, et al. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–523. | ||
Shanafelt TD, Call TG, Zent CS, et al. Phase 2 trial of daily, oral Polyphenon E in patients with asymptomatic, Rai stage 0 to II chronic lymphocytic leukemia. Cancer. 2013;119:363–370. | ||
Shanafelt TD, Call TG, Zent CS, et al. Phase I trial of daily oral Polyphenon E in patients with asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J Clin Oncol. 2009;27:3808–3814. | ||
Wang Y, Ren X, Deng C, et al. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol Rep. 2013;30:2691–2696. | ||
Yang EJ, Lee J, Lee SY, et al. EGCG attenuates autoimmune arthritis by inhibition of STAT3 and HIF-1alpha with Th17/Treg control. PLoS One. 2014;9:e86062. | ||
Tao YY, Yan XC, Zhou T, et al. Fuzheng Huayu recipe alleviates hepatic fibrosis via inhibiting TNF-alpha induced hepatocyte apoptosis. BMC Complement Altern Med. 2014;14:449. | ||
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. | ||
Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. | ||
Guo K, Searfoss G, Krolikowski D, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. | ||
Dorn GW 2nd, Diwan A. The rationale for cardiomyocyte resuscitation in myocardial salvage. J Mol Med (Berl). 2008;86:1085–1095. | ||
Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. | ||
McFarlane IG. Pathogenesis of autoimmune hepatitis. Biomed Pharmacother. 1999;53:255–263. | ||
Sass G, Heinlein S, Agli A, et al. Cytokine expression in three mouse models of experimental hepatitis. Cytokine. 2002;19:115–120. | ||
Kammouni W, Wong K, Ma G, et al. Regulation of apoptosis in fibroblast-like synoviocytes by the hypoxia-induced Bcl-2 family member Bcl-2/adenovirus E1B 19-kd protein-interacting protein 3. Arthritis Rheum. 2007;56:2854–2863. | ||
Renton JP, Xu N, Clark JJ, Hansen MR. Interaction of neurotrophin signaling with Bcl-2 localized to the mitochondria and endoplasmic reticulum on spiral ganglion neuron survival and neurite growth. J Neurosci Res. 2010;88:2239–2251. | ||
Cui J, Sim TH, Gong Z, Shen HM. Generation of transgenic zebrafish with liver-specific expression of EGFP-Lc3: a new in vivo model for investigation of liver autophagy. Biochem Biophys Res Commun. 2012;422:268–273. | ||
Zhou J, Farah BL, Sinha RA, et al. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS One. 2014;9:e87161. | ||
Yamagata K, Xie Y, Suzuki S, Tagami M. Epigallocatechin-3-gallate inhibits VCAM-1 expression and apoptosis induction associated with LC3 expressions in TNFalpha-stimulated human endothelial cells. Phytomedicine. 2015;22:431–437. | ||
Liu J, Tang Y, Feng Z, et al. Acetylated FoxO1 mediates high-glucose induced autophagy in H9c2 cardiomyoblasts: regulation by a polyphenol-(−)-epigallocatechin-3-gallate. Metabolism. 2014;63:1314–1323. | ||
Liu J, Tang Y, Feng Z, et al. (-)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic Res. 2014;48:898–906. | ||
Chen L, Ye HL, Zhang G, et al. Autophagy inhibition contributes to the synergistic interaction between EGCG and doxorubicin to kill the hepatoma Hep3B cells. PLoS One. 2014;9:e85771. | ||
Li CP, Yao J, Tao ZF, et al. Epigallocatechin-gallate (EGCG) regulates autophagy in human retinal pigment epithelial cells: a potential role for reducing UVB light-induced retinal damage. Biochem Biophys Res Commun. 2013;438:739–745. | ||
Hashimoto K, Sakagami H. Induction of apoptosis by epigallocatechin gallate and autophagy inhibitors in a mouse macrophage-like cell line. Anticancer Res. 2008;28:1713–1718. | ||
Santamarina AB, Carvalho-Silva M, Gomes LM, et al. Decaffeinated green tea extract rich in epigallocatechin-3-gallate prevents fatty liver disease by increased activities of mitochondrial respiratory chain complexes in diet-induced obesity mice. J Nutr Biochem. 2015;26:1348–1356. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.