Back to Journals » OncoTargets and Therapy » Volume 10
Enhancement of antitumor activity by using a fully human gene encoding a single-chain fragmented antibody specific for carcinoembryonic antigen
Authors Shibaguchi H, Luo N ![]() , Shirasu N, Kuroki M, Kuroki M
, Shirasu N, Kuroki M, Kuroki M
Received 21 April 2017
Accepted for publication 9 June 2017
Published 22 August 2017 Volume 2017:10 Pages 3979—3990
DOI https://doi.org/10.2147/OTT.S140174
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chiung-Kuei Huang
Hirotomo Shibaguchi,1,* Naixiang Luo,1,* Naoto Shirasu,1,* Motomu Kuroki,2 Masahide Kuroki1
1Department of Biochemistry, Faculty of Medicine, Fukuoka University, Fukuoka, Japan; 2School of Nursing, Faculty of Medicine, Fukuoka University, Fukuoka, Japan
*These authors equally contributed to this work
Abstract: Human leukocyte antigen and/or costimulatory molecules are frequently lacking in metastatic tumor cells, and thus tumor cells are able to escape from the immune system. Although lymphocytes with a chimeric antigen receptor (CAR) is a promising approach for overcoming this challenge in cancer immunotherapy, administration of modified T cells alone often demonstrates little efficacy in patients. Therefore, in order to enhance the antitumor activity of immune cells in the cancer microenvironment, we used lymphocytes expressing CAR in combination with a fusion protein of IL-2 that contained the single-chain fragmented antibody (scFv) specific for the carcinoembryonic antigen. Among a series of CAR constructs, with or without a spacer and the intracellular domain of CD28, the CAR construct containing CD8α, CD28, and CD3ζ most effectively activated and expressed INF-γ in CAR-bearing T cells. Furthermore, in comparison with free IL-2, the combination of peripheral blood mononuclear cells expressing CAR and the fusion protein containing IL-2 significantly enhanced the antitumor activity against MKN-45 cells, a human gastric cancer cell line. In conclusion, this novel combination therapy of CAR and a fusion protein consisting of a functional cytokine and a fully human scFv may be a promising approach for adoptive cancer immunotherapy.
Keywords: chimeric antigen receptor, fusion protein, human scFv, CEA, combination therapy
Introduction
In addition to the 3 primary treatment options for cancer, viz., surgery, radiotherapy, and chemotherapy, cell-based cancer immunotherapy has become a promising approach in recent years.1–4 In clinical studies, it has been reported that this treatment was efficacious in some cases, as indicated by the complete response, whereas, in other cases, the lower response rate, at least in part, due to loss/dysfunction of human leukocyte antigen (HLA) on cancer cells and/or immune suppression in the microenvironment of the tumor mass, remains an issue to be overcome.5–7 As a strategy for addressing non-HLA-restriction, cytotoxic T lymphocytes (CTL) with a chimeric antigen receptor (CAR) that has an antibody-based cellular arm for antigen recognition and the intracellular domain of a T cell receptor or Fc receptor, for cell activation, is a promising approach to cancer gene therapy. To date, many tumor-associated antigens (TAAs) have been used as a target of CAR in preclinical and clinical investigations, and the results have indicated the usefulness of CAR in cancer therapy.1,8,9
Although aspects of the gene construction of CAR, for example, the importance and length of the spacer between the cellular arm and the intracellular domain and the type of immunoreceptor tyrosine-based activating motif (ITAM) used, are debated, it is thought that the CD3ζ chain is most effective at signaling as an ITAM in CAR.10 Furthermore, it has also been reported that the existence of a costimulatory signaling domain in CAR may be important, especially for long-lasting T-cell activation in vivo.11–13 The expected effect was, however, not achieved in adoptive cancer therapy when CAR-bearing CTLs were used alone. However, recent gene therapy studies using CAR have suggested that the combination of Th2 cells expressing CAR and CAR-bearing CTLs effectively rejects a tumor mass in the mouse xenograft model and that natural killer (NK) cells expressing CAR also demonstrated a significant antitumor effect equivalent to that of CAR-bearing CTLs.14–19 Furthermore, in order to maintain a Th1-dominated immune balance in the cancer microenvironment, the antibody that provides or blocks immune signaling by binding to the target (eg, OX40, 4-1BB, or CTLA-4) helps to control the immune balance in adoptive immunotherapy.20–22 A novel strategy for integrating and enhancing antitumor activity to effect more efficient and continuous cancer immunotherapy might be as follows: 1) the use of other activation signals, such as cytokine/chemokine signals, in addition to signals 1 and 2; and 2) the use of other immune cells, such as neutrophil or NK cells, in addition to Th1 and Th2 T cells.
Previously, we have generated mouse and human antibodies specific for carcinoembryonic antigen (CEA), followed by a single-chain fragmented antibody (scFv), to investigate their usefulness in cell-based cancer immunotherapy.23–29 In the present study, we constructed a series of CAR genes with or without a spacer/hinge and a CD28 intracellular domain, in addition to CD3ζ, by using the mouse scFv gene, and investigated the efficacy of these in the activation of CAR transfectants. Subsequently, a fully human CAR gene was generated, based on the construct that provided the best results, by exchanging the mouse scFv gene for its human equivalent. Furthermore, a construct encoding a fusion protein of human scFv and human IL-2 (hIL-2), designated scFv-IL2, was also generated, and its usefulness in combination cancer therapy with CAR-bearing peripheral blood mononuclear cells (PBMCs) was investigated.
Materials and methods
Reagents
CEA or bovine serum albumin (BSA) was labeled using the allophycocyanin (APC) labeling kit-NH2 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s protocol. Mouse anti-IFN-γ-PE and anti-hIL-2-fluorescein isothiocyanate (FITC) monoclonal antibodies for flow cytometry were purchased from Miltenyi Biotech Inc. (Gladbach, Germany). Mouse anti-His and anti-hIL-2 antibodies and rabbit anti-c-myc and anti-hIL-2 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). All other reagents were commercial cell culture grade or analytical grade products.
Cancer cell lines
The acute human T cell leukemia line, Jurkat clone E6.1, and the murine cytotoxic T cell line, CTLL-2, from American Type Culture Collection (ATCC; Manassas, VA, USA) were maintained in RPMI-1640 culture medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) (JRH Bioscience, Lenexa, KS, USA), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Sigma-Aldrich). The human gastric cancer cell line, MKN-45, with or without the luciferase gene, from Cell Bank (Riken BioResource Center, Ibaraki, Japan), was grown in DMEM (Sigma-Aldrich) with supplements, as described above. The MKN-45 cells were detached and harvested after treatment with trypsin-EDTA/PBS (Sigma-Aldrich) for 5 min. The cells were maintained at 37°C in humidified 5% CO2/air, and the culture medium was changed twice a week.
Gene construction
The human genes encoding CD7 and hIL-2 were obtained from RIKEN gene bank. The CAR genes were cloned into pcDNA3.1(−) or pIRES2-EGFP, the mammalian expression vector (Thermo Fisher Scientific, Waltham, MA, USA; Figure 1A). The series of CAR genes with CD3ζ and with or without CD7/CD8α as a spacer/hinge, or CD28 as a secondary signal, were designated mCR-0, mCR-1, mCR-2, and mCR-3, respectively, as shown in Figure 1B. The cloning of mCR-2 and hCR-2 genes into pcDNA3.1(−) has been described previously (F39scFv/CIR-2 and L45scFv-CIR, respectively).23,26 The hCR-2 in pIRES-EGFP was generated by digesting mCR-2/pcDNA3.1(−) with BglII/BamHI. The purified fragment was then cloned into the pIRES-EGFP vector. The antibody-cytokine fusion protein, scFv-IL2, was constructed by splice-overlap extension (SOE)-polymerase chain reaction (PCR) using appropriate primers (Figure 2A and B and Table 1). The purified fragment was digested with NheI/HindIII and cloned into the pBAD/gIII Escherichia coli expression vector. The integrity of all plasmid constructs was confirmed with restriction digestion and DNA sequencing.
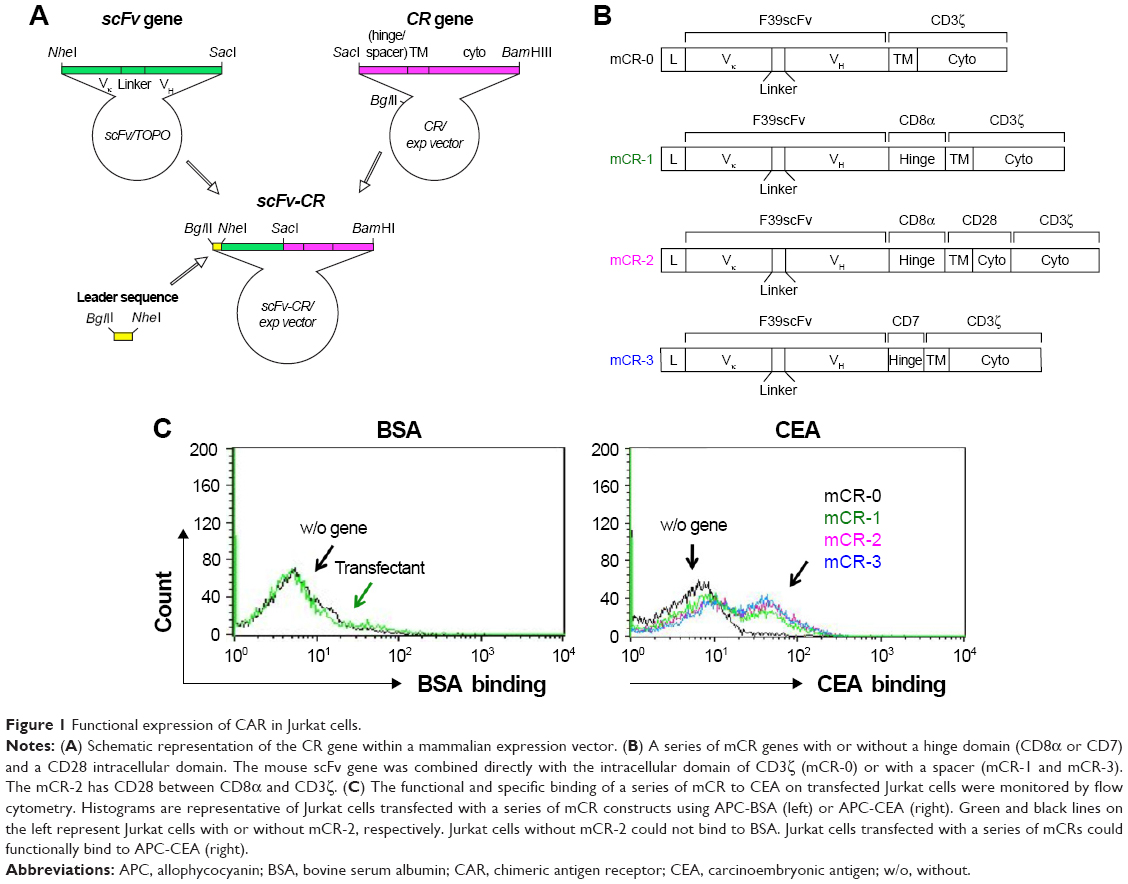 | Figure 1 Functional expression of CAR in Jurkat cells. |
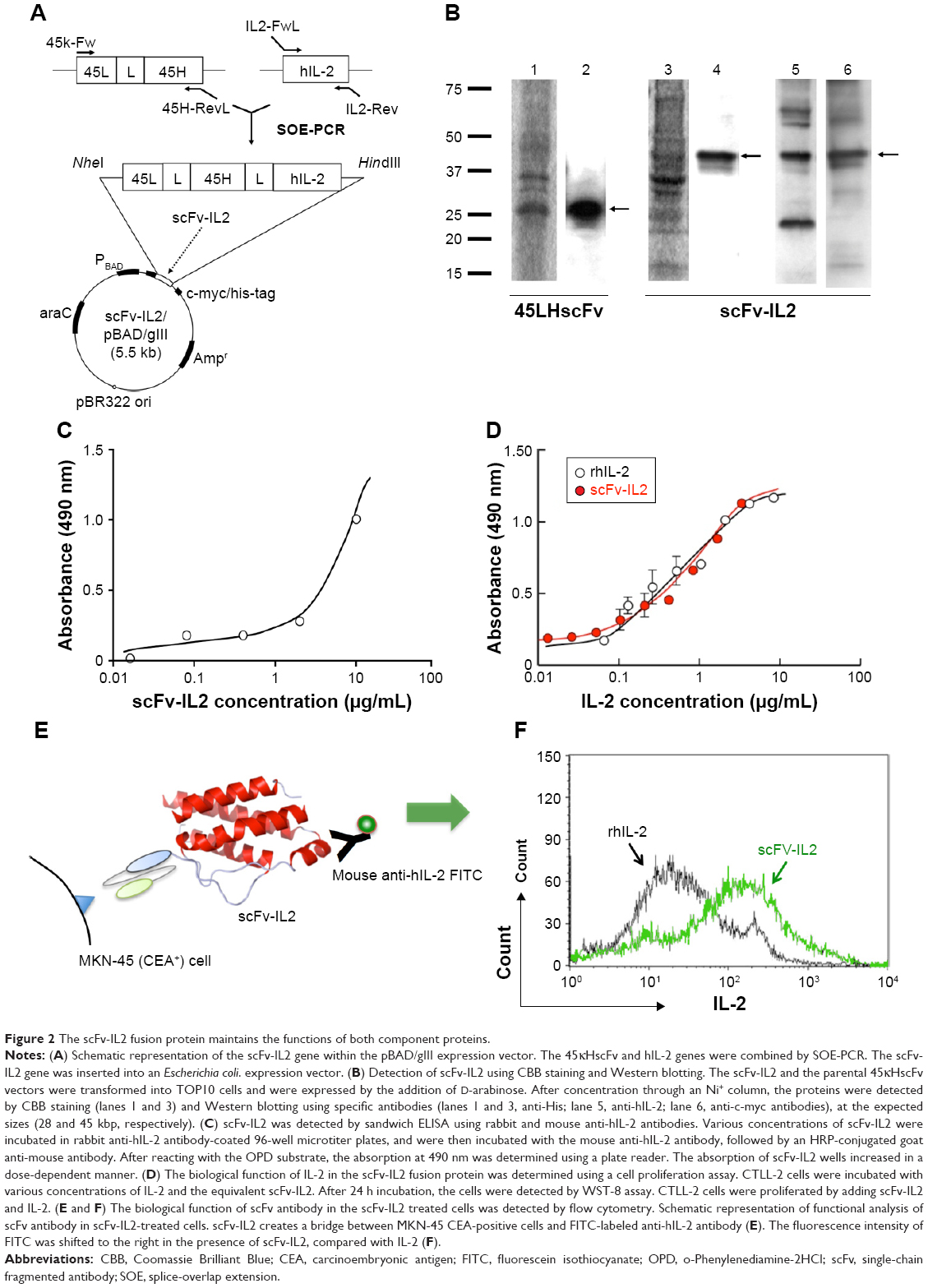 | Figure 2 The scFv-IL2 fusion protein maintains the functions of both component proteins. |
 | Table 1 The sequences of specific primers used for SOE-PCR of scFv-IL2 |
Lymphocyte isolation and T cell expansion
PBMCs were obtained from healthy volunteers after obtaining informed consent in accordance with the Helsinki Declaration of 1975 and was approved by the Institutional Review Board of Fukuoka University. The specimens were obtained using LSM® (Cappel, Aurora, OH, USA) according to the manufacturer’s protocol. After a 2 h incubation in a 10 cm culture dish, the floating cells were transferred to a new culture dish containing RPMI-1640 culture medium and the human T cell activation/expansion kit (Miltenyi Biotec). After 2 d incubation, 20 IU/mL of hIL-2 was added to the culture medium and the cells were fed twice a week (Figure S1A).
Gene transfection
Jurkat or expanded T cells (1×106) were transfected with 6 μg/mL CAR gene construct or pmaxGFP, using the Amaxa Cell Line Nucleofection kit R with program A-23, or the Amaxa Human T Cell Nucleofection kit with program U-14 (Amaxa, Lonza, Switzerland), respectively. Moreover, 1×106 expanded T cells and PBMCs were transfected with 10 μg/mL CAR gene construct or pIRES-EGFP using a NEPA21 electroporator (NepaGene, Chiba, Japan). After gene transfection, the cells were immediately placed into RPMI-1640 culture media, and were incubated for 24 h before investigating cell viability, CAR expression on the cellular surface, or the antitumor effect.
Flow cytometry
CAR expression on the cells and CEA recognition after gene transfection were detected by flow cytometry using APC-labeled CEA or BSA. APC-labeled proteins (10 μg) were mixed with 5×105 of transfected cells and were stained for 1 h on ice for specific binding to CAR. Alternatively, CAR expression on PBMCs was confirmed by detecting EGFP using the pIRES-EGFP vector. For intracellular staining of IFN-γ, the transfected T cells were incubated for 24 h with MKN-45 cells and were collected by washing with PBS. The collected T cells were then permeabilized using Inside Fix (Miltenyi Biotech Inc.) and stained with anti-IFN-γ-PE according to the manufacturer’s recommendations. For specific binding of scFv-IL2 to CEA, MKN-45 cells were stained with anti-hIL-2-FITC antibody by ensuring bridging between cells and the antibody with 1 μg/mL of scFv-IL2 or IL-2. After washing twice with PBS containing 0.5% BSA, the fluorescence intensity of the stained cells was assessed using a FACSCalibur (BD Biosciences, San Jose, CA, USA) and was analyzed using Cell Quest Pro (BD Biosciences).
Microscopic analysis
To investigate the CEA recognition and binding by CAR on the cellular surface of primary T cell transfectants, the cell interaction between CAR-bearing T cells and CEA+ cancer cells was observed using microscopy. One day before observation, a mock or mCR gene was transfected into the primary T cells, and 1×105 MKN-45 cells were cultured in a 35 mm diameter dish for 16 h under normal culture conditions. Of these transfected cells, 1×106 were then added into the dish, and were co-cultured for an additional 2 h. Before observing cell–cell interaction, cells were washed twice, gently, with PBS.
Immunoblotting
To confirm the expression of scFv-IL2 in E. coli, the purified fusion protein was analyzed by Western blotting. After adding 0.02% of L-arabinose in Luria–Bertani (LB) medium, TOP10 cells transfected with the scFv-IL2/pBAD vector were cultured for approximately 24 h at 37°C to express scFv-IL2. After collecting the supernatant of the culture medium, 6× His-tagged scFv-IL2 was purified by AKTAprime™ (GE Healthcare Japan, Tokyo, Japan) using an Ni2+ column. The purified scFv-IL2 was analyzed by SDS-PAGE, followed by Western blotting using anti-His, anti-c-myc, and anti-hIL-2 antibodies.
Cell viability
After electroporation, the cell viability of transfected cells was detected using a Countess® Automated Cell Counter (Thermo Fisher Scientific) using trypan blue staining, according to the manufacturer’s instructions. Cell viability was calculated as a percentage of the number of living cells versus the total number of cells (living cells and dead cells). In addition, the antitumor effect of the CAR-bearing PBMCs in combination with scFv-IL2 was evaluated by measuring the intensity of light produced by MKN-45 cells expressing luciferase, using a luciferase assay system (Promega, Madison, WI, USA) according to the manufacturer’s protocol. MKN-45 cells (1×104) were co-cultured with 5×104 of PBMCs transfected with the hCR-2 gene construct or PBMCs at an effector to target cell ratio (E:T) of 5:1 in the presence of scFv-IL2 or IL-2, for 24 h. After washing with PBS, 20 μL of lysis reagent was added to each well and, subsequently, the light produced was measured using a MikroWin2000 Advanced II system (Berthold Technologies, Tokyo, Japan) after adding 100 μL of luciferase assay reagent to each well. The cell viability was calculated as a percentage of the light intensity of the treated wells versus that of non-treated wells (n=3–4).
Statistical analysis
Numerical data are presented as the mean ± standard error of the mean (SEM) from at least 3 independent experiments. Significance was assessed with one-way ANOVA followed by Bonferroni’s multiple comparison tests; P-values <0.05 were considered to be statistically significant.
Results
Various CAR genes transfected by electroporation into Jurkat cells expressed and bound to CEA to the same extent
We first determined whether CAR genes of different sizes and constituent gene types could be functionally expressed in mammalian cells by using the pcDNA3.1(−) expression vector. Four kinds of genes were constructed with or without a spacer/hinge and CD28 intracellular signaling domain, and were designated CR-0 to CR-3 (Figure 1A and B). In this study, the extracellular domains of CD8α (195 bp) and CD7 (96 bp) were assessed as a spacer/hinge because of the difference in their size. Only CR-2 retained the intracellular domain of the secondary signal, CD28, but not the others. Eventually, the size of CAR genes constructed here were 1,201, 1,398, 1,533, and 1,302 bp (CR-0, CR-1, CR-2, and CR-3, respectively). The CAR genes were transfected into Jurkat cells by electroporation and their functional expression was assessed 24 h after transfection by flow cytometric analysis with an APC-labeled antigen, viz., APC-CEA or APC-BSA (see “Materials and methods” section for details). Interestingly, the expression level of the various CAR genes on Jurkat cells were similar (about 60%) in our experimental conditions, as determined by comparing their binding to CEA and BSA, as a control (Figure 1C).
The CAR gene expressing CD8α, CD28, and CD3ζ most effectively stimulated T cells
To determine which of the 4 CAR genes activated immune cells most effectively and demonstrated the highest anticancer activity, the various CAR genes were transfected into artificially expanded T cells and Jurkat cells and their functional properties were analyzed and compared. T cells from volunteers’ PBMCs were sufficiently expanded by over 15-fold over the course of 9 days (Figure S1A). After artificial T cell expansion, the majority of cells became T cells and the ratio of CD4+ T cells and CD8+ T cell was approximately 1.2:1.0 (Figure S1B). The transfection efficacy of CAR genes into these artificially expanded T cells was about 35%, and that of the EGFP control over 70%, as assessed 24 h after transfection and using our experimental conditions (Figure 3A; Table S1).
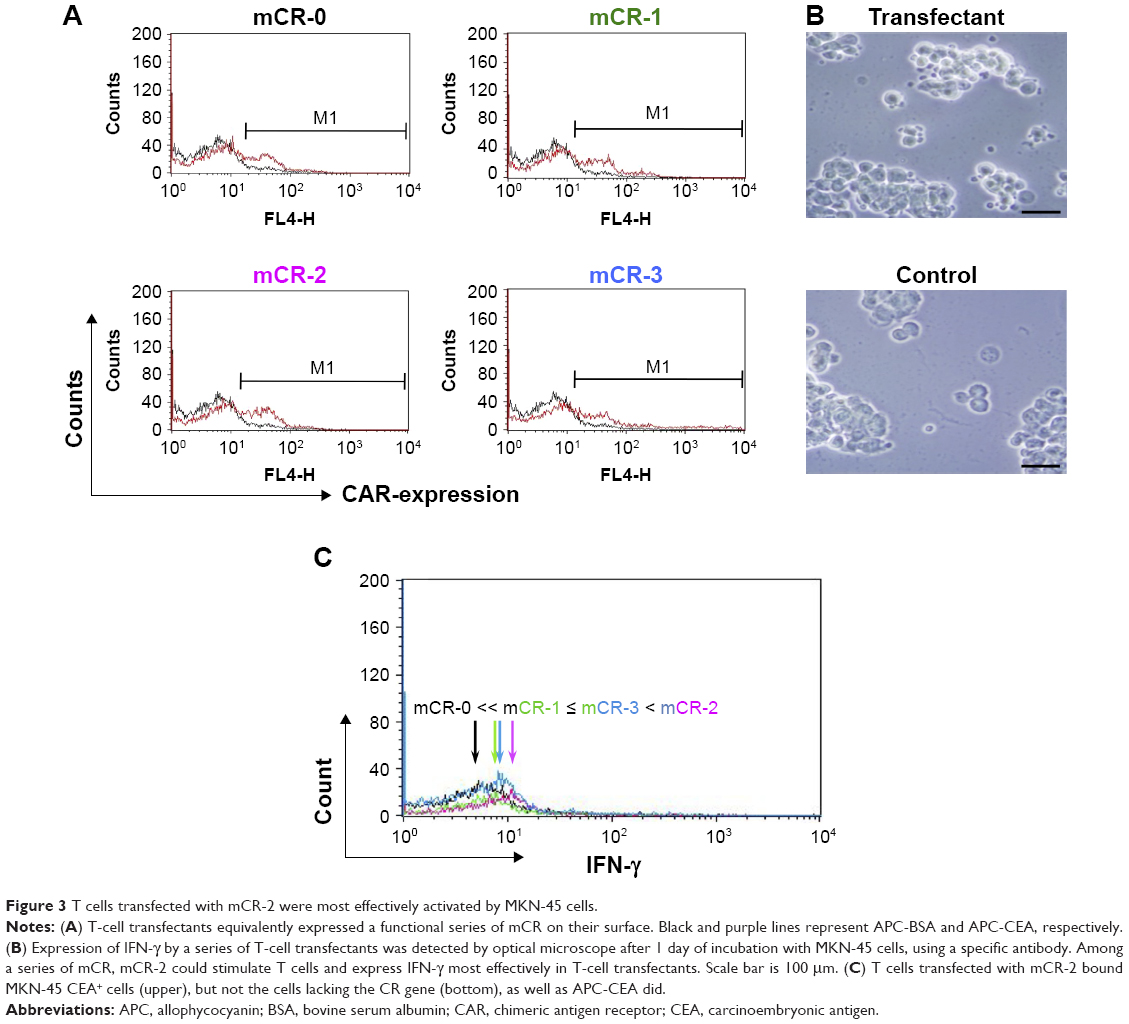 | Figure 3 T cells transfected with mCR-2 were most effectively activated by MKN-45 cells. |
The cell viability of the transfected T cells was about 50% for all 4 CAR genes, as determined by trypan blue staining. After the transfected T cells were incubated with CEA+ cancer cells (MKN-45) for an additional 24 h, the activation efficacy of CAR-bearing T cells was determined to follow the order: CR-2> CR-3≥ CR-1> CR-0, as assessed by the production of IFN-γ and analyzed using flow cytometry with a specific antibody (Figure 3B). The spacer had no effect on CAR expression on primary T cells or on CEA recognition, but it was critical in efficient activation of genetically engineered cells. After addition of the purified CEA coating onto the bottom of a 24-well plate, recognition and binding of CAR-bearing T cells to CEA expressed on the cellular surface of MKN-45 cells, a human gastric cancer cell line, was confirmed using microscopy. After the T cells transfected with CAR genes were co-cultured with MKN-45 cells for 2 h, they bound to CEA+ cancer cells, but not to the control mock gene, as shown in Figure 3C.
The scFv-IL2 fusion protein generated by fully human genes maintains the function of both constituent proteins
The gene encoding the scFv-IL2 fusion protein was generated by SOE-PCR from the human genes by using the primers shown in Figure 2A and Table 1, and was expressed in an E. coli protein expression system by adding D-arabinose to the LB medium. The scFv-IL2 and scFv proteins purified from the supernatant of the LB medium using an Ni2+ column was confirmed by Western blotting using an anti-hIL-2, anti-c-myc, or anti-His antibody; the appropriate bands were detected at the expected sizes (28 and 45 kbp, respectively; Figure 2B). Furthermore, a sandwich enzyme-linked immunosorbent assay (ELISA) was performed to verify the structure of hIL-2 in scFv-IL2 under physiological conditions (Figure 2C).
As shown in Figure 2D, the cell number of CTLL-2 cells that expressed the IL-2 receptor and proliferated in the presence of hIL-2 increased in a dose-dependent manner after 48 h incubation with scFv-IL2.
The binding properties of svFv-IL2 were investigated by flow cytometry using CEA+ MKN-45 cells and an FITC-labeled anti-hIL-2 antibody (Figure 2E and F). The fluorescent intensity of FITC increased after 1 h incubation with scFv-IL2, but not with hIL-2, indicating that scFv-IL2 bound to CEA on the cellular surface of MKN-45 cells, whereas hIL-2 did not bind MKN-45 cells (Figure 2F). Taken together, these results indicated that scFv-IL2, the fusion protein of the scFv antibody specific for CEA and hIL-2, could be successfully generated using an E. coli protein expression system, and retained their functions.
Combination of CAR-bearing PBMCs and scFv-IL2 efficiently damages MKN-45 cells
Taking into consideration that CAR and the fusion protein may be applied as combination therapy in patients, all genes used for this purpose should be of human origin. In this context, the mouse scFv gene of mCR-2, which most effectively activated primary T cells and produced INF-γ among the 4 CAR genes, was replaced with its human equivalent; this construct was designated as hCR-2, and subsequently the anticancer effect of PBMCs expressing this CAR was investigated in combination with scFv-IL2 (Figure 4A). The expression of the 4 types of CAR genes on Jurkat cells was detected by flow cytometry using APC-CEA and APC-BSA and was compared. The expression level of hCR-2 was marginally higher than that of mCR-2, whereas the different expression vectors, pcDNA3.1(−) and pIRES, did not affect the CAR level on Jurkat cells under our experimental conditions (Figure 4B).
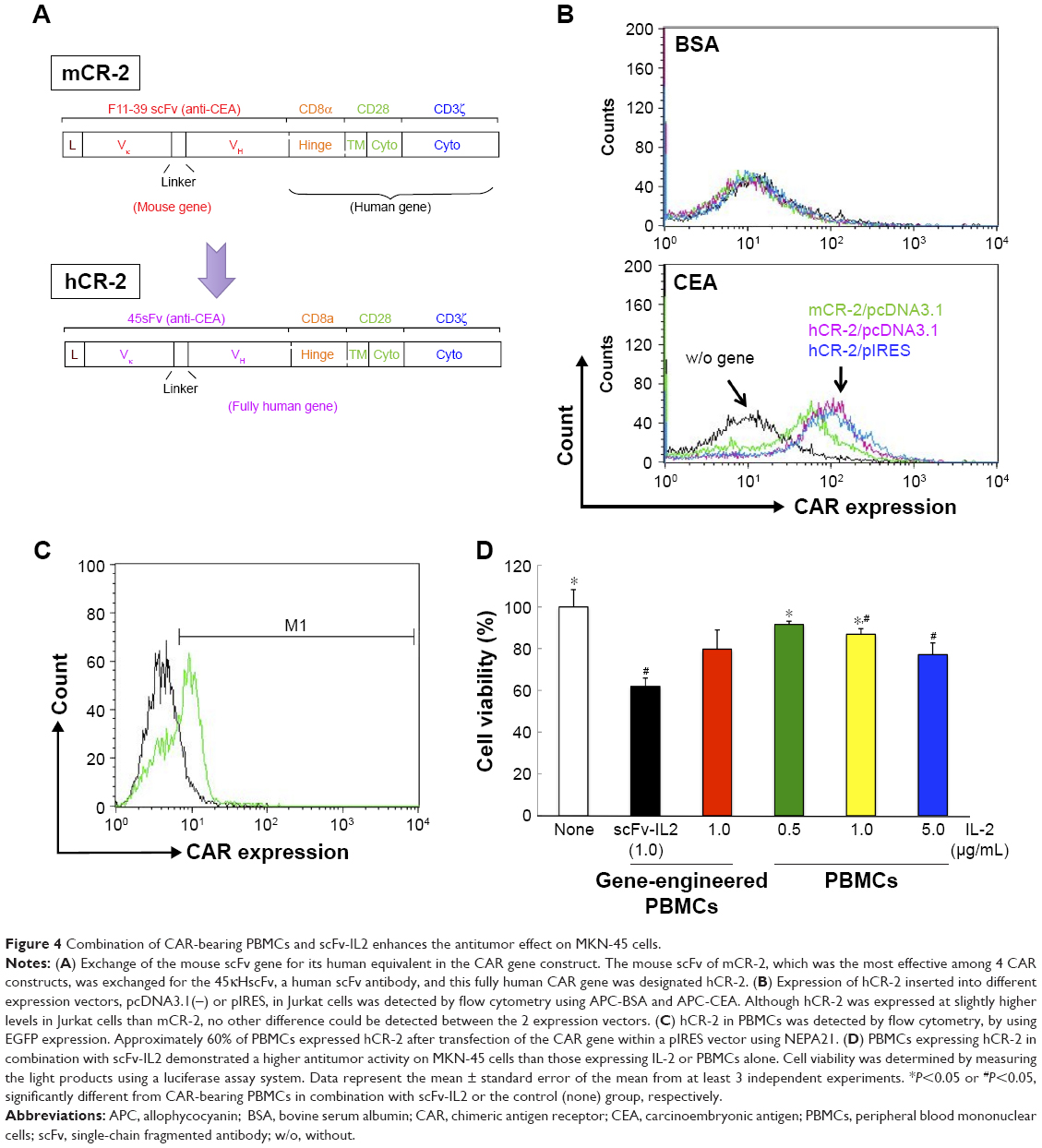 | Figure 4 Combination of CAR-bearing PBMCs and scFv-IL2 enhances the antitumor effect on MKN-45 cells. |
Although the efficacy of gene expression in T cells artificially expanded from PBMCs using the Human T cell Activation/Expansion kit (Miltenyi Biotec) reached 40% after electroporation using a Nucleofector™ (Lonza Group Ltd, Basel, Switzerland), the cell viability after a 24 h incubation was, unfortunately, <50% in our experiment. Because we used primary cells to assess the anticancer effect of the combination of scFv in CAR and in the fusion protein, it was important to increase cell viability after gene transfection. We compared 2 electroporation systems, Nucleofector and NEPA21, for transfection efficacy and cell viability: NEPA21 performed better in human T cells, as summarized in Table S1. Therefore, we decided to use NEPA21 for transfection of the hCR-2 gene into PBMCs. In the present study, PBMCs and T cells were used for CAR gene transduction, as we expected synergy in the anticancer effect of immune cells. As shown in Figure 4C the expression of hCR-2 in PBMCs after NEPA21 transfection was about 60% at 24 h after electroporation, as determined by the expression of EGFP by using flow cytometry. Under these experimental conditions, the cell viability of CAR-bearing PBMCs was about 95%.
The PBMCs were then incubated with the luciferase-expressing MKN-45 cells for 24 h in the presence of scFv-IL2 or hIL-2 to investigate the antitumor effect. The cell viability decreased in a dose-dependent manner in the presence of hIL-2 (Figure 4D). The CAR-bearing PBMCs damaged cancer cells more efficiently than did untransfected PBMCs, as expected. Comparison of the antitumor effects of genetically engineered PBMCs and parental PBMCs revealed a fivefold difference in the concentration of free hIL-2. Interestingly, the efficacy of CAR-bearing PBMCs in combination with scFv-IL2 was double that of treatment with the same amount of free-hIL-2.
Discussion
We have demonstrated the potential of a combination cancer treatment using a human scFv gene in both CAR and a fusion protein of a cytokine in this study. Furthermore, PBMCs, in addition to CLT, CD4+, and CD8+ T cells or NK cells, were found to be effective targets for gene engineering along with CARs in adoptive cancer immunotherapy. Very recently, Srivastava et al demonstrated that activated NK cells and dendric cells (DC) interacted and collaborated to enhance antitumor immunity related to CTL.30 This may be an advantage for the use of PBMCs expressing CAR in patients, without HLA restriction, in adoptive cancer immunotherapy.
To date, many genes from the variable fragment region of an antibody specific for TAA have been reported and applied in the production of CAR or fusion protein in cancer treatment in order to overcome the loss or dysfunction of HLA on the cellular surface of tumor cells.9 In combination with CAR-bearing PBMCs, the scFv-IL2 fusion protein was twice as effective in damaging MKN-45 cells as compared to the same amount of free hIL-2. This phenomenon may indicate the importance of simultaneous input of a third signal, together with the first and secondary signals for activation of T cells, because the anticancer activity of LAK cells using free hIL-2 alone or the fusion of the scFv antibody and hIL-2 indicated similar effects in adoptive immunotherapy.31 Artificial expansion of primary T cells from PBMCs may support this hypothesis. In this context, the combination of CAR and the fusion protein containing the scFv specific for TAA seems to be a promising approach for activating immune cells efficiently and maintaining long-lasting activation of cytokine production in the cancer microenvironment, in addition to the advantage of reduction in the dose of exogenous cytokines required for the treatment of the patient. Moreover, the importance of other cytokines (eg, IL-12, IL-15, and IFN-γ) or immunoregulatory antibodies (eg, anti-CTLA-4, anti-PD-1, anti-IL-10, anti-TGF-β, anti-4–1BB, and anti-OX-40) in the microenvironment has also been reported.22,32–35 Thus, the usefulness of fusion proteins of scFv and these cytokines, or bispecific antibodies, in combination with CAR-bearing PBMCs, as well as the efficacy of these molecules in an in vivo xenograft model, should be assessed in future experiments.36
In our results, the spacer between the domains of the cellular arm and the intracellular ITAM was critical for activation of CAR-bearing T cells, whereas the type of spacer may not be as important as the ITAM domain in CAR. Although there is still considerable debate as to the type of ITAM domain used to construct an effective CAR gene in adoptive cancer immunotherapy, recent reports using a third-generation of CAR genes suggest that it may be important to have signal 1 and at least 1 or 2 signal 2-related ITAM domains, with a spacer, to activate T cells expressing CAR and to maintain long-lasting stability in the patient.13 When comparing the 4 kinds of CAR genes reported here, the CR-2 gene demonstrated the desired results.
Given the efficacy of gene transfection and cell viability after 24 h, electroporation using the NEPA21 system seems to be a promising candidate for use in gene therapy. Interestingly, the hCR-2 gene may be transduced not only into T cells but also into NK cells because PBMCs constitute less than 50% of CD3+ T cells. This may be an explanation for the higher antitumor effect in CAR-bearing PBMCs than in the parental PBMCs with the same concentration of hIL-2.
A concern about using PBMCs in gene-engineered cell-based cancer immunotherapy is that regulatory T (Treg) cells expressing CAR, which activate as well as CTL and NK cells expressing CAR, will suppress the immune response in a cancer microenvironment. Fortunately, because Treg cells can be positively selected from PBMCs using a cell sorter or separator with their specific antibody, PBMCs lacking Treg cells from patients will provide adequate cell-based immunotherapy.
In conclusion, application of the CEA-specific scFv in the combination of CAR and cytokine fusion protein, resulting in the enhancement of anticancer activity of immune cells at the cancer microenvironment might be a promising candidate in adoptive cancer immunotherapy (Figure S2).
Acknowledgments
The authors are particularly grateful for the assistance given by Ms H Yamada to perform the experiments. This work was supported in part by a fund (No 086005) from the Central Research Institute of Fukuoka University. Naixiang Luo’s current affiliation is Department of Immunology, Faculty of Basic Medicine, Guilin Medical University, Guilin, China.
Disclosure
The authors report no conflicts of interest in this work.
References
Norelli M, Casucci M, Bonini C, Bondanza A. Clinical pharmacology of CAR-T cells: Linking cellular pharmacodynamics to pharmacokinetics and antitumor effects. Biochim Biophys Acta. 2016;1865(1):90–100. | ||
June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–1476. | ||
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. | ||
Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50(2–3):248–254. | ||
Garrido F, Algarra I, Garcia-Lora AM. The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol Immunother. 2010;59(10):1601–1606. | ||
Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27(45):5869–5885. | ||
Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. | ||
Chmielewski M, Hombach AA, Abken H. Antigen-specific T-Cell activation independently of the MHC: chimeric antigen receptor-redirected T Cells. Front Immunol. 2013;4:371. | ||
Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3(1):35–45. | ||
Guest RD, Hawkins RE, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. | ||
Heuser C, Hombach A, Losch C, Manista K, Abken H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003;10(17):1408–1419. | ||
Lamers CH, Gratama JW, Pouw NM, et al. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther. 2005;16(12):1452–1462. | ||
Wang J, Jensen M, Lin Y, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18(8):712–725. | ||
Gyobu H, Tsuji T, Suzuki Y, et al. Generation and targeting of human tumor-specific Tc1 and Th1 cells transduced with a lentivirus containing a chimeric immunoglobulin T-cell receptor. Cancer Res. 2004;64(4):1490–1495. | ||
Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67(10):5003–5008. | ||
Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106(5):1544–1551. | ||
Glienke W, Esser R, Priesner C, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21. | ||
Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2013;28(4):917–927. | ||
Altvater B, Landmeier S, Pscherer S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15(15):4857–4866. | ||
Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014;2(2):142–153. | ||
Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7(2):95–106. | ||
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. | ||
Arakawa F, Shibaguchi H, Xu Z, Kuroki M. Targeting of T cells to CEA-expressing tumor cells by chimeric immune receptors with a highly specific single-chain anti-CEA activity. Anticancer Res. 2002;22(6C):4285–4289. | ||
Huang J, Shibaguchi H, Zhao J, et al. IgG isotype conversion of a novel human anti-carcinoembryonic antigen antibody to increase its biological activity. Anticancer Res. 2006;26(2A):1057–1063. | ||
Imakiire T, Kuroki M, Shibaguchi H, et al. Generation, immunologic characterization and antitumor effects of human monoclonal antibodies for carcinoembryonic antigen. Int J Cancer. 2004;108(4):564–570. | ||
Shibaguchi H, Luo NX, Kuroki M, et al. A fully human chimeric immune receptor for retargeting T-cells to CEA-expressing tumor cells. Anticancer Res. 2006;26(6A):4067–4072. | ||
Shibaguchi H, Tsuru H, Kuroki M. Enhancement of the antitumor effect on combination therapy of an anticancer drug and its antibody against carcinoembryonic antigen. Chemotherapy. 2012;58(2):110–117. | ||
Shirasu N, Shibaguci H, Kuroki M, Yamada H. Construction and molecular characterization of human chimeric T-cell antigen receptors specific for carcinoembryonic antigen. Anticancer Res. 2010;30(7):2731–2738. | ||
Zhao J, Kuroki M, Shibaguchi H, et al. Recombinant human monoclonal igA antibody against CEA to recruit neutrophils to CEA-expressing cells. Oncol Res. 2008;17(5):217–222. | ||
Srivastava RM, Lee SC, Andrade Filho PA, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19(7):1858–1872. | ||
Matsumoto H, Liao S, Arakawa F, et al. Targeting of interleukin-2 to human MK-1-expressing carcinoma by fusion with a single-chain Fv of anti-MK-1 antibody. Anticancer Res. 2002;22(4):2001–2007. | ||
Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol. 2013;191(3):1011–1015. | ||
Yu P, Bamford RN, Waldmann TA. IL-15-dependent CD8+ CD122+ T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL-17 production by CD4+ T cells. Eur J Immunol. 2014;44(11):3330–3341. | ||
Cecil DL, Holt GE, Park KH, et al. Elimination of IL-10-inducing T-helper epitopes from an IGFBP-2 vaccine ensures potent antitumor activity. Cancer Res. 2014;74(10):2710–2718. | ||
Alvarez M, Bouchlaka MN, Sckisel GD, Sungur CM, Chen M, Murphy WJ. Increased antitumor effects using IL-2 with anti-TGF-beta reveals competition between mouse NK and CD8 T cells. J Immunol. 2014;193(4):1709–1716. | ||
Kermer V, Hornig N, Harder M, Bondarieva A, Kontermann RE, Muller D. Combining antibody-directed presentation of IL-15 and 4–1BBL in a trifunctional fusion protein for cancer immunotherapy. Mol Cancer Ther. 2013;13(1):112–121. |
Supplementary materials
Artificial T cell expansion
PBMCs were obtained from healthy volunteers. The specimens were obtained using LSM® (Cappel, Aurora, OH, USA) according to the manufacturer’s protocol. After a 2 h incubation in a 10 cm culture dish, the floating cells were transferred to a new culture dish containing RPMI-1640 culture medium and the human T cell activation/expansion kit (Miltenyi Biotech Inc., Gladbach, Germany). After 2 d incubation, 20 IU/mL of hIL-2 was added to the culture medium and the cells were fed twice a week with hIL-2.
Flow cytometry
The component of CD4+ and CD8+ T cells 9 days after artificial expansion of PBMCs was analyzed using a FACSCalibur system (BD Biosciences, San Jose, CA, USA) with their specific antibodies (CD4-PE and CD8-FITC, respectively; Miltenyi Biotec GmgH, Bergisch Gladbach, Germany), and was analyzed using Cell Quest Pro (BD Biosciences).
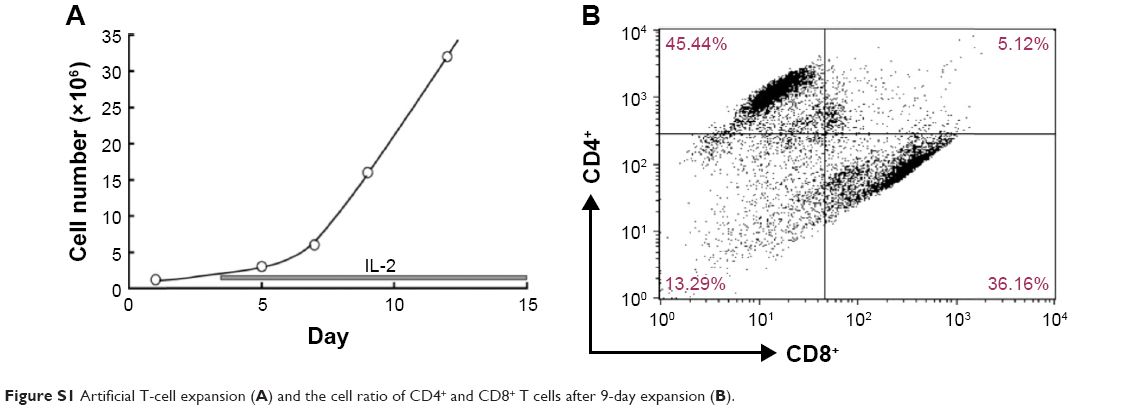 | Figure S1 Artificial T-cell expansion (A) and the cell ratio of CD4+ and CD8+ T cells after 9-day expansion (B). |
 | Table S1 Transfection efficiency of electroporation using Nucleofector™ and NEPA21 |
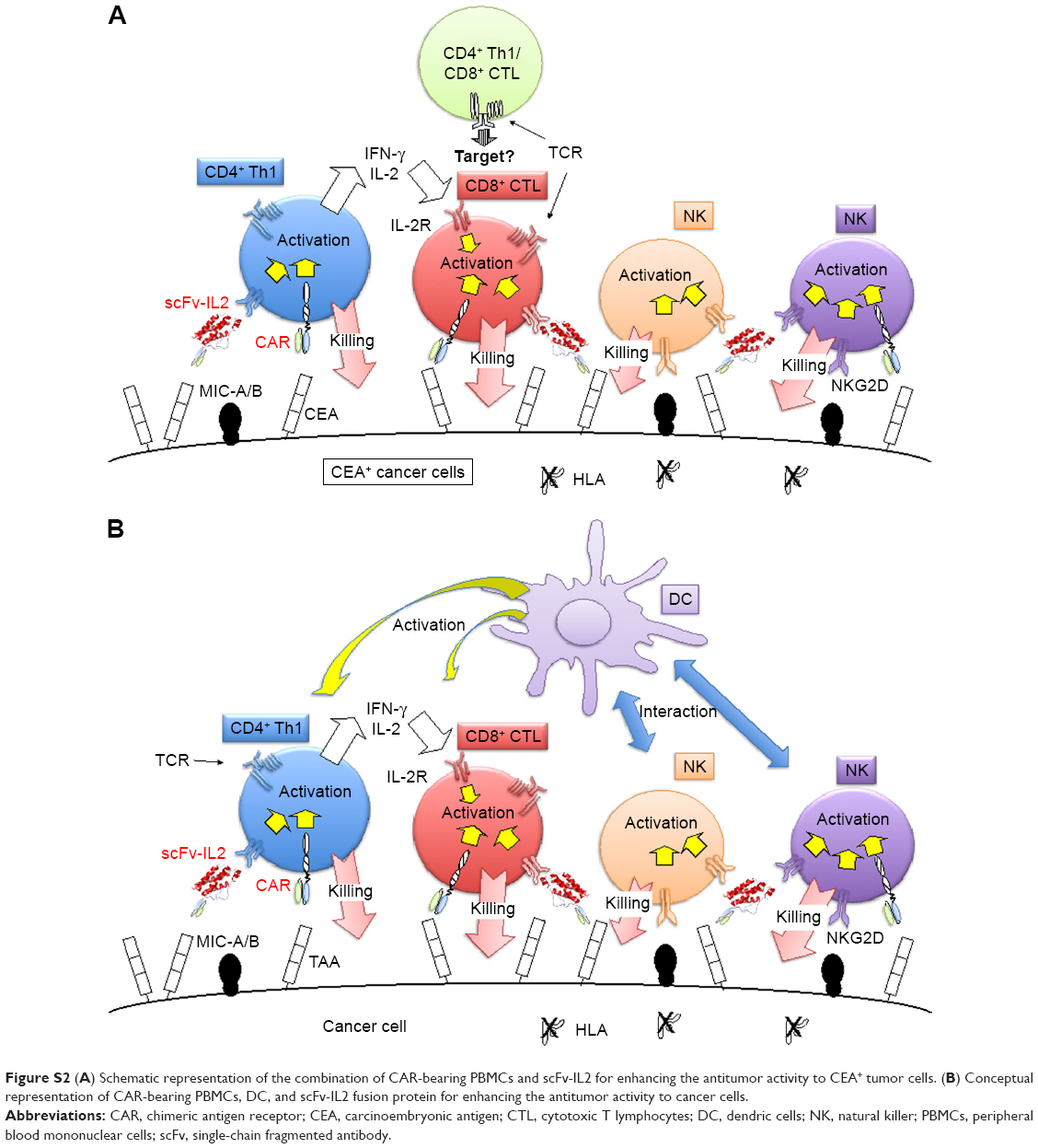 | Figure S2 (A) Schematic representation of the combination of CAR-bearing PBMCs and scFv-IL2 for enhancing the antitumor activity to CEA+ tumor cells. (B) Conceptual representation of CAR-bearing PBMCs, DC, and scFv-IL2 fusion protein for enhancing the antitumor activity to cancer cells. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.