")
Back to Journals » International Journal of Nanomedicine » Volume 12
Enhancing siRNA-based cancer therapy using a new pH-responsive activatable cell-penetrating peptide-modified liposomal system
Authors Xiang B , Jia XL, Qi JL, Yang LP, Sun WH, Yan X, Yang SK, Cao DY, Du Q, Qi XR
Received 6 December 2016
Accepted for publication 28 February 2017
Published 28 March 2017 Volume 2017:12 Pages 2385—2405
DOI https://doi.org/10.2147/IJN.S129574
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Bai Xiang,1,* Xue-Li Jia,1,* Jin-Long Qi,2 Li-Ping Yang,1 Wei-Hong Sun,1 Xiao Yan,1 Shao-Kun Yang,1 De-Ying Cao,1 Qing Du,1 Xian-Rong Qi3
1Department of Pharmaceutics, School of Pharmaceutical Sciences, 2Department of Pharmacology, Hebei Medical University, Shijiazhuang, Hebei, 3School of Pharmaceutical Sciences, Peking University, Beijing, China
*These authors contributed equally to this work
Abstract: As a potent therapeutic agent, small interfering RNA (siRNA) has been exploited to silence critical genes involved in tumor initiation and progression. However, development of a desirable delivery system is required to overcome the unfavorable properties of siRNA such as its high degradability, molecular size, and negative charge to help increase its accumulation in tumor tissues and promote efficient cellular uptake and endosomal/lysosomal escape of the nucleic acids. In this study, we developed a new activatable cell-penetrating peptide (ACPP) that is responsive to an acidic tumor microenvironment, which was then used to modify the surfaces of siRNA-loaded liposomes. The ACPP is composed of a cell-penetrating peptide (CPP), an acid-labile linker (hydrazone), and a polyanionic domain, including glutamic acid and histidine. In the systemic circulation (pH 7.4), the surface polycationic moieties of the CPP (polyarginine) are “shielded” by the intramolecular electrostatic interaction of the inhibitory domain. When exposed to a lower pH, a common property of solid tumors, the ACPP undergoes acid-catalyzed breakage at the hydrazone site, and the consequent protonation of histidine residues promotes detachment of the inhibitory peptide. Subsequently, the unshielded CPP would facilitate the cellular membrane penetration and efficient endosomal/lysosomal evasion of liposomal siRNA. A series of investigations demonstrated that once exposed to an acidic pH, the ACPP-modified liposomes showed elevated cellular uptake, downregulated expression of polo-like kinase 1, and augmented cell apoptosis. In addition, favorable siRNA avoidance of the endosome/lysosome was observed in both MCF-7 and A549 cells, followed by effective cytoplasmic release. In view of its acid sensitivity and therapeutic potency, this newly developed pH-responsive and ACPP-mediated liposome system represents a potential platform for siRNA-based cancer treatment.
Keywords: siRNA, ACPP, hydrazone, liposome, endosomal/lysosomal escape
Introduction
Currently, the treatment of proliferative disorders such as cancer mainly relies on nonspecific therapies (ie, chemotherapies). Nevertheless, accumulating clinical data suggest that chemotherapy may only be able to prolong survival in a subset of patients, and the benefit of conventional chemotherapy is generally limited due to its similar cytotoxicity to both cancerous and healthy cells.1,2 In contrast, RNA interference (RNAi) may serve as a potent therapeutic strategy with several advantages, including its higher specificity and the possibility of modulating the posttranscriptional expression of any key regulator involved in disease progression.3 To explore this potentially specific and more tolerated therapeutic approach, synthetic small interfering RNA (siRNA) has been exploited to silence the critical genes involved in tumor initiation, growth, and metastasis formation.
However, the therapeutic application of siRNAs remains a substantial challenge because of their low biological stability, unfavorable pharmacokinetics, and poor cellular uptake. To overcome these obstacles, several types of nanocarriers have been developed for the delivery of siRNA, and cationic liposomes are the most widely utilized and studied carriers to date.4 The additional modification of poly(ethylene glycol) (PEG) onto the liposome surface reduces the probable complement activation and inflammation, leading to extended circulation times and conferring the nanocarriers with the ability of passive targeting to tumors via an enhanced permeability and retention (EPR) effect. However, the EPR effect may not be sufficient to direct the accurate tissue distribution of liposomal siRNA due to its nonspecific feature.5 Previous studies have also shown that the use of PEG as a steric blockade had an obstructive effect on cellular uptake and delayed the escape from the lysosome, in which siRNA would undergo considerably high degradation.6 Therefore, a new desirable delivery platform with reinforced specificity toward the tumor, efficient cellular uptake, and endosomal escape of the nucleic acids is needed.
Cell-penetrating peptides (CPPs) such as transactivator of transcription (TAT) and polyarginine have been shown to facilitate the intracellular delivery of bioactive molecules with low membrane permeabilities.7 However, the majority of known CPPs are not cell or tissue specific, and their translocation across the cell membrane occurs in nearly all cell types by means of heparan sulfates and other glycosaminoglycans.8 With the aim of solving this dilemma, in 2004, Jiang et al9 introduced a protease-triggered system, representing the first activatable cell-penetrating peptide (ACPP), and applied it to tumor imaging. This ACPP was composed of a polycationic CPP sequence, a matrix metalloproteinase (MMP)-cleavable peptide linker, and a polyanionic inhibitory domain. The CPP-bearing payload would not be activated due to electrostatic masking of the anionic peptide. When exposed to a tumor microenvironment, the ACPP was cut off at the cleavable linker by the function of overexpressed MMP2/9, enabling the detachment of the inhibitory domain and the subsequent recovery of the polycationic CPP. Since the proposal of this innovative ACPP construct, several groups, including our own, have developed various other enzyme-triggered ACPPs for potential applications in tumor imaging and therapy.10–14 In response to certain upregulated enzymes such as MMP, prostate-specific antigen, and urokinase plasminogen activator,15,16 the inner cleavable linkage suffers from specific proteolysis, generating “off–on” switches of CPP activity. Nevertheless, there are still some considerable challenges to overcome with this strategy. For example, there is significant heterogeneity in the expression of certain enzymes, not only between patients and tumor types but also within individual tumors.17 Therefore, in comparison with enzyme-specific delivery, the development of targeting strategies with higher universality across a broad range of tumors is a primary research goal.
Cancer development and progression are related to intensified metabolic activity and hypoxia, inducing the elevated extracellular acidity that is observed in almost all human and animal tumors, which results in an extracellular pH (pHe) ranging from 6 to 7.18 This abnormal microenvironment, which is more acidic than the blood (pH 7.4), has been utilized to deliver therapeutic and imaging agents toward a wide variety of solid tumors.19 Among those developed to date, a hydrazone-based stimulus-responsive strategy represents a promising type of tumor treatment, which ensures the integrity of carriers at normal physiological pH but allows them to be effectively degraded and undergo cell uptake in the tumor microenvironment with a low pHe. For example, studies have demonstrated the steric protection of CPP by the introduction of long PEG chains onto the surface of nanocarriers via hydrazone bonds; thus, CPP could remain inert under physiological conditions (in both the blood circulation and normal tissues). Following their localization to the tumor extracellular regions by means of the EPR effect and ligand interaction, these carriers could lose their outer PEG layer after the acid-promoted cleavage of the hydrazone linker and could ultimately be transported into cells via the power of the exposed TAT peptide20–22 or oligoarginine23 moieties. In addition, diethylenetriaminepentaacetic acid (DTPA) was chosen as a shielding molecule owing to its hydrophilicity and negative charge.24 After the conjugation of DTPA at the terminus of a targeting ligand (peptide T7) through hydrazone, Jiang et al could achieve an “active/inert” switch of the ligand at different pH values outside of tumor/normal cells, which could further stimulate or deactivate the receptor-mediated endocytosis.
Here, we introduce our newly developed rational strategy to take advantage of the integration of pH-sensitive linkage and ACPP modification for creating a more selective and efficient siRNA delivery system to tumor cells (Figure 1). The pH-responsive activatable peptide includes three units, a cell-penetrating domain (octaarginine), a pH-sensitive cleavable linker (hydrazone), and a polyanionic inhibitory peptide (ehGehGehGehG). The basic concept is as follows: In the circulation (pH 7.4), both glutamic acid and histidine present in the shielding peptides remain negatively charged owing to their lower pKa values,25 and then the surface polycationic moieties in the CPPs are “shielded” by electrostatic attraction of the inhibitory domain. Following accumulation in the tumor tissues with decreased pH, the ACPPs undergo breakage due to acid-catalyzed hydrolysis of the hydrazone bonds. Moreover, histidine obtains a net positive charge in this acidic environment,26 which substantially decreases the total negative charges of the shielding domain to finally promote the detachment of the inhibitory peptide fractions with the CPP segments in the ACPP. The activated liposomal carriers are subsequently taken up by the tumor cells via the recovered CPP effect. Furthermore, after trafficking into the tumor cell, octaarginine peptides allow for the surface-modified liposomes to efficiently avoid lysosome uptake, facilitating the cytoplasm distribution of the loaded siRNAs.
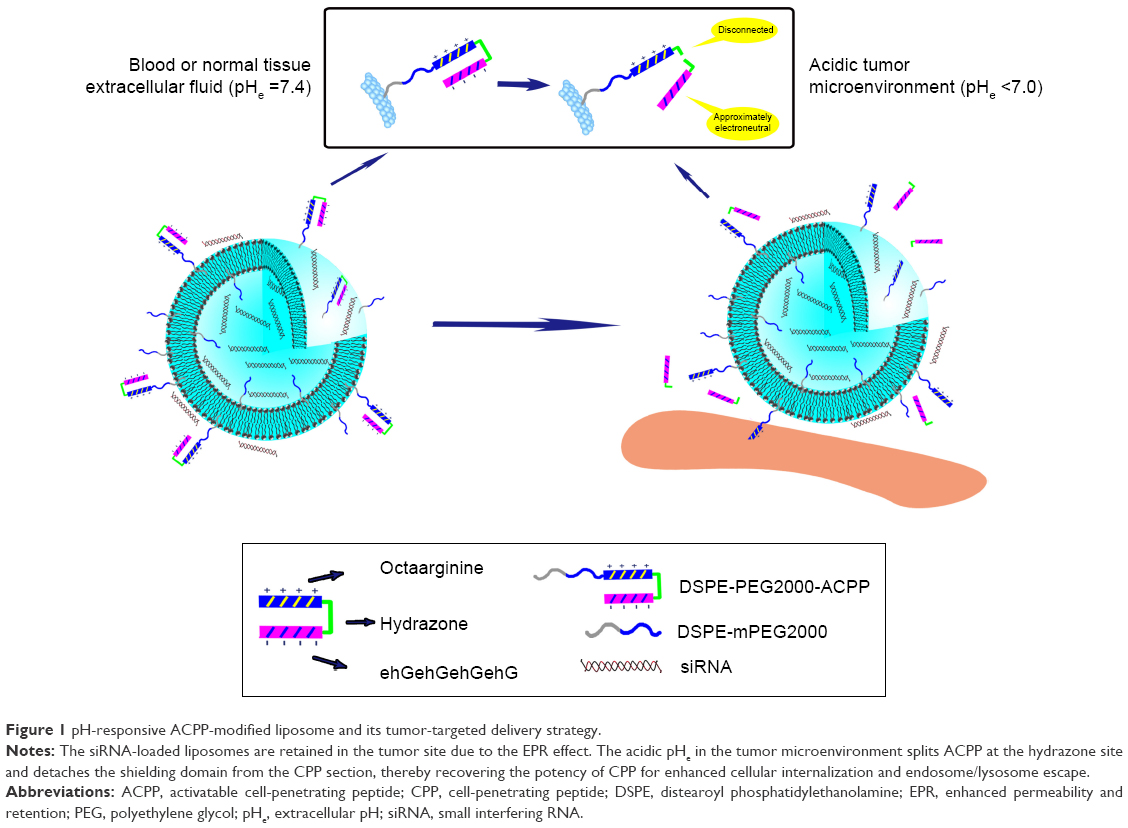 | Figure 1 pH-responsive ACPP-modified liposome and its tumor-targeted delivery strategy. |
Materials and methods
Materials
N-(4-Acetylphenyl)maleimide (NAPM) was purchased from Alfa Aesar Corp. (Tianjin, China). 3-Maleimidopropionic acid hydrazide-trifluoroacetic acid (MPH) was obtained from Balmxy Pharmaceutic Co., Ltd. (Shanghai, China). Cysteine–glycine–[(D-glutamic acid)–(D-histidine)–(D-glycine)]4 (NP) and CPP peptides (Ac-CGehGehGehGehG and GrrrrrrrrGC) were ordered in >95% purity from GL Biochem (Shanghai, China).
Soybean phosphatidylcholine (SPC) was purchased from Lipoid (Ludwigshafen, Germany), and cholesterol was obtained from Wako (Tokyo, Japan). 3β[N-(N′,N′-Dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol) was synthesized in our laboratory.27 N-(Aminopropyl polyethyleneglycol)carbamyl-distearoylphosphatidyl-ethanolamine (DSPE-mPEG2000) and 3-(N-succinimidyloxyglutaryl)aminopropyl, polyethyleneglycol-carbamyl distearoylphosphatidyl-ethanolamine (DSPE-PEG2000-NHS) were purchased from NOF (Tokyo, Japan).
Negative control siRNA (siN.C.), 6-Carboxyfluorescein-aminohexyl (FAM)-labeled negative control siRNA (FAM-siRNA) (antisense strand, 5′-ACGUGACACGUUCGGAGAATT-3′), and siRNA targeting polo-like kinase 1 (PLK-1) messenger RNA (mRNA) (siPLK-1, antisense strand, 5′-GUGAUCUUCUUCAUCAAGGdTdT-3′) were custom-synthesized by GenePharma (Shanghai, China). All primers were synthesized by AuGCT Biotechnology (Beijing, China). Fluorescent probes such as rhodamine-phalloidin, CellLight Early Endosomes-RFP BacMam 2.0, and LysoTracker Red were obtained from Invitrogen/Thermo Fisher Scientific (Waltham, MA, USA). Roswell Park Memorial Institute (RPMI) 1640 medium and fetal bovine serum (FBS) were purchased from Gibco/Thermo Fisher Scientific. Hoechst 33258 and 4% formaldehyde were supplied by Macgene (Beijing, China).
Synthesis of ACPP conjugates
The synthesis procedure of the ACPP conjugates is displayed in Figure 2. Compound NAPM–MPH (N-M) was prepared according to a previously reported procedure.28 In brief, NAPM (15.0 mg, 69.7 μmol) in MeOH (1.0 mL) was added to a stirred solution of MPH (20.7 mg, 69.7 μmol) in MeOH (1.0 mL). The mixture was agitated for 15 min at 60°C under nitrogen. The obtained precipitate was filtered, rinsed with cold MeOH, and dried under vacuum to obtain N-M, a pale yellow powder.
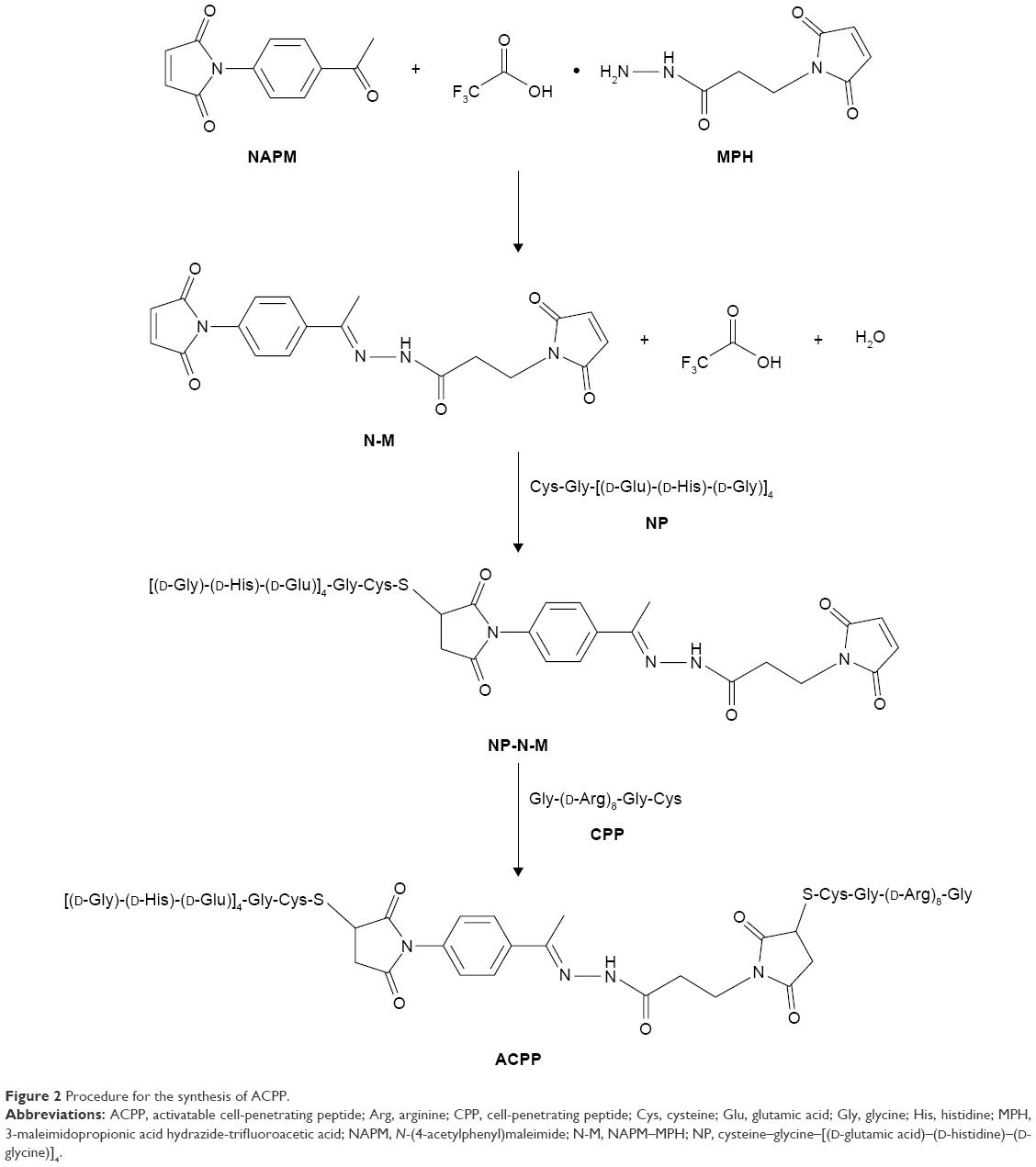 | Figure 2 Procedure for the synthesis of ACPP. |
Melting points were determined in open-glass capillaries on a BUCHI M-560 melting point apparatus (Buchi Labortechnik AG, Flawil, Switzerland) and were uncorrected. The 1H nuclear magnetic resonance (NMR) spectra were assayed on a Bruker AVANCE III-400 spectrophotometer (Bruker, Fllanden, Switzerland) using tetramethylsilane as the internal standard and deuterated dimethyl sulfoxide as the solvent. Mass spectra were recorded on an Applied Biosystems 3200 QTRAP mass spectrometer (ABSciex, Framingham, MA, USA), which were analyzed in the positive ion mode.
The following conjugates (NP-N-M and ACPP) were acquired through the covalence of thiol and maleoyl, using our previously reported method.29 To form NP-N-M, an NP (5.0 mg, 3.3 μmol) solution was prepared in N, N-dimethylformamide (DMF) and then added drop-wise to the N-M (1.3 mg, 3.3 μmol) solution. The reaction system was stirred under nitrogen at room temperature for 12 h, followed by a quick influx into cold anhydrous diethyl ether (DEE). The resultant white solid was separated, washed, and dried under vacuum.
For the synthesis of the ACPP conjugate, CPP (2.5 mg, 1.7 μmol) in DMF was mixed with NP-N-M (3.2 mg, 1.7 μmol) in the presence of a 2 molar excess of N-ethyldiisopropylamine (DIPEA). The reaction mixture was subjected to constant agitation under nitrogen at 40°C for 24 h. After precipitation of the product into cold anhydrous DEE/acetone (7:3, v/v), the resulting solid was collected by centrifugation, rinsed with the same solvent systems, and treated to dryness under reduced pressure.
Hydrolysis kinetics of the ACPPs
To verify the acidic pH-mediated degradation of the hydrazone-contained ACPP, the peptide was incubated at pH 6.2 and 7.4 in phosphate-buffered saline (PBS; 10 mM phosphate, 150 mM NaCl) in a horizontal shaker, respectively, with a final concentration of 1.37 mg/mL. During incubation in the buffer solutions at 37°C, aliquots of the mixture were sampled out and analyzed by high-performance liquid chromatography (HPLC) at discrete time points (0, 1, 3, 6, 9, 12, 24, and 48 h).
HPLC analysis was performed with a Kromasil 100-5 C18 column (250 ×4.6 mm, 5-μm particle size) on a SHIMADZU LC-20AT HPLC system (Shimadzu, Kyoto, Japan), and the chromatograms were recorded at 220 nm using acetonitrile:water (5:95) containing 0.05% trifluoroacetic acid (v/v) as a solvent, with a flow rate of 1.0 mL/min at 30°C.
Synthesis of functional conjugates
The ACPP peptide was conjugated with DSPE-PEG2000-NHS (1:1.2 molar ratio) in DMF by the nucleophilic substitution reaction according to the method of Liang et al,30 with minor modifications. In brief, the ACPP solution was added to the DSPE-PEG2000-NHS system containing a 5-fold molar amount of triethylamine (TEA). After 48 h of stirring under nitrogen protection, the resulting solution was dialyzed with a molecular weight cutoff of 3.5 kDa (Solarbio, Beijing, China) against distilled water (adjusted to pH 10.5–11.0 using 1 M NaOH) for 24 h. The final solution was lyophilized and stored at −20°C until use. The linkage of CPP with DSPE-PEG2000-NHS was conducted under a two-fold molar amount of TEA, and the subsequent dialysis was performed against distilled water. All other procedures were conducted in the same manner as described for DSPE-PEG2000-ACPP.
The molecular weight distributions of DSPE-PEG2000-ACPP and DSPE-PEG2000-CPP were determined by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Bruker Daltonics, Bremen, Germany) using ferulic acid or alpha-cyano-hydroxycinnamic acid as the matrix for mass spectrometric analysis.
Preparation of siRNA-loaded liposomes
To prepare the preformed PEGylated liposomes, a lipid composition (molar ratio) of SPC (48%), cholesterol (8%), DC-Chol (40%), and DSPE-mPEG2000 (3%) was used. A thin lipid film was formed in a round-bottomed flask by dissolving the lipids in chloroform:methanol (3:1, v/v) and removing the solvent in a rotary evaporator for 30 min at 37°C. After continued vacuum drying for 1 h at room temperature, the lipid film was hydrated using 1.5 μM of siRNA (siN.C., siPLK-1, or FAM-siRNA) solution that was pretreated with diethyl pyrocarbonate. To control the size, the lipid dispersion was extruded sequentially through polycarbonate Track-Etch membranes (Whatman, Kent, UK) with 0.4 mm (5 times) and 0.2 mm (5 times) pore sizes using an NLI TBX 001 liposome extruder device (Northern Lipids, Burnaby, BC, Canada).
Nonmodified liposomes (N-L), CPP-modified liposomes (C-L), and ACPP-modified liposomes (A-L) were formed by the postinsertion method. In brief, a lipid film of DSPE-PEG2000, DSPE-PEG2000-CPP, or DSPE-PEG2000-ACPP was prepared by rotary evaporation and further dried under vacuum for 1 h. The dried lipid film was then hydrated with diethyl pyrocarbonate-treated water for the formation of micelles and incubated for 15 min at 60°C. For the N-L, C-L, or A-L preparations, 0.25 mL of the micelle solution (DSPE-PEG2000, DSPE-PEG2000-CPP, or DSPE-PEG2000-ACPP micelles) was added to 1 mL of preformed liposomes at the required molar ratio (3% DSPE-PEG or DSPE-PEG-peptides of total lipid) and incubated for 4 h at 37°C in a water bath. All the resulting liposomes were cooled to room temperature before use.
Characterization of siRNA-loaded liposomes
Particle size and zeta potential
The size distribution and zeta potential of each formulation were determined with dynamic light scattering (DLS; Malvern Zetasizer Nano ZS 90, Malvern, UK). A 1 mL suspension was placed in a DLS cuvette and measured with detection optics arranged at 90°. Three serial measurements were performed for each sample.
Morphology
The morphology of the A-L liposomes was examined by transmission electron microscopy (TEM) and atomic force microscopy (AFM). For TEM analysis, 10 μL of the liposomal formulation was uniformly loaded on a carbon-coated copper grid for 1 min, after which the films were negatively stained with 10 μL of 1% phosphotungstic acid solution for 1 min. Excess sample and stain were blotted with filter paper, and the copper grid was dried for imaging using a HITACHI H-7500 transmission electron microscope (Hitachi, Tokyo, Japan) at 50,000× magnification. To acquire AFM topographic images, the A-L sample solution was dropped on to the surface of mica and dried under nitrogen gas for 2 h. The samples were observed with a Bioscope Resolve atomic force microscope (Bruker Nano Surfaces, Santa Barbara, CA, USA) in PeakForce quantitative nanomechanical imaging mode. The A-L suspension was imaged with a scan rate of 1 Hz. A Bruker silicon-nitride ScanAsyst Air probe with a spring constant of 0.4 N/m was used. All images were processed with the NanoScope Analysis software (Nanoscope Analysis, Bruker-AXS, Santa Barbara, CA, USA) and the Young’s modulus of A-L was calculated by fitting the retract curve using the Derjaguin–Muller–Toropov model.31
Cell culture
The human breast adenocarcinoma cell line (MCF-7 cells) and human pulmonary adenocarcinoma cell line (A549 cells) were obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultivated in RPMI 1640 medium supplemented with 2 mM L-glutamine, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10% FBS, 100 IU/mL penicillin, and 100 mg/mL streptomycin. The cells were maintained in a 37°C humidified incubator with a 5% CO2 atmosphere.
By serial passages, some cells were adapted for growth in the low pH medium (pH 6.2), adjusted with 1 M HCl.
Cellular uptake
The MCF-7 cells grown in pH 6.2 or 7.4 medium were seeded into 6-well plates at a density of 2×105 cells per well in 2 mL of the corresponding medium and cultured at 37°C in a 5% CO2 humidified atmosphere for 24 h. After rinsing with PBS, the cells were incubated with pH 6.2 or 7.4 medium containing free FAM-siRNA or various FAM-siRNA-loaded liposomes, which were subjected to 5 h of preincubation in serum-free medium at the corresponding pH. The exposed concentration of FAM-siRNA was 100 nM. Following the treatment for 2 h at 37°C, the cells were trypsinized and washed with cold PBS containing heparin (500 U/mL). After two further washes with cold PBS, the cells were filtered and subjected to flow cytometric analysis utilizing a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
To assess the cellular uptake by confocal microscopy, MCF-7 cells (1×105 cells) were seeded on to a sterile glass-bottomed dish (35×10 mm) and incubated for 48 h in complete RPMI 1640 medium at pH 6.2 and 7.4, respectively. Following five washes with PBS, the serum-free medium containing free or liposomal FAM-siRNA was introduced as described in the earlier paragraph. The final concentration of FAM-siRNA in the culture medium was 200 nM. Subsequently, the cells were incubated for 3.5 h at 37°C and washed with cold PBS containing heparin (500 U/mL) four times. Fixation with 4% formaldehyde was then performed for 10 min, followed by three 5-min rinses with cold PBS. To image the F-actin in the cells, the cells were stained with rhodamine-phalloidin for 20 min after permeabilization with 0.1% Triton X-100 in PBS. Finally, the nuclei were labeled with Hoechst 33258 for an additional 20 min at 37°C and were imaged on a Leica TCS SP8 confocal platform (Leica Microsystems Inc., Mannheim, Germany). FAM-siRNA, rhodamine-phalloidin, and Hoechst 33258 were excited using 494, 540, and 352 nm lasers, respectively.
Intracellular trafficking behavior of liposomal siRNA
The internalization and endosomal release of the liposomal FAM-siRNA were characterized by confocal laser-scanning microscopy (CLSM). MCF-7 cells were seeded in a sterile glass-bottomed dish (35×10 mm) at a density of 6×104 cells per well and cultured in complete RPMI 1640 medium at pH 6.2 and 7.4, respectively. To stain the early endosomes of the cells, the MCF-7 cells were preincubated with CellLight Early Endosomes-RFP BacMam 2.0 (20 particles per cell, as recommended by the supplier) for 24 h at 37°C. The following day, the cells were incubated with the A-L that had been pretreated under acidic or neutral conditions (as described earlier) for 2 or 6 h at pH 6.2 and 7.4, respectively. Subsequently, the cells were rinsed three times with cold PBS containing heparin (500 U/mL) and then treated for fixation and nuclear staining. The MCF-7 cells were also incubated with N-L, and the detailed procedure for staining is provided in the Supplementary materials.
To visualize the intracellular trafficking of liposomal FAM-siRNA, A549 cells were seeded on to a sterile glass-bottomed dish at a density of 10×104 cells per well and cultured in complete RPMI 1640 medium at pH 6.2. Following a 1- or 6-h incubation with acid-pretreated A-L as mentioned earlier, A549 cells were washed three times with cold PBS including heparin (500 U/mL) and were then treated as described in the earlier paragraph for the MCF-7 cells. Late endosome/lysosome labeling was performed for 0.5 h using LysoTracker Red at a concentration of 500 nM.
Confocal microscopic images were acquired using a Leica confocal platform with a 63× oil objective with an excitation wavelength of 494 nm for FAM-siRNA, 555 nm for CellLight Early Endosomes-RFP BacMam 2.0, 561 nm for LysoTracker Red, and 352 nm for Hoechst 33258.
Cell apoptosis assay
MCF-7 cells growing in pH 6.2 or 7.4 medium were cultured in 25 cm2 tissue culture flasks at 6×105 cells per flask in 5 mL of complete RPMI 1640 medium as mentioned earlier. After 24 h culture at 37°C in a 5% CO2 humidified atmosphere, the cells were washed with PBS and exposed to fresh serum-free medium containing the siRNA-loaded samples as described earlier. The final concentration of siRNA (siPLK-1 or siN.C.) used in the experiment was 100 nM. Following 6 h of incubation, the medium was replaced with complete medium for a routine culture of 72 h at 37°C. Subsequently, the cells were collected and stained with the Annexin V-FITC apoptosis detection kit (KeyGEN, Nanjing, China) according to the manufacturer’s instructions and were immediately analyzed on a BD FACSCalibur flow cytometer with 10,000 events collected (excitation 488 nm; emission 530 nm).
In vitro transfection and analysis of gene silencing
MCF-7 cells were seeded into 25 cm2 tissue culture flasks and treated with fresh serum-free medium containing siRNA-loaded samples as mentioned earlier. Following 6 h of incubation, the medium was replaced with complete medium and the cells were incubated for an additional 48 h (for the mRNA assay) or 72 h (for protein quantification) at 37°C. Subsequently, the PLK-1 mRNA and protein levels were determined using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and Western blot analysis, respectively.
In the qRT-PCR experiments, the total RNA from transfected cells was extracted using TRIzol reagent (Tiangen, Beijing, China) according to the manufacturer’s protocol. After complementary DNA (cDNA) synthesis from 2 mg of total RNA with a Quantscript RT kit (Tiangen), 4 mL of cDNA was subjected to qRT-PCR analysis targeting PLK-1 and the reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the SuperReal Premix SYBR Green kit (Tiangen). The relative gene expression was analyzed on the CFX 96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA), followed by quantitation using the 2−ΔΔCt method. Data are presented as the fold change in PLK-1 expression normalized with that of the endogenous reference, GAPDH, and relative to the expression level of the untreated cells. The primers used for PCR amplification were GAPDH forward: 5′-GGGTGTGAACCATGAGAAGT-3′, reverse: 5′-GACTGTGGTCATGAGTCCT-3′; PLK-1 forward: 5′-CGAGGTGCTGAGCAAGAAAGGGC-3′, reverse: 5′-CCACGGGGTTGATGTGCTTGGGA-3′. PCR parameters were as follows: 95°C for 15 min, followed by 40 cycles at 95°C for 10 s and 61°C for 30 s. Specificity was verified by melt curve analysis and agarose gel electrophoresis.
For Western blotting analysis, the transfected cells were first washed three times with ice-cold PBS and then lysed in radioimmunoprecipitation assay buffer (Bestbio. Co. Ltd., Shanghai, China) containing phenylmethanesulphonylfluoride. The resultant cell suspension was incubated on ice for 30 min, accompanied by vortexing every 5 min. The lysates were collected by centrifugation for 10 min at 14,000 rpm at 4°C. Subsequently, the protein concentration was determined by the bicinchoninic acid protein assay (MultiSciences Biotech, Beijing, China). Following separation by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, the total protein (50 mg) was transferred (at 250 mA for 2.5 h) to Immobilon-P membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline with Tween-20 (TBST) for 1 h at room temperature and incubated overnight at 4°C in 5% BSA in TBST with anti-PLK-1 monoclonal antibody (Cell Signaling Technology Inc., Danvers, MA, USA; 1:1,000) or rabbit anti-beta-actin (Antibody Revolution Inc., San Diego, CA, USA; 1:2,000) as the internal control. An additional incubation was performed in 5% BSA with anti-rabbit IgG horseradish peroxidase-linked antibody (Cell Signaling Technology Inc.; 1:3,000) for 1 h at ambient temperature, followed by imaging using the Molecular Imager ChemiDoc XRS + system (Bio-Rad).
Statistical analysis
Data are presented as the mean ± standard deviation. The difference between any two groups was determined by analysis of variance. P<0.05 was considered to be statistically significant.
Results
Synthesis and identification of ACPP
The ACPP was synthesized according to the steps shown in Figure 2. In the initial step, the nucleophilic addition elimination reaction of the hydrazide (MPH) with the ketone (NAPM) resulted in the hydrazone linker (N-M). The synthesis of hydrazone derivatives (NP-N-M) was carried out via the Michael addition reaction of the N-M with NP peptide. Finally, the treatment of NP-N-M with CPP afforded the target compound (ACPP) in the presence of DIPEA.
The structure of N-M was confirmed by 1H NMR spectroscopy (Figure 3A) and electrospray ionization mass spectral data (Figure 3B). In the 1H NMR spectra of this compound, the signal due to the hydrazone proton (e) was observed in the region of 10.0–11.0 ppm. The signal due to the methyl protons (d) appeared in the range of 2.2–2.3 ppm. The four protons (b, c) of the phenyl moiety resonated as two doublets in the region of 7.3–7.4 and 7.8–7.9 ppm. The appearance of signals at 6.9–7.3 ppm was assigned to the –CO–CH– protons (a, h) of maleimide.32 Other aliphatic protons (g, f) were observed at the expected regions. In the mass spectra of N-M, the [M+1]+ peak was observed in agreement with its calculated values. The molecular weight of N-M was further confirmed by the presentation of an M+Na and M+K peak at m/z 403 and m/z 419, respectively. The melting point of N-M was found to be in the range of 218°C–220°C.
 | Figure 3 Identification of ACPP and its intermediate products. |
The structure confirmation of NP-N-M was determined by analysis of the mass spectra (Figure 3C); the [M+2H]2+ (m/z 948.2) and [M+3H]3+ (m/z 632.7) peaks were consistent with the calculated molecular weight (1,893.84). Successful conjugation of ACPP was supported by its MALDI-TOF mass spectrum (Figure 3D), in which the mass: charge ratio of the major peak (3,378.93) was in accordance with the theoretical value, 3,378.61.
Characterization of ACPP
Prior to the conjugation of ACPP to the PEGylated lipid, the stability of the peptide was investigated under an acidic condition (pH 6.2). Over the period of this study, two newly formed peaks were observed in the HPLC chromatograph at the retention time of 4.9 min (corresponding to the retention time of octaarginine) and 7.3 min, which was different from that of the tested ACPP sample (~11.5 min). As expected, the results displayed the cleavage of the acid-labile hydrazone bonds in the ACPP. As shown in the degradation profile of ACPP (Figure 4), the time required for splitting half of the peptide was ~5 h. However, at the physiological pH (pH 7.4), much higher stability was illustrated in the curve, with a slower decrease and prolonged half-life of ACPP up to 32.8 h. The resultant difference between the different pH values exhibited the pronounced pH sensitivity of the designed ACPP.
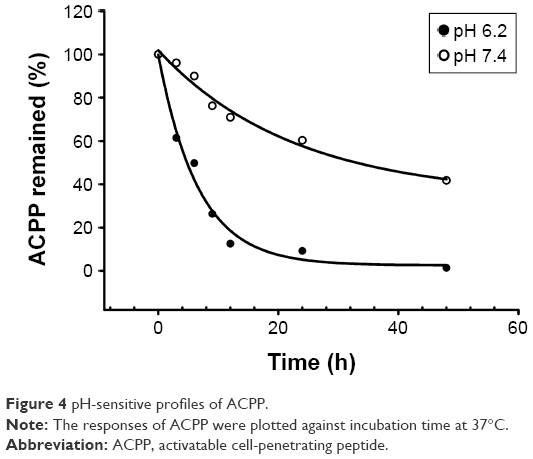 | Figure 4 pH-sensitive profiles of ACPP. |
Synthesis and identification of functional conjugates
We synthesized DSPE-PEG2000-ACPP and DSPE-PEG2000-CPP via the nucleophilic substitution reactions of the NHS group with the primary amine groups of the peptides (Figure 5A). The two major multipeaks centered at 6,231.50 (Figure 5B) and 4,380.89 (Figure 5C) mass: charge ratios, respectively, indicated that the mean molecular weights of DSPE-PEG2000-ACPP and DSPE-PEG2000-CPP were 6,230 and 4,380 Da, respectively. These data are consistent with the theoretical mean molecular weights of the corresponding conjugates (6,228 and 4,334 Da, respectively).
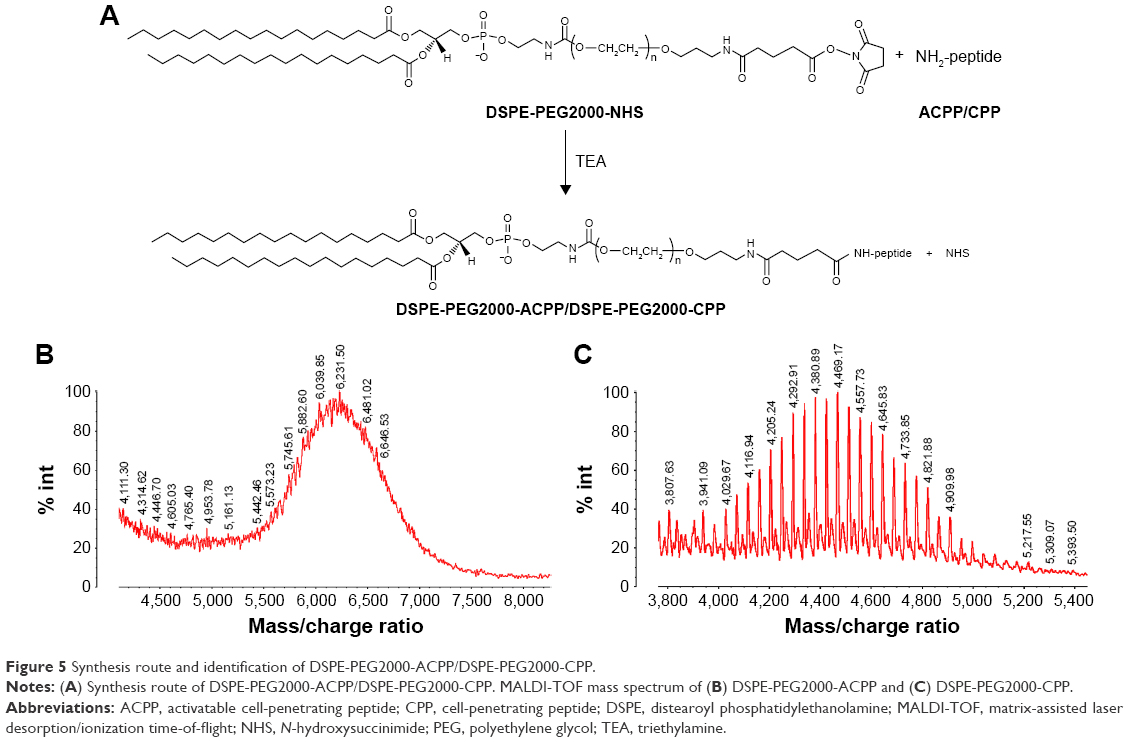 | Figure 5 Synthesis route and identification of DSPE-PEG2000-ACPP/DSPE-PEG2000-CPP. |
Preparation and characterization of liposomal formulations
In this experiment, we first developed preformed PEGylated liposomes via the thin-film hydration technique followed by extrusion. To produce N-L, C-L, and A-L, the preformed liposomes were individually subjected to further surface modifications of PEG, CPP, and ACPP, respectively, using postinsertion, by which the DSPE-based lipid conjugate was anchored to the outer layer of the liposomal membrane. Thus, three types of liposomes were formed as follows: N-L (the polymer was 6% DSPE-PEG2000, molar ratio of total lipids), C-L (3% DSPE-PEG2000 and 3% DSPE-PEG2000-CPP, molar ratio of total lipids), and A-L (3% DSPE-PEG2000 and 3% DSPE-PEG2000-ACPP, molar ratio of total lipids).
Size measurements were conducted by DLS, which indicated that all the liposome types had small particle sizes (200–220 nm) and a narrow size distribution with a polydispersity index range of 0.2–0.3 (Table 1). The zeta potential represents the electrostatic charge of the particle surface, which serves as a repulsive energy barrier resisting the proximity of particles and promoting the stability of dispersion. As indicated in Table 1, C-L showed the highest positive charges among all the studied formulations, with an average zeta potential up to +45 mV. By contrast, A-L showed a reduced zeta potential value (+39 mV), which was close to that of N-L (+35 mV).
 | Table 1 Physicochemical properties of liposomes carrying siRNA |
Morphology
The TEM micrographs revealed that the liposomes displayed a spherical morphology (Figure 6A). The observed liposomes had sizes ~100 nm, which was smaller than the results of the DLS measurements. The AFM images showed uniform and spherical-shaped vesicles with a smooth surface (Figure 6B and C), and the size of ~200 nm was in rather good agreement with the data obtained from the DLS analysis.
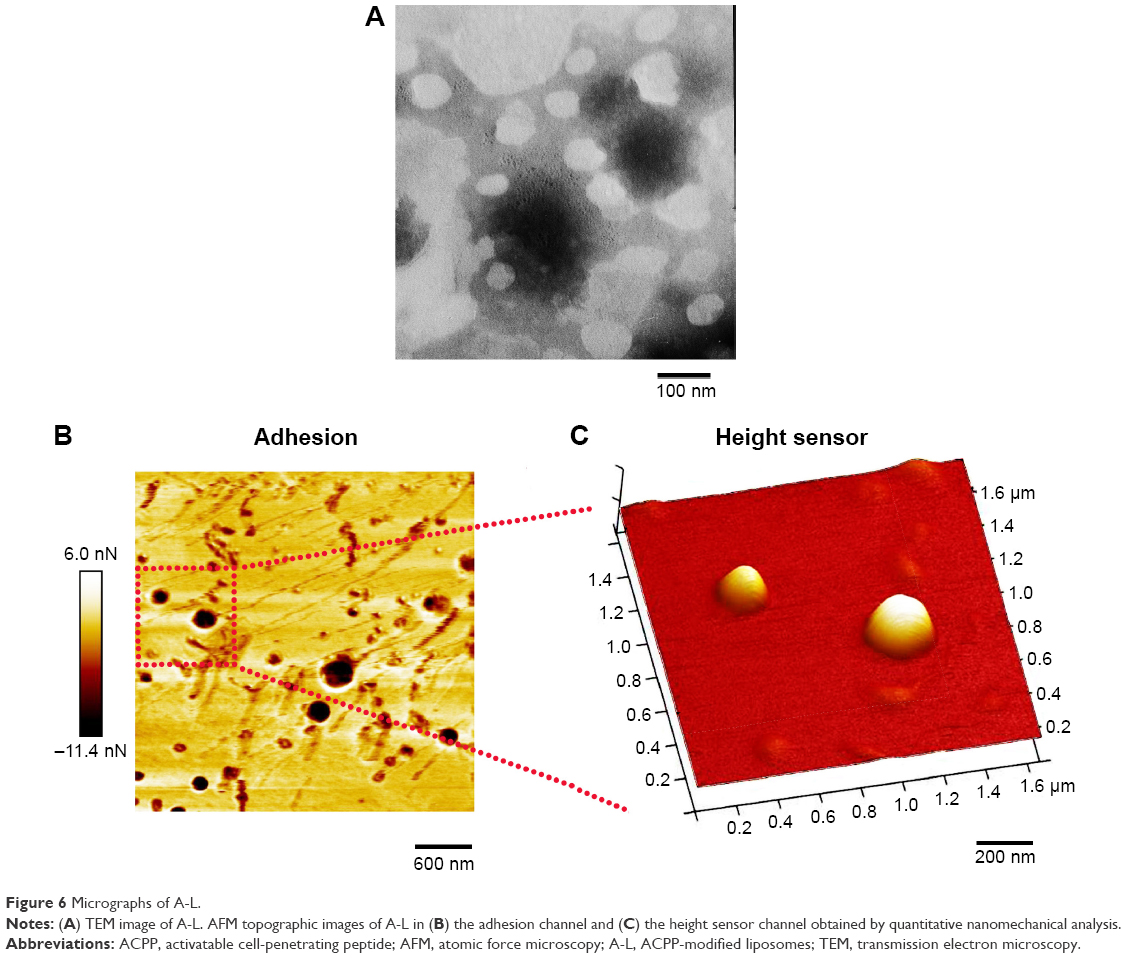 | Figure 6 Micrographs of A-L. |
Cellular internalization
To systematically investigate the effects that the various liposomal formulations might have on the cell uptake of siRNA, we incubated MCF-7 cells with the various liposomes (N-L, C-L, and A-L) at pH 6.2 and 7.4, respectively. The greatest amount of FAM-siRNA entry (Figure 7) was found in the C-L treatment among all groups, indicating the potency of CPP. However, relative to the exposure of C-L at pH 7.4, a significantly decreased mean fluorescence intensity was detected in cells treated with A-L (P<0.0001), which suggested the neutralization of the positive charges on CPP by the shielding domain. In contrast with the result observed at pH 7.4, only the A-L group exhibited a significant promotion in cellular uptake at pH 6.2 (P<0.001), suggesting the intended activation of the inert CPP. The evaluation of liposomal siRNA internalization via CLSM (Figure 8) showed that among all groups cultured at pH 6.2, only administration of A-L significantly enhanced the cell internalization of FAM-siRNA compared with the corresponding degree observed at pH 7.4, which is consistent with the result of the flow cytometry assessment described earlier.
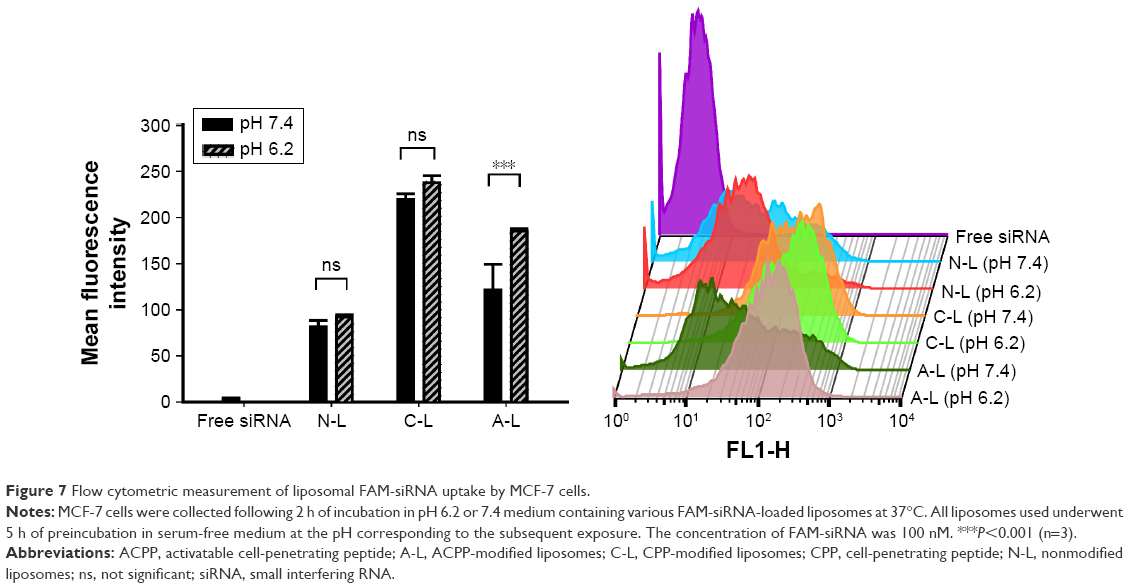 | Figure 7 Flow cytometric measurement of liposomal FAM-siRNA uptake by MCF-7 cells. |
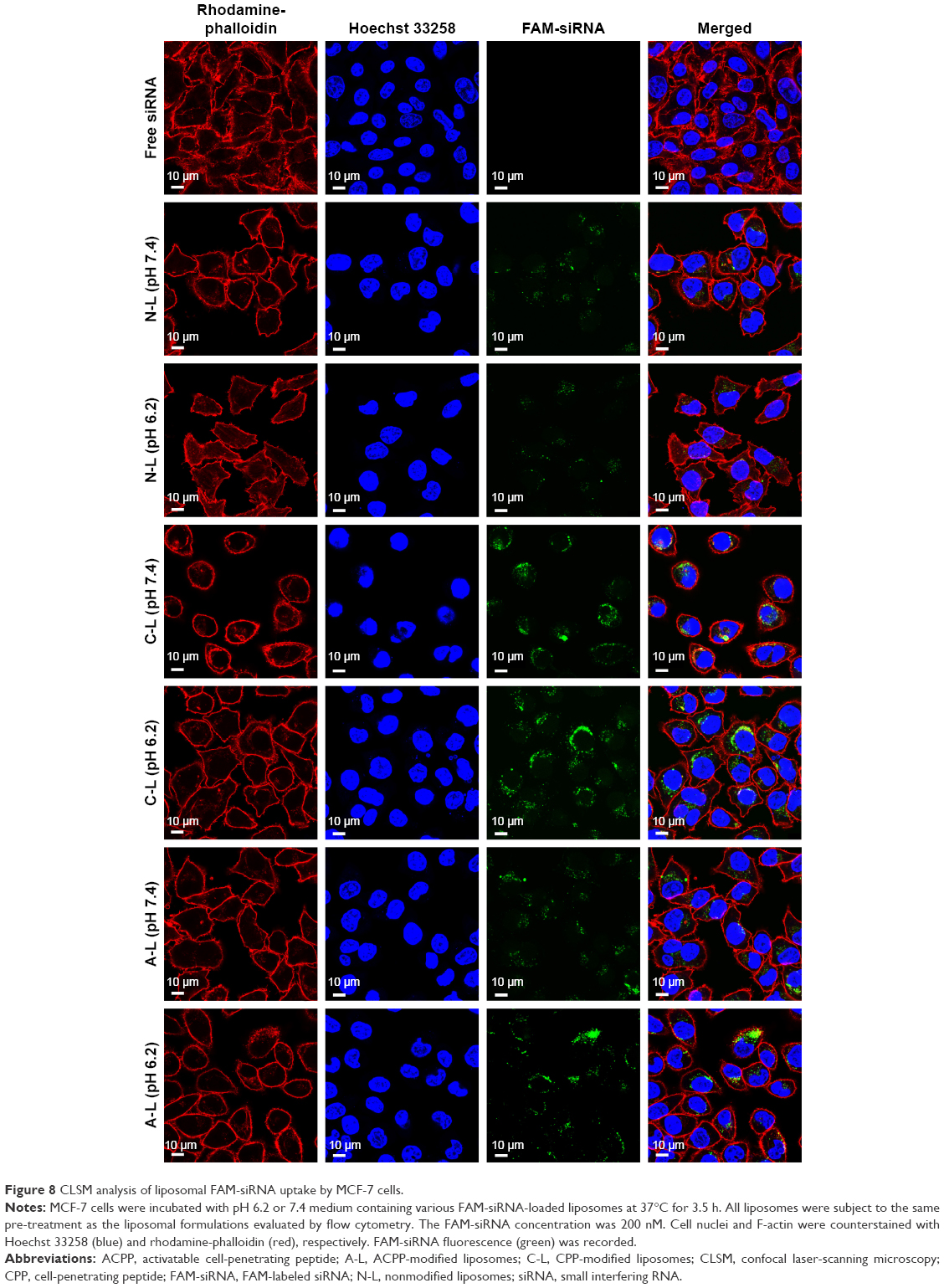 | Figure 8 CLSM analysis of liposomal FAM-siRNA uptake by MCF-7 cells. |
Endosomal/lysosomal escape
After both 2 and 6 h of the routine culture of MCF-7 cells treated with A-L at pH 7.4 (Figure 9A), the intracellular FAM-siRNA (green) was partly colocalized with the CellLight Early Endosomes-RFP-labeled organelles (red), indicating that the liposomal FAM-siRNA entered the MCF-7 cells via endocytosis. In contrast to the image acquired at 2 h of incubation, a longer treatment period (6 h) led to a portion of scattered FAM-siRNA, which suggested the occurrence of endosome/lysosome escape. The intracellular trafficking of siRNA was also investigated under the acidic condition, at pH 6.2. After 2 h of treatment, only a very minor fraction of internalized siRNA was found in the early endosomes. However, following 6 h of incubation, the image showed barely any colocalization of siRNA with the early endosomal marker, and most of the intracellular siRNA was dispersedly distributed. Overall, as the acidity increased from pH 7.4 to pH 6.2, the endosome/lysosome escape of liposomal siRNA was promoted. As exhibited in MCF-7 cells with treatment of N-L (Figure S1) at pH 7.4, the intracellular FAM-siRNA (green) was entrapped in the early endosomes in part and exclusively present in condensed form, suggesting a slight liposomal siRNA avoidance of endosome/lysosome. As the acidity shifted to pH 6.2, no obviously improved escape and dissociation of the internalized FAM-siRNA were observed.
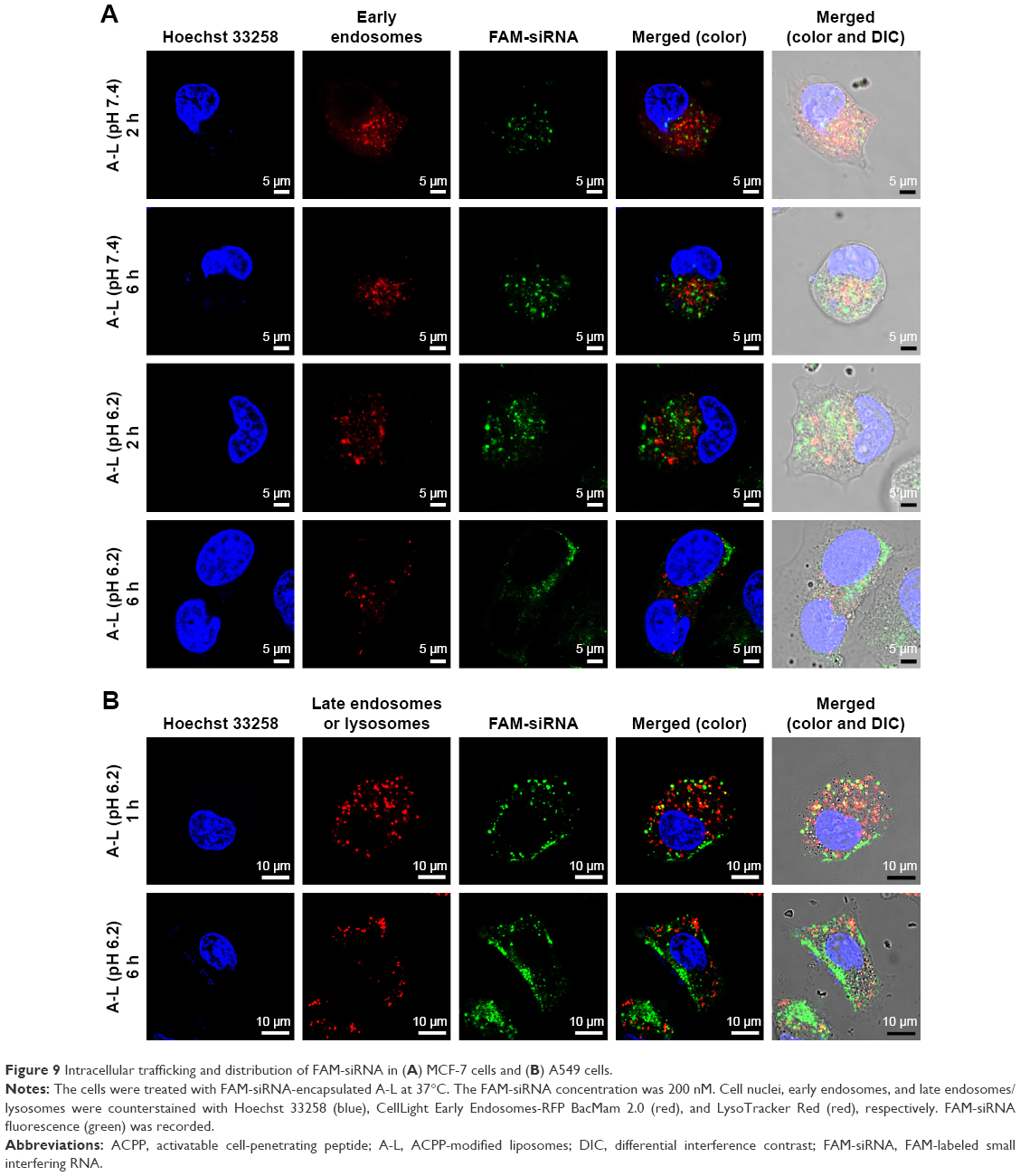 | Figure 9 Intracellular trafficking and distribution of FAM-siRNA in (A) MCF-7 cells and (B) A549 cells. |
By means of the fluorescence staining of late endosomes/lysosomes, the intracellular transportation of siRNA loaded in A-L was also detected in A549 cells. Partially overlapping images between siRNA and late endosomes/lysosomes were observed following 1 h of incubation at pH 6.2 (Figure 9B). However, the subsequent 5 h of exposure showed that only a negligible quantity of siRNA remained colocalized with the LysoTracker Red-labeled compartments, and the internalized siRNA was largely diffused in the cytoplasm (Figure 9B). These data revealed that 6 h of treatment provided more efficient lysosomal avoidance for the liposomal siRNA as well as greater intracellular release compared with the images recorded at the earlier time point (1 h).
Cell apoptosis assay
We next assessed how modifications of the liposomes affected their apoptotic activity and whether the induction of apoptosis by liposomes exhibited pH sensitivity. As shown in Figure 10, the cells exposed to siPLK-loaded C-L presented the highest level of induced apoptosis (~50% of apoptotic cells). In contrast, transfection with siPLK-1-incorporated A-L at pH 7.4 resulted in a significantly decreased percentage (>30%) of apoptotic cells, which indicated that the enhanced induction of cell apoptosis by CPP was notably inhibited due to the conjunction of the shielding peptide. As the acidity increased by the change in pH from 7.4 to 6.2, no obvious change in the apoptotic rate was observed following the exposure to N-L or C-L. However, the induction of cell apoptosis by A-L was significantly promoted (P<0.05), with the percentage of apoptotic cells exceeding 40%, suggesting activation of the shielded CPP.
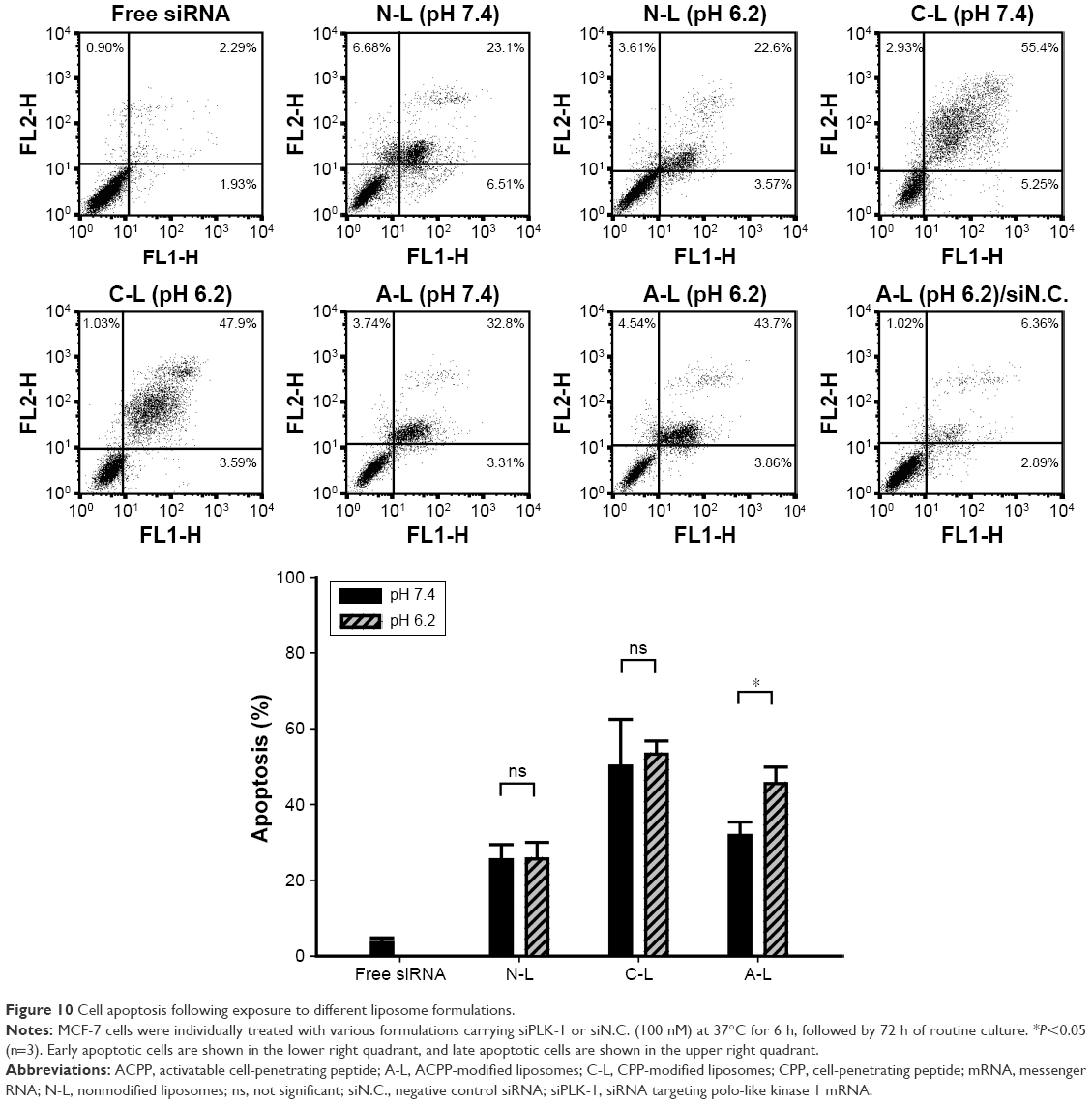 | Figure 10 Cell apoptosis following exposure to different liposome formulations. |
Gene silencing analysis
To investigate whether this induction of cell apoptosis described earlier was due to downregulation of the target gene PLK-1 by the siRNA-loaded liposomes, we performed qRT-PCR and Western blot analyses in MCF-7 cells. As presented in Figure 11A, the delivery of siPLK-1 by the C-L resulted in the lowest PLK-1 mRNA expression level among all treatments, which is similar to the protein expression profile shown in Figure 11B. However, the exposure to siPLK-1-contained A-L at pH 7.4 resulted in much higher levels of PLK-1 mRNA and protein expression in comparison to the treatment with C-L (Figure 11A and B). As the pH was decreased to 6.2, markedly strengthened silencing of PLK-1 mRNA was found in the cells incubated with A-L (P<0.05) (Figure 11A), which was also in agreement with the effects on PLK-1 protein expression (Figure 11B). These results supported that the decrease in PLK-1 mRNA is responsible for the downregulated expression of PLK-1 protein in the MCF-7 cells and the consequent cell apoptosis.
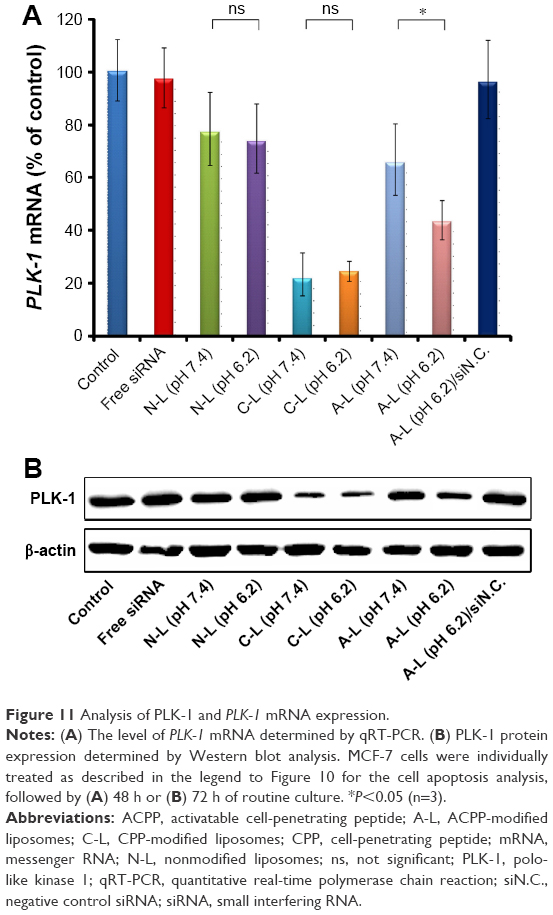 | Figure 11 Analysis of PLK-1 and PLK-1 mRNA expression. |
Discussion
Previous studies from our own and other laboratories have demonstrated that cell internalization of delivered cargo could be significantly enhanced by mediation of polyarginine-based CPP33–35 and the promoted uptake suffered from marked inhibition after the conjugation of a negatively charged peptide (EGG)4.13,14 Based on this background, a new type of pH-sensitive ACPP was designed and developed, in which octaarginine is connected to a modified shielding peptide, (ehG)4, via a hydrazone bond. The present CPP-incorporated peptide is the first hydrazone-based ACPP structure reported. Using a liposomal carrier, we constructed an ACPP-modified nano-sized vehicle for delivering siRNA into tumor cells. Use of this ACPP-guided delivery system possesses several advantages for applications in targeted RNAi-based anticancer therapy. First, the reported activation of ACPPs based on electrostatic inhibition was exclusively induced by proteolysis,15 which is associated with a considerable challenge given the inherent heterogeneous expression of the targeted proteases. In the present strategy, the creative incorporation of an acid-triggered cleavability property of the ACPPs provides more effective and universal selectivity to the tumor microenvironment. Second, our group has previously utilized two types of anionic peptides (DGG)n11,12 and (EGG)n,13,14 both of which resulted in a noticeable electrostatic shielding effect on oligoarginine. However, the electrostatic interaction between the guanidiniums in arginine and the negatively charged groups in the shielding peptides might lead to unsatisfactory detachment of the shielding domain after the splitting of the enzyme-cleavable linker, thereby reducing the functional recovery of CPP. From this perspective, we here performed partial substitution of the glycines in the shielding peptides with histidines to fabricate an acid-labile ACPP. With a pKa of ~6.5, the histidine residues obtain a net positive charge by the imidazole ring protonation in the acidic tumor environment,36,37 which could greatly decrease the total negative charges in the shielding domain and eventually be conducive to the effective dissociation of these two oppositely charged fractions in ACPP. Third, according to our previous investigation12 and other reports,38 oligoarginine peptides can acquire the ability of efficient endosome escape after trafficking into a tumor cell, owing to the ion-pair effect. Consequently, this polycationic peptide facilitated the cytoplasm distribution of siRNA, which lays the foundation for subsequent targeted posttranscriptional gene silencing.
Once subjected to lysosomal trapping after getting into the cell, the delivered siRNA would undergo degradation from the activities of the inner enzymes.39 Accordingly, effective endosomal/lysosomal escape of these small oligonucleotides is essential for accurate posttranscriptional gene silencing in the cytoplasm. For the A-L that was pretreated under the acidic condition (pH 6.2), subcellular localization in the MCF-7 cells was analyzed after staining with LysoTracker Red, a marker for late endosomes and lysosomes. Consequently, the signal of the internalized siRNA did not overlap with that of the late endosomes/lysosomes at either 2 or 6 h but was instead seemingly located in the cytoplasm at these two time points (data not shown). To further clarify the intracellular trafficking of the cargo, the superposition of siRNA with the early endosome marker was examined. The colocalization of siRNA with early endosomes, which was found only at the early time point (2 h), suggested that a portion of the liposomal particles would enter cells through an endocytic mode that triggers the formation of endosome and that the loaded siRNA efficiently escaped from the endosomal vesicles.
Upon the exposure of octaarginine anchored on to the liposome surface, there are probably two major mechanisms by which the liposomal siRNA can avoid the lysosome to reach the cytosol. As a membrane-disruptive peptide, oligoarginine has been reported to trigger the endosomal release of the attached cargo. The charge–charge interaction between multiple cationic guanidiniums and anionic groups in the lipid components of the endosomal membrane is conducive to both the ion-pair formation during acidification of the endosome and the following destabilization of the endosomal membrane due to exclusion of the surface-bound water.38 Besides the ion-pair effect, the ability of oligoarginines such as octaarginine (R8) to avoid the lysosome may also be ascribed to adjustment of the uptake mechanism to a more efficient pathway (ie, macropinocytosis34,40), so as to favorably control the subsequent intracellular trafficking of siRNA-loaded particles. As reported by Zhou and Chau,41 modification with tetraarginines (R4) allowed for the delivered particles to be taken up via lipid raft-dependent endocytosis, whereas R8-modified particles were internalized by different modes, including macropinocytosis. Following entry to the cell, only those particles modified by R8 showed the capacity to escape from endosomes/lysosomes to enter the cytosol, whereas the R4-incorporated carriers did not. Another investigation of the cell uptake of R8-modified liposomes revealed that macropinocytosis was also involved in the internalization of R8-attached cargo, and this type of route could be less susceptible to lysosomal degradation compared with entry routes involving clathrin-mediated endocytosis.42 In this study, the siRNA was not colocalized with the late endosomes/lysosomes of MCF-7 cells, and only a small fraction of the cargo showed overlapping with the early endosomes at the early stage (2 h) (Figure 9A). These facts suggest that a portion of intracellular escape would occur as part of an entry mode that circumvents both the early endosome and the lysosomal compartment. In light of the two studies mentioned earlier, it can be inferred that the present carriers may possibly be taken up by a mechanism involving nonspecific macropinocytosis, an internalization process that does not rely on the formation of the early endosome.43 In this manner, the intracellular escape of cargo is facilitated and efficient owing to the leaky macropinosomes formed.5 The proposed mechanism for siRNA-loaded A-L transportation across the cellular membrane and escape into cytoplasm was illustrated in Figure 12.
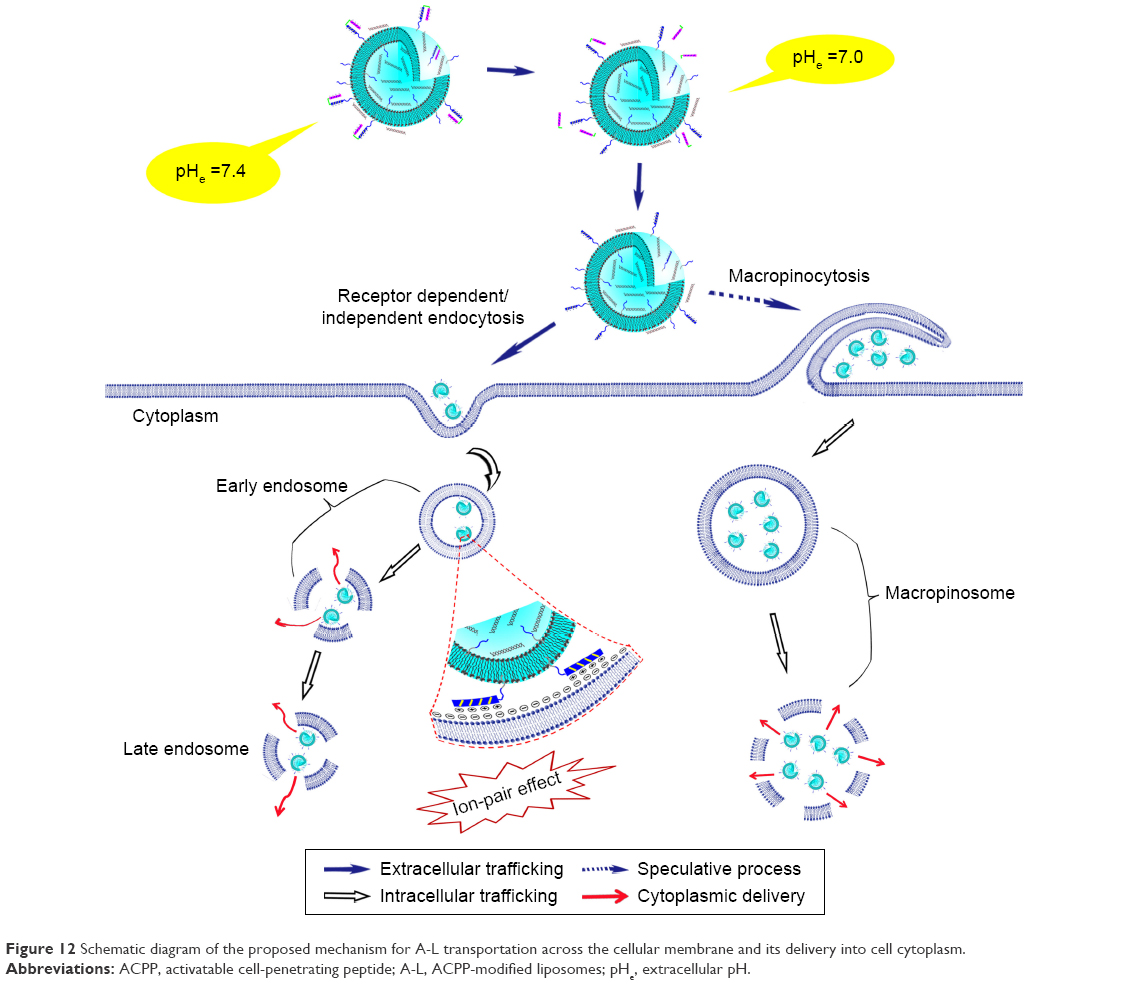 | Figure 12 Schematic diagram of the proposed mechanism for A-L transportation across the cellular membrane and its delivery into cell cytoplasm. |
In summary, in this study, we developed a novel type of hydrazone-based delivery system to enhance the selectivity of CPP toward the acidic tumor microenvironment. When pretreated under an acidic condition (pH 6.2), this siRNA-loaded liposomal system resulted in a significantly elevated level of cellular uptake (Figures 7 and 8) and apoptosis (Figure 10) in vitro compared with those incubated at pH 7.4. These data were consistent with the significant changes in PLK-1 expression (Figure 11) under the same pH condition, reflecting the expected response of the liposomal carriers to the acidic pH in tumor microregions. However, there are still some limitations to overcome with this siRNA-delivered system. The results of cell uptake, apoptosis, and gene expression analyses under the acidic condition for the A-L group were not comparable to those of the CPP-modified liposome group. This discrepancy indicates that the potency of the original CPP could not be fully activated under the present test conditions, which might be due to the insufficient cleavage of the hydrazone linkage in A-L. Further research is required to identify and select other acid-labile linkers such as 2,3-dimethylmaleic amide44,45 with higher sensitivity to the mildly acidic pHe of the tumor microenvironment (pH 6.5–6.817).
Conclusion
Herein, we designed and constructed a novel type of acidic pH-sensitive ACPP to improve the selectivity of CPP toward the tumor microenvironment. Following the synthesis of DSPE-PEG2000-ACPP, an acid-responsive and ACPP-mediated liposomal system (A-L) was developed with surface modification of this ACPP-incorporated conjugate. In contrast to siRNA-encapsulated control liposomes (non-modified and CPP-modified), such efficient ACPP-decorated liposomes presented a noticeable response to an acidic pH, with significantly improved uptake, downregulated PLK-1 expression, and reinforced apoptosis in MCF-7 cells. Furthermore, investigation of the intracellular behavior of A-L demonstrated the favorable avoidance of the lysosome and effective release in the cytoplasm. In short, the present ACPP-modified liposome is a potential delivery system for siRNA-based cancer therapy.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No 81302725, No 21402039, No 81473156, and No 81673365), the National Science and Technology Major Projects of New Drugs (No 2014ZX09507001), and the Natural Science Foundation of Hebei Province, China (No H2015206356).
Disclosure
The authors report no conflicts of interest in this work.
References
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. | ||
Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. 2016;85:152–167. | ||
Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14(12):843–856. | ||
Singh Y, Tomar S, Khan S, et al. Bridging small interfering RNA with giant therapeutic outcomes using nanometric liposomes. J Control Release. 2015;220(pt A):368–387. | ||
Xia Y, Tian J, Chen X. Effect of surface properties on liposomal siRNA delivery. Biomaterials. 2016;79:56–68. | ||
Jhaveri A, Torchilin V. Intracellular delivery of nanocarriers and targeting to subcellular organelles. Expert Opin Drug Deliv. 2016;13(1):49–70. | ||
Zhang D, Wang J, Xu D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J Control Release. 2016;229:130–139. | ||
Tai W, Gao X. Functional peptides for siRNA delivery. Adv Drug Deliv Rev. Epub 2016 Aug 13. | ||
Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101(51):17867–17872. | ||
Nguyen QT, Tsien RY. Fluorescence-guided surgery with live molecular navigation – a new cutting edge. Nat Rev Cancer. 2013;13(9):653–662. | ||
Shi NQ, Gao W, Xiang B, Qi XR. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int J Nanomedicine. 2012;7:1613–1621. | ||
Xiang B, Dong DW, Shi NQ, et al. PSA-responsive and PSMA-mediated multifunctional liposomes for targeted therapy of prostate cancer. Biomaterials. 2013;34(28):6976–6991. | ||
Gao W, Xiang B, Meng TT, Liu F, Qi XR. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34(16):4137–4149. | ||
Gao W, Meng T, Shi N, Zhuang H, Yang Z, Qi X. Targeting and microenvironment-responsive lipid nanocarrier for the enhancement of tumor cell recognition and therapeutic efficiency. Adv Healthc Mater. 2015;4(5):748–759. | ||
He H, Sun L, Ye J, et al. Enzyme-triggered, cell penetrating peptide-mediated delivery of anti-tumor agents. J Control Release. 2016;240:67–76. | ||
Zhang B, Zhang Y, Liao Z, et al. UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials. 2015;36:98–109. | ||
Du J, Lane LA, Nie S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J Control Release. 2015;219:205–214. | ||
Jhaveri A, Deshpande P, Torchilin V. Stimuli-sensitive nanopreparations for combination cancer therapy. J Control Release. 2014;190:352–370. | ||
Karimi M, Ghasemi A, Sahandi Zangabad P, et al. Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem Soc Rev. 2016;45(5):1457–1501. | ||
Sawant RM, Hurley JP, Salmaso S, et al. “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17(4):943–949. | ||
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160(2):264–273. | ||
Apte A, Koren E, Koshkaryev A, Torchilin VP. Doxorubicin in TAT peptide-modified multifunctional immunoliposomes demonstrates increased activity against both drug-sensitive and drug-resistant ovarian cancer models. Cancer Biol Ther. 2014;15(1):69–80. | ||
Ding Y, Sun D, Wang GL, et al. An efficient PEGylated liposomal nanocarrier containing cell-penetrating peptide and pH-sensitive hydrazone bond for enhancing tumor-targeted drug delivery. Int J Nanomedicine. 2015;10:6199–6214. | ||
Han L, Guo Y, Ma H, et al. Acid active receptor-specific peptide ligand for in vivo tumor-targeted delivery. Small. 2013;9(21):3647–3658. | ||
Zhang Q, Gao H, He Q. Taming cell penetrating peptides: never too old to teach old dogs new tricks. Mol Pharm. 2015;12(9):3105–3118. | ||
Zhang W, Song J, Zhang B, Liu L, Wang K, Wang R. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconjug Chem. 2011;22(7):1410–1415. | ||
Zhang Y, Rong Qi X, Gao Y, Wei L, Maitani Y, Nagai T. Mechanisms of co-modified liver-targeting liposomes as gene delivery carriers based on cellular uptake and antigens inhibition effect. J Control Release. 2007;117(2):281–290. | ||
Leamon CP, Reddy JA, Vlahov IR, Kleindl PJ, Vetzel M, Westrick E. Synthesis and biological evaluation of EC140: a novel folate-targeted vinca alkaloid conjugate. Bioconjug Chem. 2006;17(5):1226–1232. | ||
Dong DW, Xiang B, Gao W, Yang ZZ, Li JQ, Qi XR. pH-responsive complexes using prefunctionalized polymers for synchronous delivery of doxorubicin and siRNA to cancer cells. Biomaterials. 2013;34(20):4849–4859. | ||
Liang DS, Su HT, Liu YJ, Wang AT, Qi XR. Tumor-specific penetrating peptides-functionalized hyaluronic acid-d-alpha-tocopheryl succinate based nanoparticles for multi-task delivery to invasive cancers. Biomaterials. 2015;71:11–23. | ||
Derjaguin BV, Muller VM, Toporov YP. Effect of contact deformations on the adhesion of particles. J Colloid Interface Sci. 1975;53(2):314–326. | ||
Christie RJ, Anderson DJ, Grainger DW. Comparison of hydrazone heterobifunctional cross-linking agents for reversible conjugation of thiol-containing chemistry. Bioconjug Chem. 2010;21(10):1779–1787. | ||
Ma DX, Shi NQ, Qi XR. Distinct transduction modes of arginine-rich cell-penetrating peptides for cargo delivery into tumor cells. Int J Pharm. 2011;419(1–2):200–208. | ||
Komin A, Russell LM, Hristova KA, Searson PC. Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: mechanisms and challenges. Adv Drug Deliv Rev. Epub 2016 Jun 13. | ||
Jeong EJ, Choi M, Lee J, Rhim T, Lee KY. The spacer arm length in cell-penetrating peptides influences chitosan/siRNA nanoparticle delivery for pulmonary inflammation treatment. Nanoscale. 2015;7(47):20095–20104. | ||
Oude Blenke EE, van den Dikkenberg J, van Kolck B, Kros A, Mastrobattista E. Coiled coil interactions for the targeting of liposomes for nucleic acid delivery. Nanoscale. 2016;8(16):8955–8965. | ||
Zaro JL, Fei L, Shen WC. Recombinant peptide constructs for targeted cell penetrating peptide-mediated delivery. J Control Release. 2012;158(3):357–361. | ||
Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61(9):721–731. | ||
Shi NQ, Qi XR, Xiang B, Zhang Y. A survey on “Trojan Horse” peptides: opportunities, issues and controlled entry to “Troy”. J Control Release. 2014;194:53–70. | ||
Gilleron J, Querbes W, Zeigerer A, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638–646. | ||
Zhou J, Chau Y. Different oligoarginine modifications alter endocytic pathways and subcellular trafficking of polymeric nanoparticles. Biomater Sci. 2016;4(10):1462–1472. | ||
Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281(6):3544–3551. | ||
Zhang J, Liu D, Zhang M, et al. The cellular uptake mechanism, intracellular transportation, and exocytosis of polyamidoamine dendrimers in multidrug-resistant breast cancer cells. Int J Nanomedicine. 2016;11:3677–3690. | ||
Du JZ, Sun TM, Song WJ, Wu J, Wang J. A tumor-acidity-activated charge-conversional nanogel as an intelligent vehicle for promoted tumoral-cell uptake and drug delivery. Angew Chem Int Ed Engl. 2010;49(21):3621–3626. | ||
Du JZ, Du XJ, Mao CQ, Wang J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J Am Chem Soc. 2011;133(44):17560–17563. |
Supplementary materials
Intracellular trafficking behavior of FAM-siRNA delivered by N-L
MCF-7 cells were seeded in a sterile glass-bottomed dish (35×10 mm) at a density of 6×104 cells per well and cultured in complete RPMI 1640 medium at pH 6.2 and 7.4, respectively. To stain the early endosomes of the cells, the MCF-7 cells were preincubated with CellLight Early Endosomes-RFP BacMam 2.0 (20 particles per cell, as recommended by the supplier) for 24 h at 37°C. The following day, the cells were incubated with pH 6.2 or 7.4 medium containing FAM-siRNA-loaded N-L, which was subjected to 5 h of preincubation in serum-free medium at the corresponding pH. The exposed concentration of FAM-siRNA was 350 nM. Subsequently, the cells were rinsed three times with cold PBS containing heparin (500 U/mL) and then treated for fixation and nuclear staining.
Confocal microscopic images were acquired using a Leica confocal platform with a 63× oil objective with an excitation wavelength of 494 nm for FAM-siRNA, 555 nm for CellLight Early Endosomes-RFP BacMam 2.0, and 352 nm for Hoechst 33258.
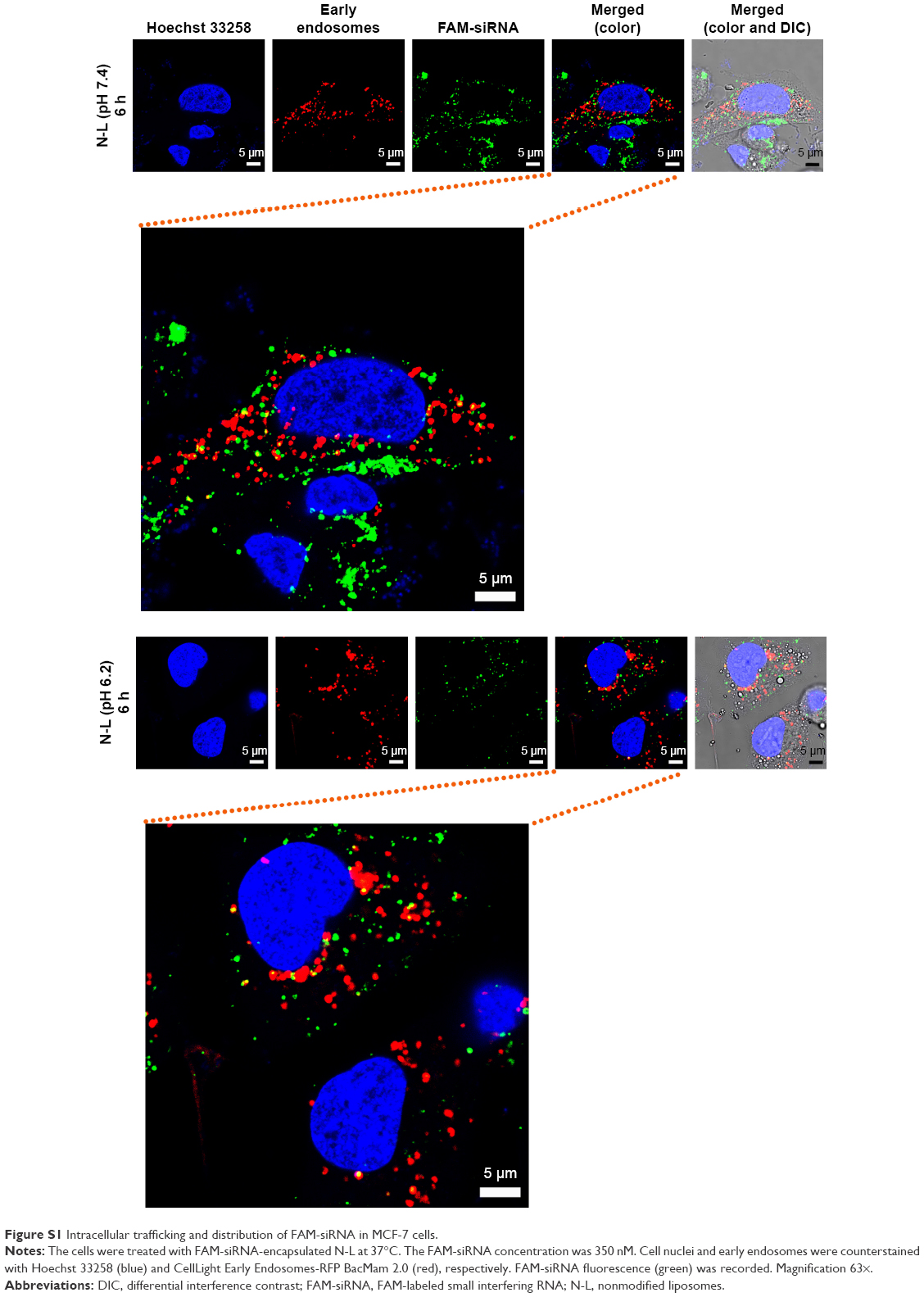 | Figure S1 Intracellular trafficking and distribution of FAM-siRNA in MCF-7 cells. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.