")
Back to Journals » OncoTargets and Therapy » Volume 13
Entire ABL1 Gene Deletion Without BCR/ABL1 Rearrangement in a Female Patient with B-Cell Precursor Acute Lymphoblastic Leukemia
Authors Jiang Y, Zhang J, Guo D, Zhang C , Hong L , Huang H , Liu H
Received 12 November 2019
Accepted for publication 9 January 2020
Published 24 January 2020 Volume 2020:13 Pages 783—790
DOI https://doi.org/10.2147/OTT.S238336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Yijing Jiang,1,* Jie Zhang,2,* Dan Guo,1 Chenlu Zhang,1 Lemin Hong,1 Hongming Huang,1 Haiyan Liu1
1Department of Hematology, The Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu, People’s Republic of China; 2Department of Oncology, Affiliated Tumor Hospital of Nantong University, Nantong 226361, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hongming Huang; Haiyan Liu
Department of Hematology, The Affiliated Hospital of Nantong University, 20 Xisi Road, Nantong 226001, Jiangsu, People’s Republic of China
Email [email protected]; [email protected]
Abstract: Acute lymphoblastic leukemia (ALL) is a malignant disease characterized by lymphocytic B-line or T-line cells abnormally proliferating in the bone marrow or extramedullary sites. BCR/ABL1 fusion protein in patients with ALL accounts for acts in 15– 30% of B-lineage ALL cases, usually in adolescence. However, entire ABL1 gene deletion without BCR/ABL1 rearrangement is a rare phenomenon in ALL patients. Here we describe the first case of entire ABL1 gene deletion without BCR/ABL1 rearrangement in a female B-ALL patient. Relevant literature is reviewed to explain the association between ABL1 deletion and the pathogenesis/prognosis of this disease. ABL gene deletion can repress the activation of p53 and p73, and disrupt TGF-β signaling pathway to allow malignant cells to invade the normal tissue. The clinical significance of ABL gene deletion needs to be further explored.
Keywords: acute lymphoblastic leukemia, ALL, ABL1 deletion, pathogenesis, prognosis
Introduction
Acute lymphoblastic leukemia (ALL) is a malignant disease in which lymphocytic B-line or T-line cells proliferate abnormally in the bone marrow and extramedullary sites. The diagnosis of ALL mainly relies on cell morphology, immunology, cytogenetics and molecular biology (MICM). According to the antigens on the surface of leukemic cell, ALL can be diagnosed and divided into different subtypes. The markers of acute B-lymphoblastic leukemia (B-ALL) include CD19, cytoplasmic CD79a, cytoplasmic CD22, CD10, surface CD22, CD24, PAX5, and TdT.1 Philadelphia (Ph) chromosome is involved in 90–95% of CML patients. BCR/ABL fusion protein is produced by t(9;22) fused by BCR 22q11.2 and ABL1 of 9q34.2,3 In the world, this is the first reported B-ALL female with entire ABL1 gene deletion but not BCR/ABL1 fusion.
Case Report
A 51-year-old woman was transferred to our hospital from a local clinic. Bone marrow biopsy had confirmed the presence of B-cell precursor lymphoblastic leukemia in the local hospital. At our hospital, bone marrow cytology showed that lymphoblasts and prolymphocytes accounted for 87% of bone marrow cells (Figure 1). Flow cytometry revealed that blasts were positive for CD34, TdT, CD19, CD10. Few cells showed weakly positive and scattered expression of cytoplasmic CD79a, and MPO, Lyso, CD3 and CD138, but no expression of cytoplasmic CD56. Initial complete blood counts were as follows: a hemoglobin level of 56 g/L, a platelet count of 30*109/L and a white blood cell count of 1.5*109/L, 10.3% segmented neutrophils, 85.6% lymphocytes, 2.7% monocytes. PET/CT revealed that multiple lymphadenopathy, hepatosplenomegaly, and extensive bone marrow infiltration with high FDG. The 24 hrs unstimulated culture of bone marrow aspirates showed that all the metaphase cells, though not massive, were normal karyotypes (Figure 2). Fluorescence in situ hybridization (FISH) signals using Ph-like B-ALL probes, including IGH/CRLF2, PDGFRB, JAK2, BCR/ABL1, ABL1 and EPOR revealed that of the 100 interphase cells analyzed, 36 (36%) showed ABL1 gene deletion but no rearrangement, and the rest showed no abnormality. The FISH analysis using BCR/ABL1 and ABL1 probes confirmed the presence of nuc ish (BCR×2, ABL1×1)[35/100], (ABL1×1)[36/100] (Figure 3). The signals indicating BCR-ABL fusion did not appear.
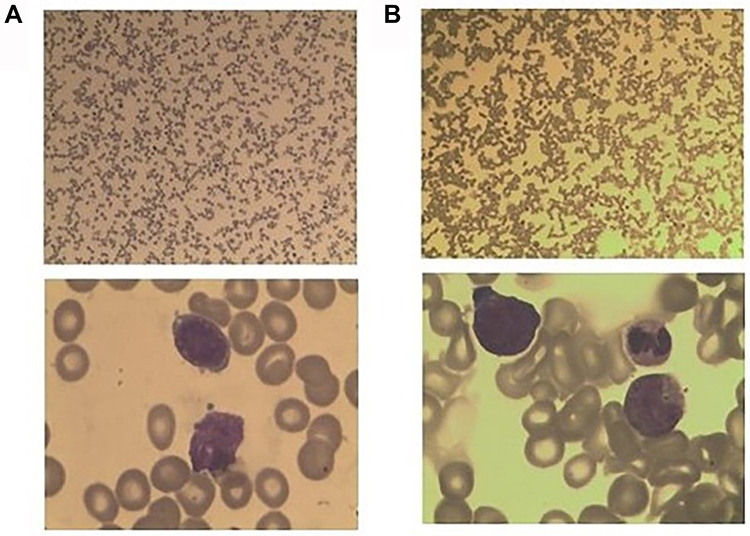 |
Figure 1 (A) The new diagnosis of bone marrow smears showed that lymphoblast and prolymphocyte accounted for 87%. (B) The Myelogram showed no blast cell after the systemic chemotherapy. |
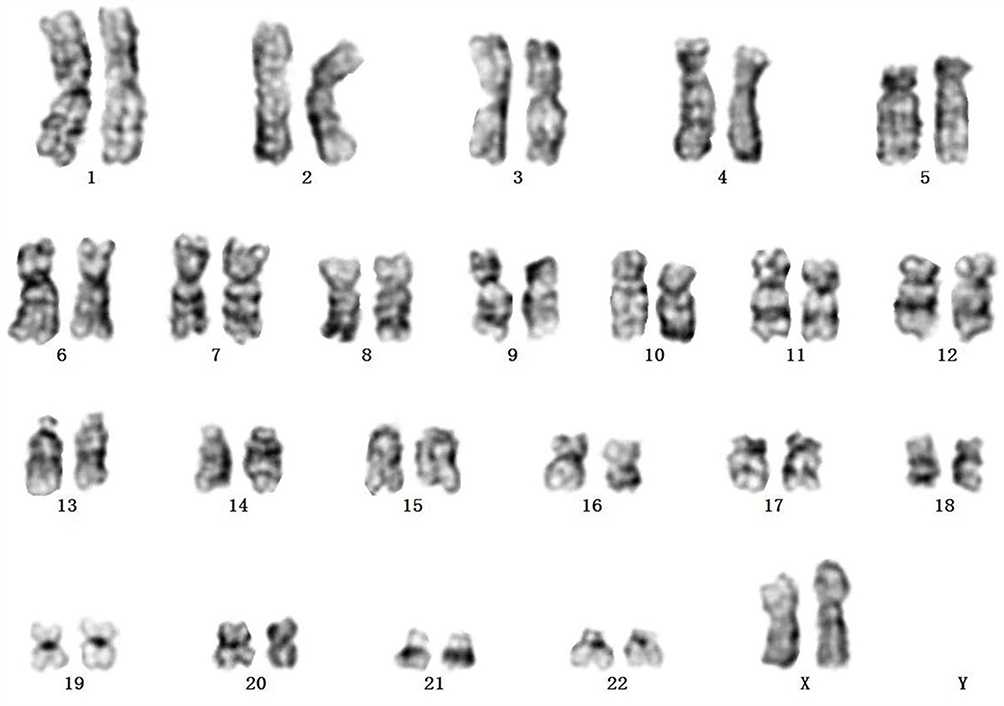 |
Figure 2 Chromosome study showing a normal female karyotype, 46,XX. |
 |
Figure 3 (A) FISH using BCR-ABL dual-color, dual-fusion translocation probe at diagnosis. Two green signals (BCR gene) are present, but only one red signal (ABL gene) is found. (B) FISH using ABL1 probe at diagnosis. Green signal represents 5ʹABL1 and red signal represents 3ʹABL1. |
Before beginning therapy, the second round of FISH analysis using BCR/ABL1 probe identified ABL1 deletion in up to 80% of the cells. Then, we proceeded copy number variations (CNV) analysis based on next-generation sequencing (NGS), belong to CNV-Seq, which covered the targeted region of 118 sequenced genes with a total length of 215kb. CNV analysis found the reduction in the copies of ABL1 gene on the long arm of chromosome 9, EVT6 gene on the short arm of chromosome 12 and DLEU2 gene on the long arm of chromosome 13; the increase in the copies of RET, PTEN, NT5C2 and SMC3 on the long arm of chromosome 10 (Figure 4). The further analysis of CNV according to the sequencing depth of NGS indicated about 90% deletion of ABL1 gene and combined with FISH considerations, ABL1 deletion is loss of heterozygosity. It is worth mentioning that the deletions of exons are 2–10 (corresponding transcript: NM_005157), that is, all exons of ABL1 involved in this panel.
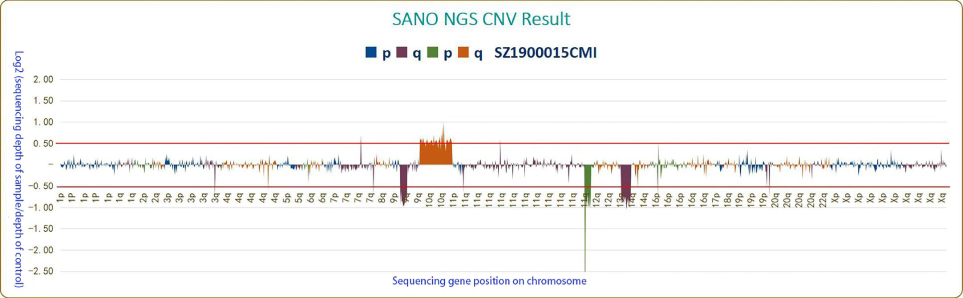 |
Figure 4 Copy number variations (CNV) analysis based on next-generation sequencing (NGS). Log2>0.5: gene copy number significantly increased; Log2<-0.5: gene copy number significantly deleted. The reduction in the copies of ABL1 gene on the long arm of chromosome 9, EVT6 gene on the short arm of chromosome 12 and DLEU2 gene on the long arm of chromosome 13; the increase in the copies of RET, PTEN, NT5C2 and SMC3 on the long arm of chromosome 10. |
Subsequently, the patient received one cycle of IVCLP chemotherapy (Idarubicin, Vindesine, Cyclophosphamide, Pegaspargase and Dexamethasone), achieving obvious clinical remission and negative results in bone marrow biopsy (Figure 1). After six regimens of chemotherapy, the patient has been in continuous remission till now.
Discussion
Unlike ABL1 deletion on derivative chromosome 9 generated by t(9;22),4–7 entire ABL1 gene deletion without BCR/ABL1 rearrangement is still a rare phenomenon. Up to now, only five cases have been reported, all in males aged 7–37 years (Table 1).8–11 The five previously reported patients did not perform CNV based on NGS; however, it is not difficult to speculate that their ABL1 deletion should also be loss of heterozygosity. Of note, this is the first reported female case. Whether males are more prone to entire ABL1 deletion needs further verifications. Although the effect of ABL1 deletion on leukemogenesis is still unclear, we speculate that ALL may occur with the mutation of ABL1 gene and the probable pathogenesis can be analyzed as follows according to the previous literature.
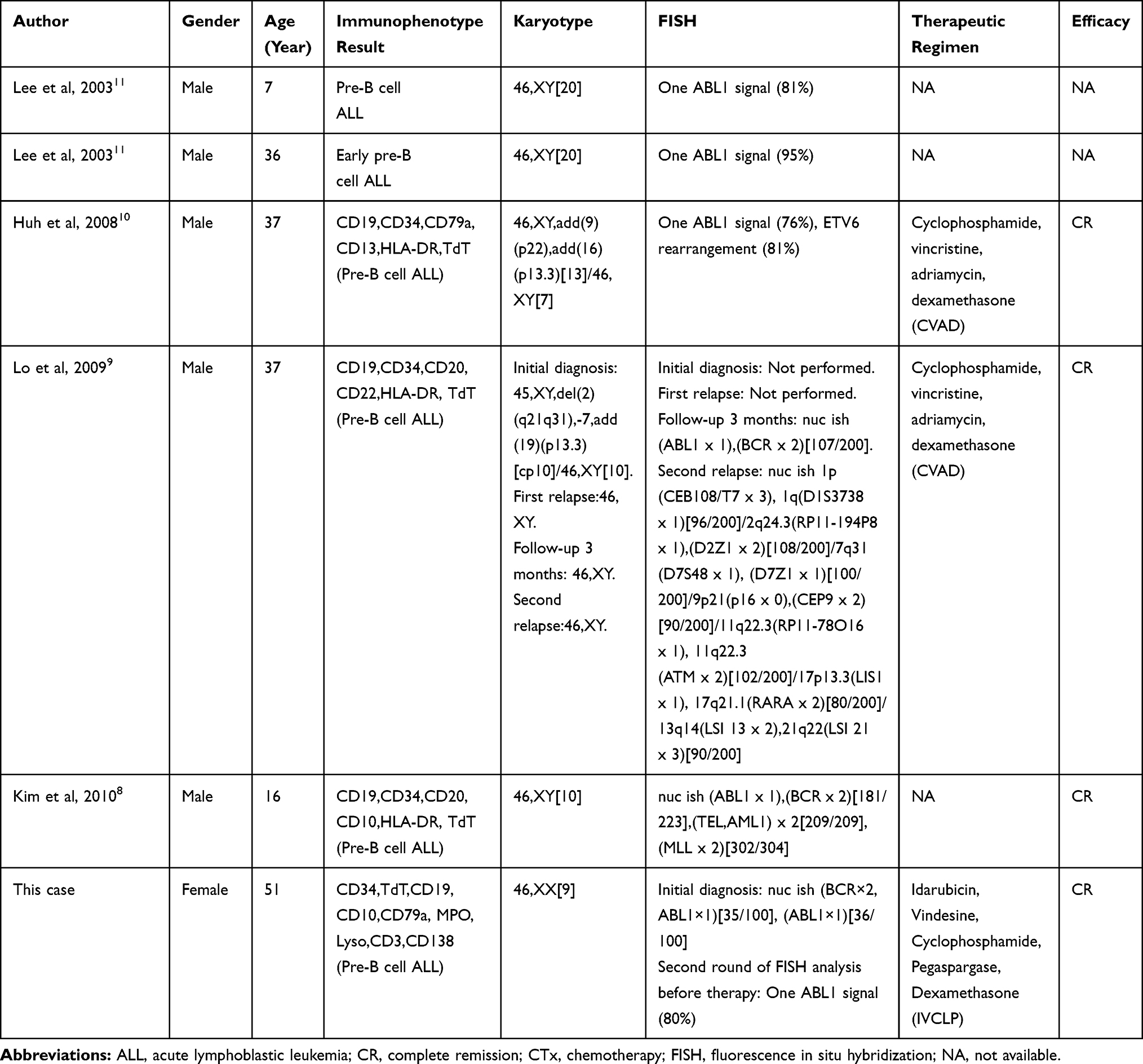 |
Table 1 Reported Cases of ABL1 Deletion Without BCR/ABL1 Rearrangement in Precursor B-Cell Acute Lymphoblastic Leukemia |
c-Abl, one member of ABL family, can be provoked through a variety of cellar signals in response to stimuli.12,13 In normal physiological conditions, c-Abl is retained in the cytoplasm to interact with other protein. Upon DNA-mediated breaks, c-Abl is activated in the cell nucleus, a process that requires the ATM checkpoint kinase to be phosphorylated at c-Abl Ser465.14,15 Activated c-Abl protects p53 from proteasomal degradation through interactions with HDM2.16 HDM2, human Mdm2, is a primary negative regulator of p53 through its E3 ligase activity.17,18 Actually, c-Abl counteracts the degradation of p53 by HDM2 and inhibit the transcription and apoptosis of p53.19 In this mechanism, the phosphorylation of c-Abl is indispensable. c-Abl interacts with HDM2 to phosphorylate HDM2 at Tyr394. In turn, the phosphorylation of HDM2 Tyr394 disrupts the ability of HDM2, leading to stabilization of p53 and p53-mediated tumor apoptosis.20,21 These findings revealed that the phosphorylation of HDM2 by c-Abl activated kinase alters the signal transduction of Hdm2-p53, promoting the stabilization and activation of P53 (Figure 4). In addition, RING finger proteins HDMX originally isolated as a protein interacting with the tumor suppressor p53, shares significant structural and functional similarity with HDM2.22 Although HDMX also binds to p53, HDMX does not induce p53 degradation directly, due to the lack of E3 ubiquitin ligase activity on p53. c-Abl phosphorylation of HDM2 increases the formation of heterodimer HDM2-HDMX proximal to the RING domain of HDM2, and creates a junction between HDMX and HDM2 N-termini. This junction switches HDM2-mediated ubiquitination away from p53 to heterodimer formation, ultimately promoting p53 activation23–25 (Figure 5). Therefore, ABL1 deletion in B-ALL may repress the activity and stability of tumor suppressor gene.
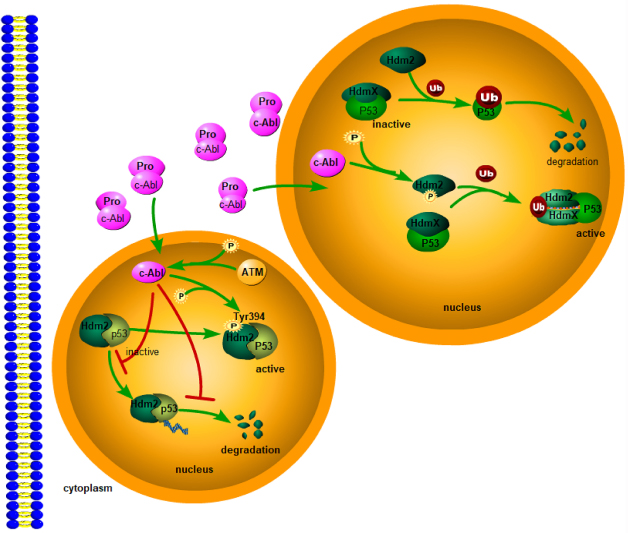 |
Figure 5 On one hand, the phosphorylation of HDM2 by c-Abl activated kinase alters the signal transduction of Hdm2-p53, promoting the stabilization and activation of P53. On the other hand, c-Abl phosphorylation of Hdm2 increases Hdm2-HdmX complex formation and promotes MDM2-directed MDMX ubiquitination, promoting p53 activation. |
Apart from p53, c-Abl also regulates p73, a structural and functional homologue of p53, through the signaling path of ATM–cAbl–YAP1–p73.13 The activated c-Abl phosphorylates Yes-Associated Protein 1(YAP1), which is a key co-activator of p73. YAP1 transfers its own co-activator function from proliferation-driving TEAD transcription factors to p73, and then promote the transcription of apoptosis-driving genes (Figure 6). YAP1 can also keep p53 from Tyr357 phosphorylation.26 The specific mechanism remains to be explored.
 |
Figure 6 The phosphorylation of YAP1 by c-Abl activated kinase transfers its co-activator function from proliferation-driving TEAD transcription factors to p73, and then promote the transcription of apoptotic target genes. |
ABL also suppresses tumor development through contextually regulating oncogenic transforming growth factor-β (TGF-β) signaling. TGF-β is either facilitative or inhibitive for tumorigenesis. In tumor microenvironment, TGF-β becomes tumor-facilitative.27 There are findings proposing that ABL activation can block tumor-promoting microenvironmental signals and restore a tumor-suppressing microenvironment. It is well known that matrix metalloproteinases (MMPs) can modify the extracellular matrix (ECM) to allow malignant cells to penetrate into the normal tissue.28 TGF-β up-regulates MMPs significantly, in especial MMP-9 and MMP-13, to enhance the invasion of tumor cells.29,30 ABL activation, however, can suppress the expression and secretion of MMPs through inhibiting TGF-β signaling (Figure 7). Therefore, this may be another mechanism to explain the occurrence of malignant tumors in patients with ABL1 deletion.
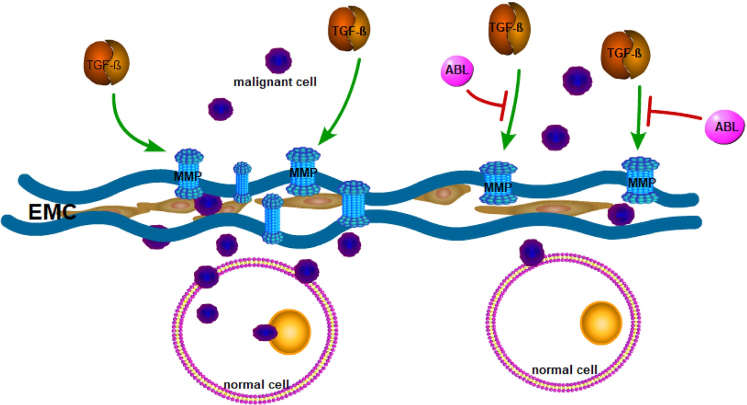 |
Figure 7 TGF-β signaling stimulates the secretion and activation of matrix metalloproteinases (MMPs) to remodel the extracellular matrix (ECM), then allowing malignant cells to penetrate the normal tissue. However, ABL activation can suppress the expression and secretion of MMPs through inhibiting TGF-β signaling and protect tumor cells from invading the tissue. |
Furthermore, ABL deletion on derivative chromosome 9 is associated with poor prognosis of patients with CML.31 The patients with ABL or BCR deletion showed shorter survival than those with normal ABL and BCR in CML.7 The mechanisms are not well known. An England research reported that the BCR 3ʹregion encodes a GTPase-activating protein for p21rac (a Ras-related GTP-binding protein) to promote the proliferation of cells associated with RAS. The activity of GTPase-activating protein is inhibited by the combination with p21rac and GTPase-activating protein.32 Thus, deletion of BCR 3ʹregion may promote the proliferation of tumor cells. These researches also speculated that certain tumor-related genes may locate near the ABL breakpoint and play a role in the inhibition of tumor progression.33 This may explain the short survival in patients with ABL deletion. On the other hand, the remaining alleles are susceptible to injure and the function of the tumor suppression may be disrupted after ABL deletion, so disease progression can be promoted as a result. In the present case, the patient’s long-term prognosis should be followed up, although she is now in complete remission.
In summary, entire ABL1 deletion without BCR/ABL1 fusion is a rare phenomenon in ALL patients, especially in females. ABL1 deletion correlates with the pathogenesis and prognosis of ALL. On one hand, ABL deletion negatively influences the activation of P53 and P73, and allows tumor cells to invade normal tissues through TGF-β signaling pathway. However, the clinical significance of ABL1 deletion in ALL development remains to be clarified with more cases.
Ethics Approval and Consent to Participate
This study was approved by the ethics committee of the Affiliated Hospital of Nantong University. The patient agreed and submitted a written informed consent to allow publication of this report and the accompanying images. Institutional approval was not required to publish this manuscript.
Acknowledgment
The authors gratefully thank Sano Suzhou Precision Medicine Co., Ltd for technical support.
Funding
This review did not receive any specific funding from any public, commercial, or nonprofit funding agencies.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102(3):83–87.
2. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36(1):93–99. doi:10.1016/0092-8674(84)90077-1
3. Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–1630. doi:10.1182/blood-2008-03-144790
4. Calabrese G, Fantasia D, Di Gianfilippo R, Stuppia L, Di Lorenzo R, Palka G. Fluorescence in situ hybridization analysis of minimal residual disease and the relevance of the der(9) deletion in imatinib-treated patients with chronic myeloid leukemia. Haematologica. 2006;91(7):994–995.
5. Zagaria A, Anelli L, Albano F, et al. Molecular cytogenetic characterization of deletions on der(9) in chronic myelocytic leukemia. Cancer Genet Cytogenet. 2006;167(2):97–102. doi:10.1016/j.cancergencyto.2006.01.011
6. Yoong Y, VanDeWalker TJ, Carlson RO, Dewald GW, Tefferi A. Clinical correlates of submicroscopic deletions involving the ABL-BCR translocation region in chronic myeloid leukemia. Eur J Haematol. 2005;74(2):124–127. doi:10.1111/ejh.2005.74.issue-2
7. Lee YK, Kim YR, Min HC, et al. Deletion of any part of the BCR or ABL gene on the derivative chromosome 9 is a poor prognostic marker in chronic myelogenous leukemia. Cancer Genet Cytogenet. 2006;166(1):65–73. doi:10.1016/j.cancergencyto.2005.08.028
8. Kim MJ, Yoon HS, Lim G, et al. ABL1 gene deletion without BCR/ABL1 rearrangement in a young adolescent with precursor B-cell acute lymphoblastic leukemia: clinical study and literature review. Cancer Genet Cytogenet. 2010;196(2):184–188. doi:10.1016/j.cancergencyto.2009.09.018
9. Lo A, Gu G, Liu M, Dev VG. ABL deletion without associated BCR-ABL in precursor B-cell acute lymphoblastic leukemia. Leuk Res. 2009;33(8):e98–e103. doi:10.1016/j.leukres.2009.01.038
10. Huh J, Moon H, Chung W. A case of ABL deletion in a patient with precursor B cell lymphoblastic leukemia. Ann Hematol. 2008;87(3):239–241. doi:10.1007/s00277-007-0374-7
11. Lee DS, Lee YS, Yun YS, et al. A study on the incidence of ABL gene deletion on derivative chromosome 9 in chronic myelogenous leukemia by interphase fluorescence in situ hybridization and its association with disease progression. Genes Chromosomes Cancer. 2003;37(3):291–299. doi:10.1002/gcc.10197
12. Zhu J, Wang JY. Death by Abl: a matter of location. Curr Top Dev Biol. 2004;59:165–192.
13. Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73(15):2829–2850. doi:10.1007/s00018-016-2130-4
14. Baskaran R, Wood LD, Whitaker LL, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387(6632):516–519. doi:10.1038/387516a0
15. Shafman T, Khanna KK, Kedar P, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387(6632):520–523. doi:10.1038/387520a0
16. Levav-Cohen Y, Goldberg Z, Zuckerman V, Grossman T, Haupt S, Haupt Y. C-Abl as a modulator of p53. Biochem Biophys Res Commun. 2005;331(3):737–749. doi:10.1016/j.bbrc.2005.03.152
17. Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13(1):49–58. doi:10.1016/S1044-579X(02)00099-8
18. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275(12):8945–8951. doi:10.1074/jbc.275.12.8945
19. Sionov RV, Moallem E, Berger M, et al. c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem. 1999;274(13):8371–8374. doi:10.1074/jbc.274.13.8371
20. Goldberg Z, Vogt Sionov R, Berger M, et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 2002;21(14):3715–3727. doi:10.1093/emboj/cdf384
21. Dias SS, Milne DM, Meek DW. c-Abl phosphorylates Hdm2 at tyrosine 276 in response to DNA damage and regulates interaction with ARF. Oncogene. 2006;25(50):6666–6671. doi:10.1038/sj.onc.1209671
22. Steegenga WT, Shvarts A, Riteco N, Bos JL, Jochemsen AG. Distinct regulation of p53 and p73 activity by adenovirus E1A, E1B, and E4 or f6 proteins. Mol Cell Biol. 1999;19(5):3885–3894. doi:10.1128/MCB.19.5.3885
23. Leslie PL, Ke H, Zhang Y. The MDM2 RING domain and central acidic domain play distinct roles in MDM2 protein homodimerization and MDM2-MDMX protein heterodimerization. J Biol Chem. 2015;290(20):12941–12950. doi:10.1074/jbc.M115.644435
24. Malbert-Colas L, Ponnuswamy A, Olivares-Illana V, Tournillon AS, Naski N, Fåhraeus R. HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol Cell. 2014;54(3):500–511. doi:10.1016/j.molcel.2014.02.035
25. Medina-Medina I, García-Beltrán P, de la Mora-de la Mora I, et al. Allosteric interactions by p53 mRNA govern HDM2 E3 ubiquitin ligase specificity under different conditions. Mol Cell Biol. 2016;36(16):2195–2205. doi:10.1128/MCB.00113-16
26. Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14(4):743–751. doi:10.1038/sj.cdd.4402063
27. Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62. doi:10.1016/j.bbcan.2006.06.004
28. Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696–1701. doi:10.2741/1915
29. Kominsky SL, Doucet M, Thorpe M, Weber KL. MMP-13 is over-expressed in renal cell carcinoma bone metastasis and is induced by TGF-beta1. Clin Exp Metastasis. 2008;25(8):865–870. doi:10.1007/s10585-008-9202-2
30. Sinpitaksakul SN, Pimkhaokham A, Sanchavanakit N, Pavasant P. TGF-beta1 induced MMP-9 expression in HNSCC cell lines via Smad/MLCK pathway. Biochem Biophys Res Commun. 2008;371(4):713–718. doi:10.1016/j.bbrc.2008.04.128
31. Sinclair PB, Nacheva EP, Leversha M, et al. Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood. 2000;95(3):738–743. doi:10.1182/blood.V95.3.738.003k21_738_743
32. Diekmann D, Brill S, Garrett MD, et al. Bcr encodes a GTPase-activating protein for p21rac. Nature. 1991;351(6325):400–402. doi:10.1038/351400a0
33. Huntly BJ, Bench AJ, Delabesse E, et al. Derivative chromosome 9 deletions in chronic myeloid leukemia: poor prognosis is not associated with loss of ABL-BCR expression, elevated BCR-ABL levels, or karyotypic instability. Blood. 2002;99(12):4547–4553. doi:10.1182/blood.V99.12.4547
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.