Back to Journals » Drug Design, Development and Therapy » Volume 8
Enhanced bioavailability and anthelmintic efficacy of mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose
Authors de la Torre-Iglesias PM, García-Rodriguez JJ, Torrado G, Torrado S, Torrado-Santiago S ![]() , Bolás-Fernández F
, Bolás-Fernández F
Received 5 April 2014
Accepted for publication 7 June 2014
Published 18 September 2014 Volume 2014:8 Pages 1467—1479
DOI https://doi.org/10.2147/DDDT.S65561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Paloma Marina de la Torre-Iglesias,1,2 Juan José García-Rodriguez,3 Guillermo Torrado,4 Susana Torrado,1,2 Santiago Torrado-Santiago,1,5 Francisco Bolás-Fernández3
1Department of Pharmaceutical Technology, Faculty of Pharmacy, Complutense University, Madrid, Spain; 2Institute of Industrial Pharmacy, Complutense University, Madrid, Spain; 3Department of Parasitology, Faculty of Pharmacy, Complutense University, Madrid, Spain; 4Department of Pharmaceutical Technology, Faculty of Pharmacy, Alcala University, Alcala de Henares (Madrid), Spain; 5Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD), Madrid, Spain
Background: Mebendazole (MBZ) is an extremely insoluble and therefore poorly absorbed drug and the variable clinical results may correlate with blood concentrations. The necessity of a prolonged high dose treatment of this drug increases the risk of adverse effects.
Methods: In the present study we prepared redispersible microparticles (RDM) containing MBZ, an oral, poorly water-soluble drug, in different proportions of low-substituted hydroxypropylcellulose (L-HPC). We investigated the microparticulate structures that emerge spontaneously upon dispersion of an RDM in aqueous medium and elucidated their influence on dissolution, and also on their oral bioavailability and therapeutic efficiency using a murine model of infection with the nematode parasite Trichinella spiralis.
Results: Elevated percentages of dissolved drug were obtained with RDM at 1:2.5 and 1:5 ratios of MBZ: L-HPC. Thermal analysis showed an amorphization of MBZ in the RDM by the absence of a clear MBZ melting peak in formulations. The rapid dissolution behavior could be due to the decreased drug crystallinity, the fast dissolution time of carriers as L-HPC, together with its superior dispersibility and excellent wetting properties. RDM-1:2.5 and RDM-1:5 resulted in increased maximum plasma concentration and area(s) under the curve (AUC)0–∞ values. Likewise, after oral administration of the RDM-1:2.5 and RDM-1:5 the AUC0–∞ were 2.67- and 2.97-fold higher, respectively, compared to those of pure MBZ. Therapeutic activity, assessed on the Trichinella spiralis life cycle, showed that RDM-1:5 was the most effective in reducing the number of parasites (4.56-fold) as compared to pure MBZ, on the encysted stage.
Conclusion: The MBZ: L-HPC RDM might be an effective way of improving oral bioavailability and therapeutic activity using low doses of MBZ (5 mg/kg), which implies a low degree of toxicity for humans.
Keywords: benzimidazole carbamates, redispersible microparticles, in vitro dissolution, pharmacokinetics, anthelmintic activity, Trichinella spiralis
Introduction
Mebendazole (MBZ), methyl-5-benzoyl benzimidazole-2-carbamate, a broad-spectrum anthelmintic drug of the benzimidazole class effective against a number of nematodal and cestodal species under oral administration as tablets or suspension, is recommended for the treatment of non-surgical cases and as a supplementary treatment prior to and post-surgery in hydatid disease patients.1
MBZ has limited water solubility. Poor aqueous solubility has been a major issue in achieving adequate oral bioavailability. Therefore, a high dose of MBZ is required for helminthic infections causing many adverse effects. So, high doses of this drug can cause anemia and liver damage. Moreover, studies have shown evidence of teratogenic effects of MBZ in rats and mice, it should therefore be avoided during pregnancy and lactation.2
In order to overcome poor aqueous solubility, a commonly used approach involves size reduction of the drug which increases the specific surface area, enhancing the dissolution rate of the drug. Freeze drying has been widely used to convert micro-suspensions to dried microparticles and finally to various solid products. However, any form of drying of the micro-suspensions generally causes the microparticles to aggregate due to liquid removal leading to loss of the large surface area of microparticles and need to be stabilized.3 Polymers of low molecular weight and high water uptake such as hydroxypropyl methylcellulose or low-substituted hydroxypropylcellulose (L-HPC) render more wettable drug microparticles with slight aggregation.3,4
In recent years, systems employing hydrophilic cellulose polymers have attracted considerable interest.3–6 The high dispersibility of L-HPC causes the spontaneous apparition of microparticulate structures in aqueous medium and also enhances drug dissolution rate by several procedures: increasing the specific surface area, wetting process, and a transition to an amorphous stage.5–8 In addition, several authors have shown how solid dispersions of L-HPC improve intestinal absorption of drugs with low solubility.5,9 In the current work we have developed L-HPC redispersible microparticles (RDM) which would allow us to achieve similar anthelmintic effects with lower doses. Furthermore, in recent years it has been sought to use lower doses of anti-parasite treatments in order to diminish the adverse effects.10 Thus, Matadamas-Martínez et al11 had shown how low-dose formulations of benzimidazole derivatives with cyclodextrins enhance anti-parasitic efficacy against Trichinella spiralis muscle larvae. Similar anthelmintic efficacy improvements have been observed by our research group and others by increasing the solubility of different benzimidazole carbamates.12
We have followed the MBZ dissolution rate study of Swanepoel et al13 using the method of the United States Pharmacopeia dissolution without sodium lauryl sulfate achieving a reduction of the dissolution profiles. In our opinion, this same method without sodium lauryl sulfate might be more appropriate to calculate various dissolution parameters such as drug percent (DP) dissolved and dissolution efficiency (DE) of the raw material and MBZ microparticles.14,15
The improved absorption of anthelmintics by means of RDM could be used for optimizing the anthelmintic efficacy with lower doses,12,16 therefore, these formulations could enhance MBZ concentrations in muscle and within cysts for inoperable or disseminated cases of hydatidosis or neurocysticercosis.
The objectives of the present study were: i) development of RDM, and ii) to elucidate their influence on increasing drug bioavailability and to study how these changes will affect the anthelmintic efficacy against the different states of T. spiralis life cycle.
Material and methods
The materials used were as follows: MBZ (Sigma-Aldrich Co., St Louis, MO, USA) and L-HPC (LH-31, hydroxypropoxy content: 11%, Shin-Etsu®, Tokyo, Japan). All other chemical reagents were of pharmaceutical grade or better.
Preparation of formulations
RDM containing drug to carrier proportions of 1:1; 1:2.5, and 1:5 (w/w) (RDM-1:1, RDM-1:2.5, and RDM-1:5 respectively) were prepared by incorporating this poorly water soluble drug into L-HPC microparticles via freeze drying. The required amount of MBZ (50.0 mg) was dissolved in formic acid and after that added onto L-HPC water dispersion (50 mg, 125 mg, and 250 mg of L-HPC in 0.224 mL, 0.56 mL, and 1.11 mL of water respectively). Thus, the RDM were obtained in a minimal volume of a water:formic acid mixture (66/33, v/v). Then formulations were frozen at −40°C. The frozen mixtures were freeze-dried using a Flexi-Dry FD-3-85A-MP (Kinetics group Inc., NY, USA). Then the different formulations were pulverized manually using a mortar and a pestle and sieved to obtain a particle size fraction of 0.125–0.500 mm. After this procedure the vials were capped within 5 minutes (minutes) and stored at room temperature (22°C–24°C) in a desiccator containing silica gel until used.
The physical mixture (PM) containing drug to carrier proportion of 1:1 (w/w) (PM-1:1) was prepared by manually mixing the appropriate amount of 0.125–0.500 mm particle size fractions of MBZ and carrier in a ceramic bowl using a polymeric spatula. Recrystallized MBZ was obtained by the same preparation method used for the RDM but without any carrier.
These formulations were directly used for the differential scanning calorimetric (DSC) analysis.
Preparation of reconstituted formulations
Prior to carrying out the in vitro dissolution test and the in vivo experiments (pharmacokinetics and anthelmintic activity), the following formulations: MBZ raw material, recrystallized MBZ, RDM-1:1, RDM-1:2.5, and RDM-1:5 were reconstituted in a 0.75% carboxymethylcellulose (CMC) solution in order to obtain an MBZ concentration of 0.5 mg/mL. This concentration was chosen to achieve the dose of 5 mg/kg when administering 0.3 mL of each formulation to mice by bucco-gastric tube.
SEM characterization
In order to evaluate by scanning electron microscopy (SEM) the morphology of recrystallized MBZ, RDM-1:5 and its blank formulation (RDM-0:5) were each suspended in two media: the first medium was organic (carbon tetrachloride) and the second was distilled water. These media presented an MBZ concentration of 0.5 mg/mL, similar to the reconstituted formulations. Afterwards, all samples were ultrasonicated for 10 minutes at 120 W (Branson 8210; Branson Ultrasonics Co, Danbury, CT, USA) and vacuum dried.
The morphology and particle size of the recrystallized MBZ, RDM at 1:5 ratios of MBZ:L-HPC, and its blank formulation (RDM-0:5) were characterized by SEM (JSM-6400, JEOL, Tokyo, Japan), with the samples being gold-plated prior to imaging. All micrographs were the product of secondary electron imaging used for surface morphology identification at the same magnification of 3,000× and an accelerating voltage of 20 kV.
Determination of drug substance content of the dried products and solubility studies
MBZ content of the dried products was determined by dissolving the equivalent of 10 mg of MBZ in 20 mL of formic acid in a 50 mL volumetric flask and it was sonicated for 10 minutes. The volume was adjusted to 50 mL with 0.1 M hydrochloric acid. The solution was filtered and diluted if necessary. Then, solutions were filtered using 0.45 μm filter (PALL Acrodisc, Pall, Cornwall, UK). The quantity of MBZ was determined at 286.8 nm using a UV-VIS 1,700 spectrophotometer (Hitachi Ltd., Tokyo, Japan), following the procedures described by García-Rodríguez et al.17 This wavelength did not interfere with the carrier (L-HPC). The amount of MBZ was determined from the appropriated calibration curve. Three samples of each formulation were used to determine the content.
Solubility experiments were performed by dissolving an excess amount of MBZ raw material, recrystallized MBZ, and RDM in 50 mL of 0.1 M hydrochloric acid. The flasks were vortex-mixed for 3 minutes and they were shaken at 37°C on an orbital shaker at a maximum speed of 10 rpm over a period of 72 hours to guarantee equilibrium. Three milliliters of samples were withdrawn and filtered through a 0.45 μm filter (PALL, Acrodisc). Then, 100 μl of the filtrates were diluted appropriately and measured using a UV-VIS 1,700 spectrophotometer (Hitachi Ltd) at 286.8 nm. All experiments were performed in triplicate.
Determination of particle size
Particle size of the recrystallized MBZ, RDM at 1:5 ratios of MBZ:L-HPC and its blank formulation (RDM-0:5) was assayed by laser diffraction (Microtrac S3500, Microtrac Inc., Montgomeryville, PA, USA). Before the analysis, each formulation was suspended in distilled water to obtain an MBZ concentration of 0.5 mg/mL as in the reconstituted formulations. Mean size (μm) was determined based on size distribution in number. Five runs of 60 seconds per sample were carried out.
DSC
DSC analysis was used to characterize the thermal behavior of: pure MBZ, recrystallized MBZ, L-HPC, PM-1:1, and MBZ redispersible microparticles RDM-1:1, RDM-1:2.5, and RDM-1:5. DSC thermograms were obtained using an automatic thermal analyzer system (Mettler Toledo TC 15, TA controller, Greifensee, Switzerland). Temperature calibration was performed using Indium Calibration Reference Standard (transition point: 156.60°C). Samples of about 2 mg were placed into aluminum pans, then hermetically sealed with aluminum lids and heated from 25°C to 375°C at a heating rate of 10°C/min under constant purging of dry nitrogen 20 mL/min. An empty pan, sealed in the same way as the sample, was used as a reference.
In vitro drug release
These studies were carried out in a dissolution bath (Vankel® VK 700; VanKel Industries, Edison, NJ, USA). A United States Pharmacopeia Apparatus 2 (paddle) was set up with a rotational speed of 75 rpm and 900 mL of dissolution medium (0.1 M hydrochloric acid). The temperature was maintained at 37.0°C±0.1°C throughout dissolution study. Thirty-six milliliters of each reconstituted formulation freshly prepared equivalent to 18 mg of MBZ were introduced into the vessel. At predetermined time points, a sample of 5 mL was withdrawn and filtered through a 0.45 μm filter (PALL, Acrodisc). The quantity of MBZ was determined at 286.8 nm using a UV-VIS 1,700 spectrophotometer (Hitachi Ltd.) following the procedures described by García-Rodríguez et al.17 This wavelength did not interfere with the carrier (L-HPC). The cumulative amount of MBZ released from the system was determined from the appropriated calibration curve. Every determination at each time point was performed in triplicate and the error bars on the graphs represented the standard deviation. DE was calculated from the area under the dissolution curve at time t and expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time.14,15
Oral bioavailability study
Swiss CD-1 mice weighing 30–35 g were employed. Food and water were supplied ad libitum. This animal experiment was approved by the Animal Ethics Committee at Complutense University Madrid, Spain. The formulations were administered orally via bucco-gastric tube. Each reconstituted formulation freshly prepared (raw material, recrystallized MBZ and RDM 1:1; 1:2.5, and 1:5) was administered at a dose of 5 mg/kg. Following the procedures described by Torrado et al18 and Beinborn et al19 after drug administration, six mice per formulation were euthanized by 15% urethane solution at each of the predetermined time points (0.25, 0.5, 0.75, 1.5, 3, 6, and 24 hours). Whole blood (approximately 1.5 mL) was collected by cardiac puncture into heparinized vials and centrifuged at 9,000 rpm for 15 minutes to obtain plasma. After the extraction process, the blood samples were frozen until high-performance liquid chromatography (HPLC) analysis.
Plasma sample analysis
Plasma aliquots of 500 μL were thawed and mixed with 2 mL of methanol in a vortex mixer for 15 s and then centrifuged at 5,000 rpm for 10 minutes. The supernatant phase was filtered through a Millipore HVLP filter of 45 μm (Millipore Iberica, Madrid, Spain) and the MBZ plasma concentrations assayed by HPLC. The quantity of MBZ was determined by HPLC method, consisting of a UV detector (Jasco® UV-1575 Intelligent UV/VIS Detector; Jasco Products Company, Oklahoma City, OK, USA), a pump (Jasco PU-1580 Intelligent HPLC pump; Jasco Products Company), a degasser (Jasco DG-2080–53; Jasco Products Company) and an automatic injector (Jasco AS-2050 Intelligent Sampler; Jasco Products Company). The selected wavelength was 291 nm. The column used was a Mediterranea Sea 5 μm C18 (4.6 mm ×20 cm, Teknokroma, Barcelona, Spain) and was maintained at room temperature. The flow rate was 1.0 mL per minute. The mobile phase consisted of a mixture of methanol and 0.05 M monobasic ammonium phosphate (60:40, v/v), and pH was adjusted to 5.5 with 0.1 M phosphoric acid, following the procedures described by Daniel-Mwambete et al.12 MBZ in plasma, with a retention time of 11 minutes, was estimated from the appropriated calibration curve. Each determination at each time point was performed in triplicate and the error bars on the graphs represented the standard deviation.
Bioavailability parameters
Tmax (time taken to achieve Cmax) and Cmax (maximum plasma concentration) were estimated as the mean values of Tmax for the six mice used per group and time interval with the highest MBZ plasma concentration for each administered formulation. The area(s) under the curve (AUC)0→∞ was calculated as the sum of AUC0→24 and AUC24→∞. Also, the AUC0→24 was calculated by the trapezoidal rule method and AUC24→∞ was estimated as the quotient between plasma concentration at 24 hours (C24) and elimination constant (Ke). The Ke was calculated as the slope, from the final phase of the log concentration-time curves. Comparative statistical studies on the bioavailability among the different formulations were performed by paired Student’s t-test. A P-value of less than 0.05 was considered as significant.
Evaluation of the efficacy of MBZ dosage forms on a T. spiralis murine model
The GM-1 isolate of T. spiralis was used. The isolate was isoenzymatically identified as T. spiralis (Reference Centre for Trichinellosis, Istituto Superiore di Santá, Rome) and kept under the code MFEL/ES/S2 GM-1-ISS48.20 In order to evaluate the anthelmintic activity of the formulations, groups of ten Swiss CD1 mice per treatment were orally infected with 300±50 muscle larvae (L1) isolated from infected mouse carcasses, following an artificial digestion. Groups of ten mice each were respectively administered with the same doses (5 mg/kg) of either reconstituted formulation of MBZ raw material or RDM-1:5, both of them freshly prepared, by bucco-gastric tube. The use of this low dose of MBZ (5 mg/kg) would allow assessing whether there is a relationship between the rate of dissolution of different formulations (MBZ raw materials and RDM-1:5) and its effectiveness against the different stages of the T. spiralis life cycle. Ten mice were kept as control per parasite stage, and were administered with vehicle alone (0.75% CMC solution). An additional group of ten mice was kept as infection control. Treatments were applied at four different stages (pre-adult, migrating larvae, encysted larvae, and adult) of the parasite. For the pre-adult parasite stage, the formulations were administered 24 hours post-infection (pi). The effectiveness of the treatment was assessed on day seven pi, after sacrificing the mice (previously anesthetized by pi injection of ketamine/xylazine) by cervical dislocation. The numbers of adults remaining in the gut were collected and counted in accordance with the method described by García et al.20 For treatment of the migrating larvae, first of all, it was necessary to remove the adults remaining in the gut without affecting the migratory new born larvae.20 This was achieved by treating both the control and experimental groups on day nine pi, with trichophon (Neguvon, Bayer AG, Leverkusen, Germany) at 90 mg/kg administered orally plus one intramuscular injection of atropine sulfate (Bayer SA), at 1 mg/kg. Later on, the MBZ formulation and MBZ raw material were administered on days 13, 14, and 15 pi. Treatment against encysted larvae was given on days 34, 35, and 36 pi. Finally, against adult stages the formulations were applied on day six pi.
The effectiveness of these treatments was assessed on day eight pi against adult stages and on day 46 pi against migrating and encysted larvae, after sacrificing the mice (previously anesthetized) by cervical dislocation. The worms remaining in the intestine and striated muscles were recovered and later counted following the procedures described by Wakelin and Lloyd.21 The efficacy of each drug treatment was determined as the percentage of reduction from the average worm burden of untreated controls. This animal study was approved by the Animal Ethics Committee at Complutense University Madrid, Spain. The comparative statistical anthelmintic efficiency studies among the different group formulations were performed by paired Student’s t-test. A P-value of less than 0.05 was considered as significant.
Results and discussion
SEM analysis, determination of drug content, solubility studies, and particle size
SEM was used to determine the morphology of different samples (recrystallized MBZ, MBZ redispersable microparticles RDM-1:5, and its blank formulation RDM-0:5), all of them at the same magnification (×3,000). Figures 1A, C, and E, show dried formulations previously dispersed in an organic medium (carbon tetrachloride). This medium allows a good dispersion of particles avoiding swelling of carrier (L-HPC). Recrystallized MBZ has two types of particles (Figure 1A): the first one consists of a great number of small rounded particles, with a smooth surface (indicated by white arrows) and the second one consists of larger and irregular surface microaggregates with round edges. At a higher magnification (data not shown) most of the microaggregates are formed by clusters of small rounded MBZ particles. Moreover, RDM-1:5 (Figure 1C) shows a change in the morphology of particles. A lower number of small round particles (that may be mainly attributed to MBZ) are observed per field. Besides, the irregular microaggregates with no rounded edges are attributed to L-HPC as their structure is similar to the L-HPC particles of formulation RDM-0:5 observed in Figure 1E. The surface of the formulations after immersion in aqueous medium and posterior vacuum drying was also studied (Figures 1B, D, and F). Figure 1B shows that recrystallized MBZ particles tend to agglomerate in big MBZ particles (5 μm) although some small rounded particles also may be observed (≤1 μm). Microparticles RDM-1:5 (Figure 1D) and the blank formulation containing only L-HPC (RDM-0:5, Figure 1F) show large swollen translucent particles with attenuated non-rounded edges, which can be attributed to the carrier (L-HPC), and small particles of less than 1 μm which can be attributed to MBZ or L-HPC.
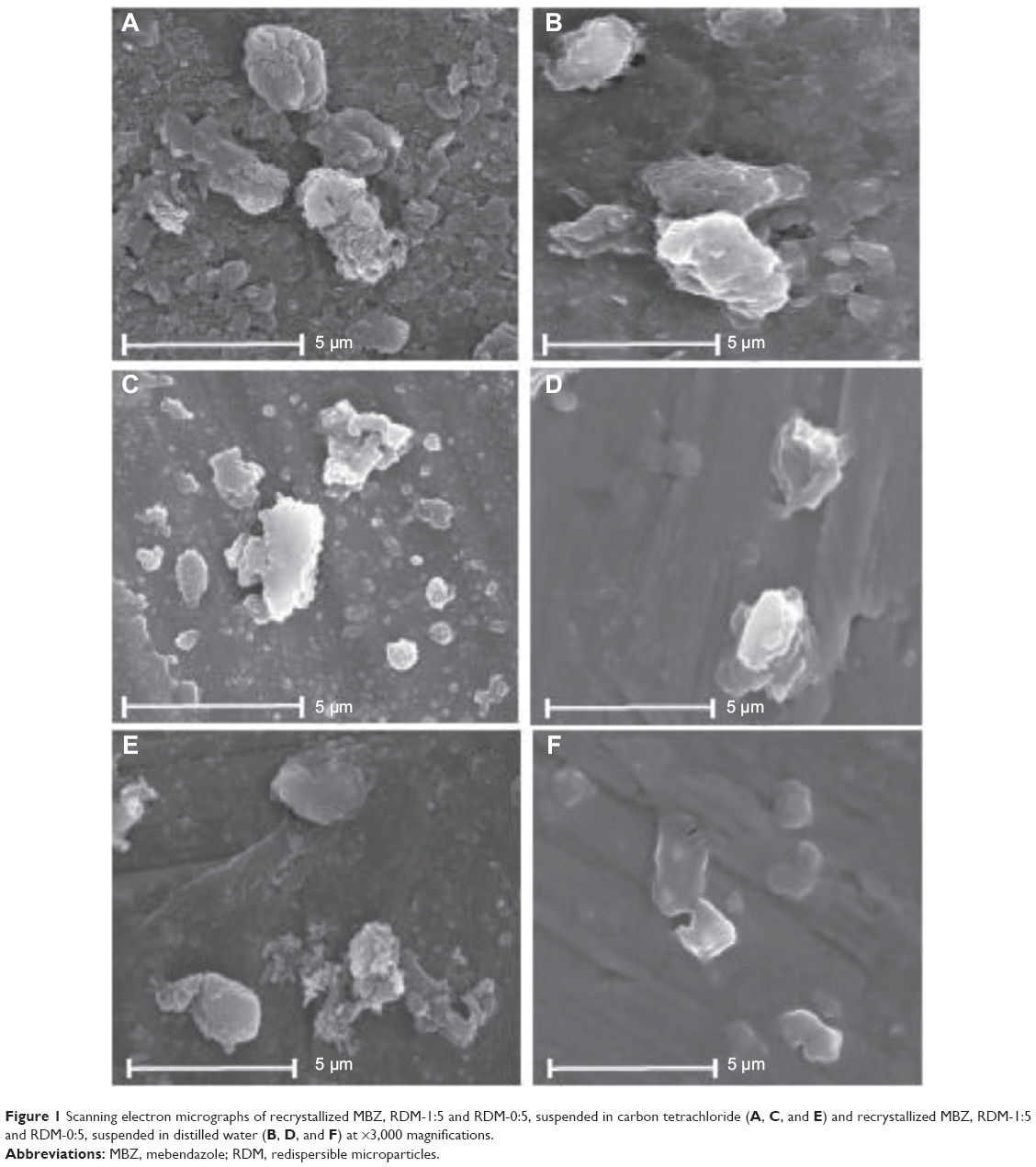 | Figure 1 Scanning electron micrographs of recrystallized MBZ, RDM-1:5 and RDM-0:5, suspended in carbon tetrachloride (A, C, and E) and recrystallized MBZ, RDM-1:5 and RDM-0:5, suspended in distilled water (B, D, and F) at ×3,000 magnifications. |
Regarding drug substance content, all freeze-dried products contained between 93.63%±4.18%–96.40%±4.91%, close to 100% of the expected theoretical drug content. Similar results have been obtained by other authors using a similar freeze drying process.22
In the solubility studies, recrystallized MBZ and formulations with lesser L-HPC proportions (RDM-1:1) showed the following results: 19.79±0.98 and 19.87±0.89 μg/mL respectively. Similar MBZ solubility coefficients were achieved by other authors at this pH.13
However, when the proportion of cellulose in the formulation is increased there is a raise in solubility and dispersibility of the system, so microparticles RDM-1:2.5 and RDM-1:5 showed values of 21.88±0.87 and 25.22±0.46 mg/mL respectively. These good results were also obtained by other authors using a similar freeze drying process for microparticles of L-HPC.4
Laser diffraction analysis is shown in Figure 2. In this figure it is shown that recrystallized MBZ presents two different populations: the first one consists of small particles under 3.89 μm (37.09%) and a second group of microaggregate particles with sizes between 3.89–13.08 μm. Other authors reported that similar small particles with high specific surfaces, tend to aggregate.23 Blank formulation RDM-0:5 only presents one population of larger particles between 3.89–13.08 μm. The formulation with only L-HPC (RDM-0:5), presented a value of polydispersity index of 0.298, values ≤0.3 means particles are quite monodisperse.24 RDM-1:5 shows two populations as recrystallized MBZ: the first one with small particles under 3.89 μm, is attributed to small MBZ particles. The higher polydispersity index (0.396) of this formulation (RDM-1:5) may possibly be attributed to the existence of these two different populations.
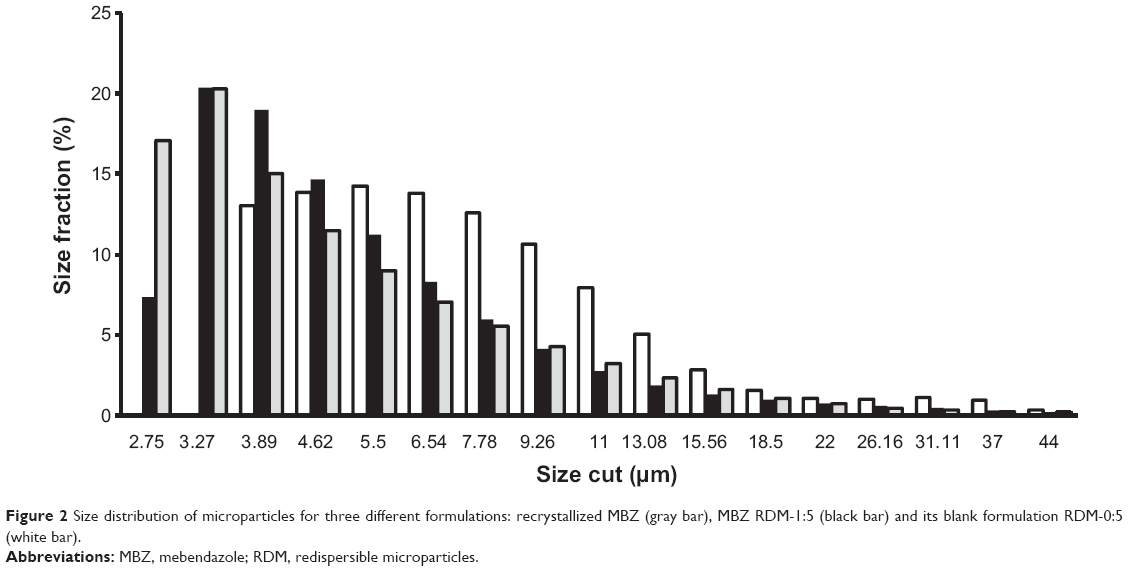 | Figure 2 Size distribution of microparticles for three different formulations: recrystallized MBZ (gray bar), MBZ RDM-1:5 (black bar) and its blank formulation RDM-0:5 (white bar). |
The inclusion of high proportions of dispersing agent in the RDM-1:5 enhances particles’ redispersion when decreasing the portion of larger particles between 3.89–7.78 μm compared to both recrystallized MBZ and RDM-0:5. A similar result was obtained by Cerdeira et al22 in microparticles of microcrystalline cellulose since a reduction was observed in the size of the microparticles when they included miconazole.
DSC analysis
Figure 3 shows thermograms of the L-HPC carrier, MBZ raw material, recrystallized MBZ, MBZ redispersible microparticles RDM-1:1, RDM-1:2.5, and RDM-1:5. MBZ raw material presents a wide endothermic peak at 253.86°C (192.54 J/g), followed by a final endothermic peak at 310.82°C (168.34 J/g), corresponding to melting of the substance. In the recrystallized MBZ thermogram a clearer peak was obtained at 247.68°C (151.58 J/g), showing a similar final endothermic melting peak at 310.82°C (182.28 J/g). Similar enthalpy values of both products, allow us to consider that the process of freeze-drying does not affect the crystalline form. According to the work of De Villiers et al25 first endothermic peak may be related to polymorph C (with sharply defined endotherm at 253°C). At this temperature the polymorph contains only an endothermic/exothermic transition between 250°C–255°C. These authors showed that the second endothermic peak around 315°C could be related to both polymorphs C and A.25 Thus, both endothermic peaks, for MBZ and recrystallized MBZ, can be attributed to a mixture of polymorph C, major, and polymorph A, minor.25
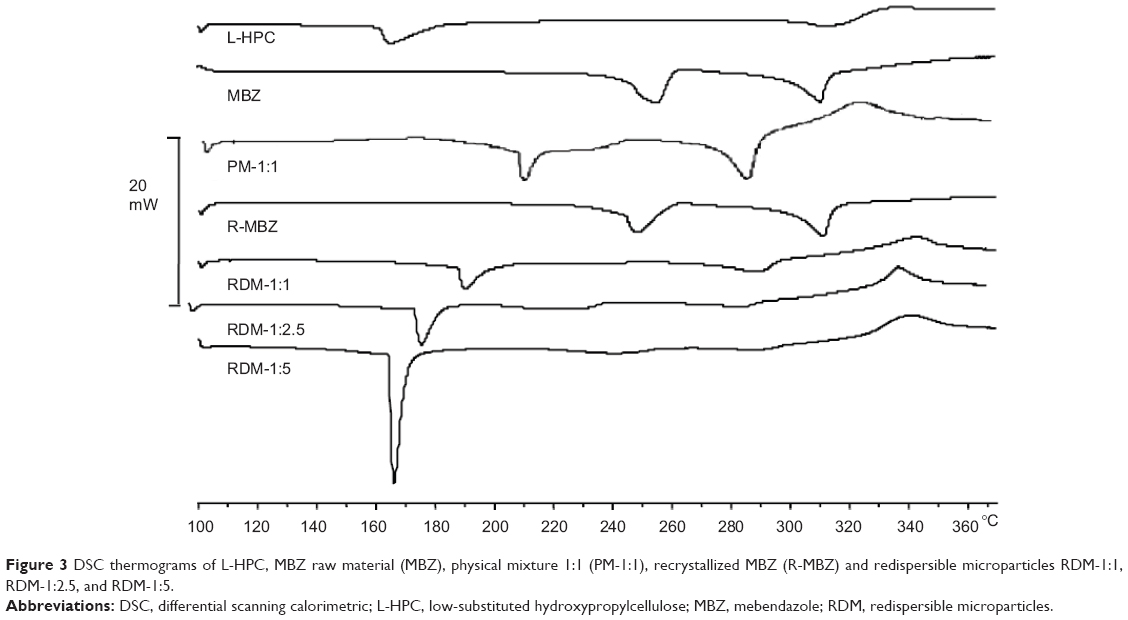 | Figure 3 DSC thermograms of L-HPC, MBZ raw material (MBZ), physical mixture 1:1 (PM-1:1), recrystallized MBZ (R-MBZ) and redispersible microparticles RDM-1:1, RDM-1:2.5, and RDM-1:5. |
In the thermogram of the carrier (L-HPC), a melting peak was observed at 167.92°C (83.96 J/g), and a slight endothermic/exothermic transition (300.68°C–333.36°C). In view of this enthalpy value this substance could present a semi-crystalline structure.
The PM-1:1 shows a shift to the left of the two MBZ endothermic peaks at 209.12°C and 284.42°C. However, the endothermic peak of L-HPC is not observed. Besides, a major endothermic/exothermic event appears in this scan (313.22°C–333.06°C) which suggests a possible solid–solid interaction between the two components, which does not necessarily indicate pharmaceutical incompatibility.26,27
All MBZ:L-HPC microparticles (RDM-1:1, RDM-1:2.5, and RDM-1:5) show a first endothermic peak, which could correspond to the semi-crystalline structure of L-HPC. When the amount of L-HPC increases in the formulation, its peak is shifted to the left (with values ranging from 189.21°C for RDM-1:1 to 166.10°C for RDM-1:5). On the other hand, in all RDM, the endothermic peak of MBZ is not observed. However, only RDM-1:1 shows a slight endothermic change close to 286.89°C. Moreover, all RDM formulations show a characteristic endothermic/exothermic event (327.69°C–353.42°C). These results could be due to the interaction between the polymer chains of the L-HPC with the drug, and to the formation of amorphous species of MBZ within the semi-crystalline structure of L-HPC.28
In vitro dissolution assay
The dissolution profiles of MBZ raw material, the recrystallized product, and the different RDM are illustrated in Figure 4. MBZ raw material and the recrystallized product exhibited similar poor dissolution rates at pH 1.2. However, formulations RDM-1:2.5 and RDM-1:5 show faster release profiles. The results of dissolution tests expressed in terms of DP dissolved at 30 minutes and 2 hours, DE at 30 minutes and 4 hours, and relative dissolution rate (RDR) at 5 minutes are summarized in Table 1.
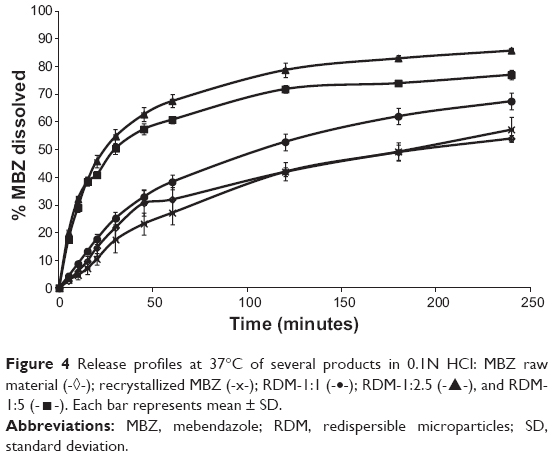 | Figure 4 Release profiles at 37°C of several products in 0.1N HCl: MBZ raw material (-◊-); recrystallized MBZ (-x-); RDM-1:1 (-•-); RDM-1:2.5 (-▴-), and RDM-1:5 (-◼-). Each bar represents mean ± SD. |
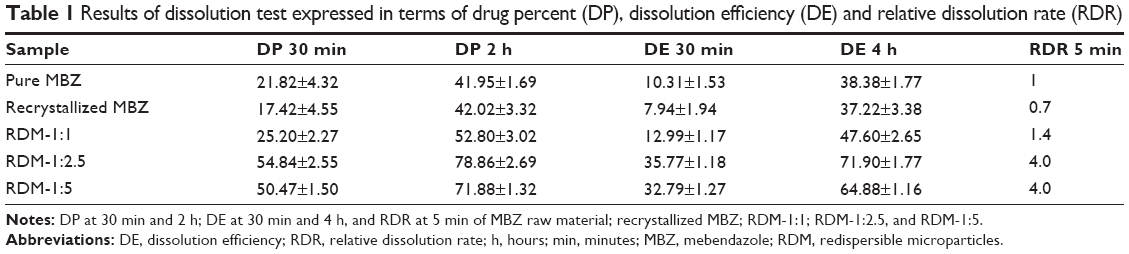 | Table 1 Results of dissolution test expressed in terms of drug percent (DP), dissolution efficiency (DE) and relative dissolution rate (RDR) |
Samples with lower percentages dissolved: MBZ raw material and recrystallized drug showed the lowest values at 30 minutes (21.82%±4.32% and 17.42%±4.55%, respectively) and 2 hours (41.95%±1.69% and 42.02%±3.32%, respectively). These percentages at 2 hours are intermediate compared to the dissolution results at the same time (2 hours) for polymorph C (50%) or polymorph A (almost 20%) obtained by Froehlich and Gasparotto29 under the same conditions. Thus, our dissolution rate data confirm the results of DSC indicating that both pure and recrystallized MBZ, present a mixture of MBZ polymorph C (major) and MBZ polymorph A (minor).25
RDM with the lowest amount of hydrophilic polymer (RDM-1:1) showed low percentages dissolved at 30 minutes (25.20%±2.27%) and 2 hours (52.80%±3.02%). However, samples with higher amounts of hydrophilic L-HPC (RDM-1:2.5 and RDM-1:5) showed the higher values of DP 30 minutes and DP 2 hours (54.84%±2.55% and 50.47%±1.50%, respectively for DP 30 minutes and 78.86%±2.69% and 71.88%±1.32%, respectively for DP 2 hours). The high values of dissolution for both formulations, RDM-1:2.5 and RDM-1:5, may be related to the MBZ structure, mostly amorphous, shown in DSC studies. These results are in agreement with those obtained by Moes et al30 where the apparition of microaggregates increased the apparent solubility of the active substance by several factors: firstly the physical state of MBZ, as amorphous, has a higher dissolution rate than the crystalline state and secondly, the polymer, partially soluble, functions as a wetting agent for the drug. When the ratio of hydrophilic polymer over MBZ was decreased to 1:1 w/w (RDM-1:1), their low dissolution rates could be related to the lower proportion of L-HPC as wetting agent of this formulation. Formulations with lesser amounts of carrier have the lowest dissolution profiles.31
To evaluate the different formulations in terms of RDR, the largest differences are observed at initial times (5 minutes).32,33 Table 1 indicates that RDM-1:1 presents only 1.4-fold compared to pure MBZ, whereas in formulations with higher amounts of hydrophilic carrier (L-HPC) such as RDM-1:2.5 and RDM-1:5, there is a 4-fold increase in MBZ dissolution rate from both RDM-1: 2.5 and RDM-1:5 as compared to that from the pure drug. The fast disintegration time of hydrophilic carriers such as L-HPC, together with its excellent wetting properties are some of the reasons for this outstanding dissolution behavior. Similar results were obtained by Loh et al4 who studied the effect of L-HPC on the solubility and dissolution parameters of norfloxacin, a poorly water-soluble drug.
MBZ raw material and recrystallized MBZ, showed the lowest DE values. Both these low DE 30 minutes and 4 hours results (Table 1) could be attributed to the low drug solubility.15 The low amount of hydrophilic polymers (L-HPC) as carrier (RDM-1:1), produces a small increase in the DE values (12.99%±1.17% for DE 30 minutes and 47.60%±2.65% DE 4 hours). This low efficiency of dissolution might be due to the low humectability. By contrast, MBZ:L-HPC 1:2.5 and 1:5 showed higher DE 30 minutes (35.77%±1.18% and 32.79%±1.27%, respectively) and DE 4 hours (71.90%±1.77% and 64.88%±1.16%, respectively). The improvement in this dissolution parameter is related to the higher wettability caused by the major increase of L-HPC amount. A similar increase of these final DE values were obtained by other formulations of norfloxacin, which used the same hydrophilic polymer, L-HPLC, as a carrier.4
Oral bioavailability study
Figure 5 shows the MBZ plasma concentrations following oral administration of MBZ raw material, recrystallized MBZ, and MBZ redispersible microparticles RDM-1:1, 1:2.5, and 1:5 at 5 mg/kg. It is clear from this Figure that RDM improve MBZ’s oral bioavailability in relation to both MBZ raw material and recrystallized MBZ. The effect of L-HPC and other excipients, such as povidone and cyclodextrins, used as complexing agents to improve oral bioavailability of low soluble drugs, such as MBZ, have been previously reported.12,18,19
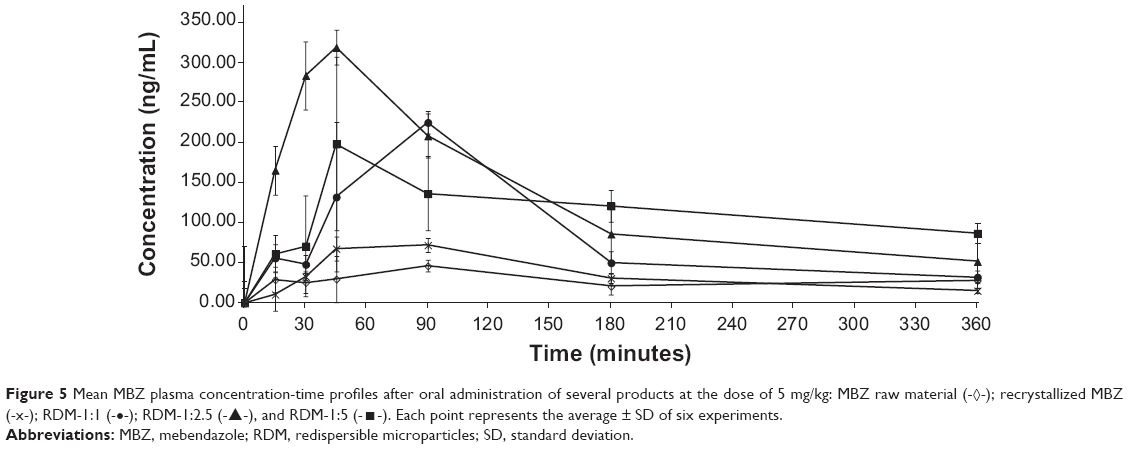 | Figure 5 Mean MBZ plasma concentration-time profiles after oral administration of several products at the dose of 5 mg/kg: MBZ raw material (-◊-); recrystallized MBZ (-x-); RDM-1:1 (-•-); RDM-1:2.5 (-▴-), and RDM-1:5 (-◼-). Each point represents the average ± SD of six experiments. |
The MBZ pharmacokinetic parameters for all these treatments are shown in Table 2. The pharmacokinetic profile of MBZ raw material was comparable to that of recrystallized MBZ with the mean AUC0−∞ values of 36.043±8.867 μg min/mL and 30.542±5.896 μg min/mL (P>0.05), respectively. However, Cmax was higher following the administration of recrystallized MBZ (0.073±0.009 μg/mL) versus MBZ alone (0.052±0.014 μg/mL), this difference being statistically significant (P=0.016).
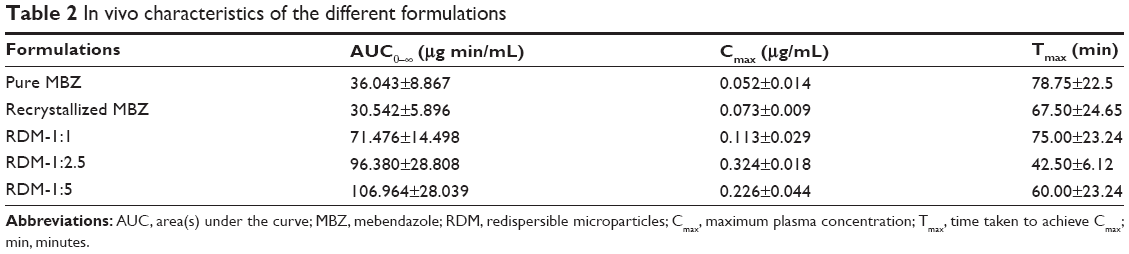 | Table 2 In vivo characteristics of the different formulations |
High variability in plasma MBZ concentrations was observed after oral administration. In this study, the low solubility of the drug has limited its absorption and kinetics after oral administration under these dosage forms. A similar result was observed when similar drugs of low solubility and permeability were administered.2,34
When MBZ was administered as L-HPC RDM, drug plasma concentrations became significantly higher (P<0.001) than for MBZ raw material. A similar enhanced bioavailability of a poorly water soluble drug, fenofibrate, was achieved by amorphous solid forms produced by a similar freeze drying process.35
Cmax =0.113±0.029 μg/mL) was reached at 75.00±23.24 minutes (Tmax) after RDM-1:1 administration and it was higher than when the pure drug or recrystallized MBZ were administered (Table 2). However, in this case, after reaching Cmax, plasma concentrations decreased sharply, thus achieving a value of 0.032±0.014 μg/mL at 6 hours, similar to those obtained at the same time point after administration of MBZ raw material (0.028±0.011 μg/mL) and about two times higher than those after administration of recrystallized MBZ (1:0) (0.015±0.006 μg/mL). Consequently, the MBZ AUC0−∞ value was greater for RDM-1:1 than for MBZ raw material or recrystallized MBZ (1.98 and 2.34-fold increase respectively). The use of an amorphous form of poorly water-soluble drugs has widened the window of achievable pharmacokinetic performance enormously.36
For formulations RDM-1:2.5 and RDM-1:5, MBZ levels showed a high increase in their maximum concentration values in comparison to mice treated with RDM-1:1. Thus, a Cmax of 0.324±0.018 μg/mL was achieved at 42.5±6.12 minutes (Tmax) for RDM-1:2.5 whereas a Cmax of 0.226±0.044 μg/mL was achieved at 60.00±23.24 minutes (Tmax) for RDM-1:5. Furthermore, as the L-HPC proportion increased in RDM from (1:2.5) to (1:5), MBZ plasma concentrations remained at levels higher than 0.050 μg/mL up to 6 hours (Figure 5). Therefore, MBZ Cmax and AUC0−∞ values were greater for both RDM-1:2.5 (2.87 and 1.35-fold increase respectively) and RDM-1:5 (2.00 and 1.50-fold increase respectively) with respect to RDM-1:1. If the comparison is with the MBZ raw material the differences for both Cmax and AUC0−∞ are greater (6.23 and 2.67-fold increase for RDM-1:2.5 and 4.35 and 2.97-fold increase for RDM-1:5 respectively). Thus, the slower dissolution profiles of RDM-1:1, pure MBZ, and recrystallized MBZ have probably been the rate-limiting factor in studies of absorption after oral administration of MBZ.2
It must be noted that in the case of animals receiving RDM-1:5, although the Cmax was lower than in those receiving RDM-1:2.5, plasma concentrations were maintained at constant levels higher than in the other formulations and for more than 6 hours (0.087±0.020 μg/mL at 6 hours). Therefore, a prolonged absorption time was seen post administration of RDM-1:5 and its AUC0−∞ was about 3.50-fold greater than following administration of recrystallized MBZ. A similar MBZ prolonged absorption time was obtained by Krishnaiah et al34 with a sustained release formulation.
Both in vitro parameters: RDR at 5 minutes and DE at 30 minutes could be related to a rapid MBZ intestinal absorption. These values are in a good correlation with Cmax and AUC0−∞ results respectively. Thus, RDR 5 minutes and Cmax for RDM-1:5 were 4.00 and 4.35-fold higher, respectively, than that obtained by pure MBZ. Besides, DE 30 minutes and AUC0−∞ values for RDM-1:5 were 3.18 and 2.97-fold higher, respectively, than that obtained for MBZ raw material.
In view of these results, RDM-1:5 has been selected to carry out studies on anthelmintic activity due to its suitable bioavailability properties.
Evaluation of the efficacy of MBZ dosage forms on a T. spiralis mouse model
In the T. spiralis life cycle, low doses of MBZ (5 mg/kg) are used against early adult and adult worms stages.20–37 However, high doses of MBZ raw material (among 20–100 mg/kg) are usually utilized for reducing the encysted parasite.38,39 Nevertheless, the use of low doses of MBZ (5 mg/kg) in formulations with a rapid dissolution rate are selected in the present work for assessing whether the rapid dissolution profile of this low dose (5 mg/kg), increases the pharmacokinetic values and the anthelmintic activity of MBZ against the different stages of the T. spiralis life cycle.
Figure 6 shows the anthelmintic efficacy for the following formulations at the same dose: control (0.75% CMC), raw material and L-HPC RDM 1:5, against the four stages of the T. spiralis life cycle. The effectiveness of MBZ RDM-1:5 against early adult stages of T. spiralis was compared with that of pure MBZ at 5 mg/kg. For all infected mice treated with RDM-1:5 a higher efficacy was obtained, achieving 87.92% worm reduction (actual worm number: 19.30±9.52). At this parasitic stage, and at the dose of 5 mg/kg, the anti-parasitic effect of the formulation became 4.44-fold greater than that of MBZ raw material (worm number: 122.00±10.86). This finding might be explained by better dissolution properties of the RDM when compared to MBZ pure drug as assessed above.
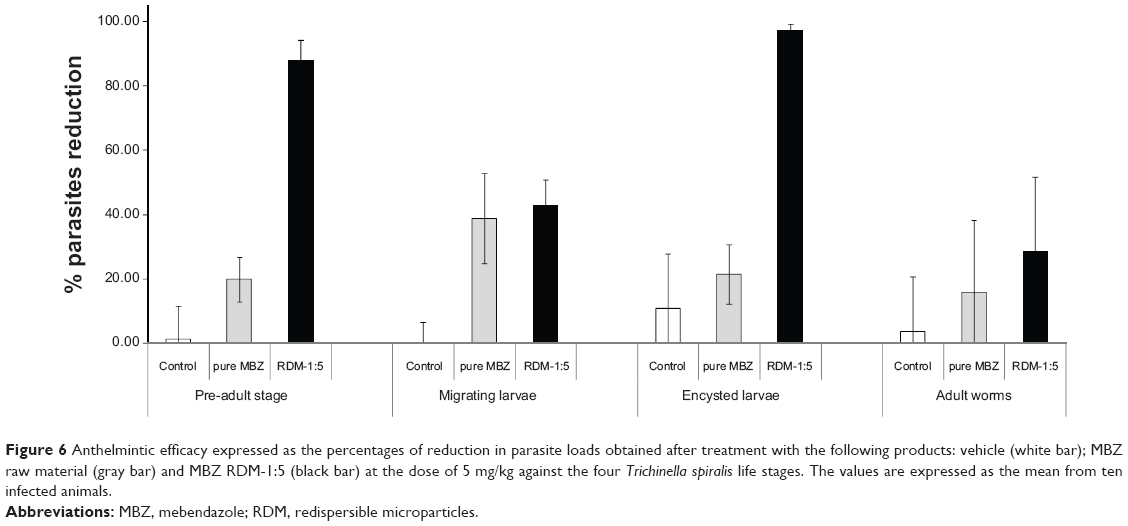 | Figure 6 Anthelmintic efficacy expressed as the percentages of reduction in parasite loads obtained after treatment with the following products: vehicle (white bar); MBZ raw material (gray bar) and MBZ RDM-1:5 (black bar) at the dose of 5 mg/kg against the four Trichinella spiralis life stages. The values are expressed as the mean from ten infected animals. |
Both MBZ products presented similar percentages of worm reduction when administered against migrating larvae (38.80% and number of worms 39,094.60±8,940.82 for pure MBZ and 42.82%, worm number 36,428.40±5,116.56 for RDM-1:5, respectively), in relation to control (0.10% and worm number 45,931.33±2,940.12). In this parenteral stage, the activity of RDM-1:5 showed minor changes as compared with the drug alone, as previously reported with albendazole.20
Against encysted larvae, MBZ raw material registered 10% higher activity (21.36% and number of worms 50,116.50±5,704.03) in relation to untreated control (10.70% and worm number 56,910.71±10,877.76) whereas the RDM-1:5 was significantly (P>0.05) more active (97.36%, worm data 1,220.83±646.40) as compared with pure MBZ (21.36%). Comparison of the efficacy results of pure MBZ and MBZ redispersable microparticles RDM-1:5 (4.56-fold greater) enabled us to assess the influence of different pharmacokinetic parameters as Cmax (4.35-fold greater) and AUC (2.97-fold greater) on this stage of the T. spiralis life cycle. The good results of efficacy with this low dose (5 mg/kg) against this stage of the parasite can be attributed to greater absorption of RDM-1:5 which allows high plasma concentrations during the 3 days of treatment. This extraordinary increase in anthelmintic activity of MBZ may render this formulation very suitable for treatment of inoperable or disseminated cases of other systemic helminthiasis such as hydatidosis or neurocysticercosis.40
In the adult worm stage, both MBZ raw material and RDM-1:5 registered reduction percentages of 15.83% (worm number 77.57±20.52) and 28.53% (worm number 65.43±21.27), respectively, in relation to untreated control (3.50% and worm number 87.75±15.80) The effectiveness of MBZ RDM-1:5 against adult worms was significantly lower (28.53%) than against any other parasite stages (P>0.05). The decline in effectiveness of albendazole and other benzimidazole-carbamates as the parasite matured from the pre-adult to the adult stage has been previously verified.41 Overall, the results of these experiments show that T. spiralis is a good model for the evaluation of new MBZ formulations against either enteral or parenteral helminthic infections.
Conclusion
MBZ RDM in L-HPC carrier showed a rapid dissolution behavior and an enhanced oral bioavailability by formation of amorphous drug forms and improved wettability (for RDM-1:2.5 and RDM-1:5 the AUC0−∞ were 2.67- and 2.97-fold higher, respectively, compared to those of pure MBZ). Efficacy studies showed that at a very low dose of MBZ (5 mg/kg), these systems showed very good effectivity against the different stages of T. spiralis. RDM system (RDM-1:5) was significantly (P>0.05) more active (4.56-fold), as compared with pure MBZ, against the encysted larvae stage. Therefore, these RDM may provide a more secure drug system for oral poorly water soluble drugs in the clinical treatment of systemic helminthic infections with low drug doses.
Acknowledgment
This work has been supported by a project from Complutense University of Madrid (Project number: CC G07-UCM/BIO-2824).
Disclosure
The authors report no conflicts of interest in this work.
References
Hemphill A, Müller J. Alveolar and cystic echinococcosis: towards novel chemotherapeutical treatment options. J Helminthol. 2009;83(2): 99–111. | ||
Dayan AD. Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta Trop. 2003;86(2–3): 141–159. | ||
Sievens-Figueroa L, Bhakay A, Jerez-Rozo JI, et al. Preparation and characterization of hydroxypropyl methyl cellulose films containing stable BCS Class II drug nanoparticles for pharmaceutical applications. Int J Pharm. 2012;423(2):496–508. | ||
Loh GOK, Tan YT, Peh KK. Hydrophilic polymer solubilization on norfloxacin solubility in preparation of solid dispersion. Powder Technology. 2014;256:462–469. | ||
Onoue S, Sato H, Ogawa K, et al. Improved dissolution and pharmacokinetic behavior of cyclosporine. A using high-energy amorphous solid dispersion approach. Int J Pharm. 2010;399(1–2):94–101. | ||
Lim SC, Paech MJ, Sunderland B, Liu Y. In vitro and in vivo evaluation of a sublingual fentanyl water formulation. Drug Des Devel Ther. 2013;7:317–324. | ||
Dinunzio J C, Brough C, Hughey J R, Miller DA, Williams RO, McGinity JW. Fusion production of solid dispersions containing a heat-sensitive active ingredient by hot melt extrusion and Kinetisol dispersing. Eur J Pharm Biopharm. 2010;74(2):340–351. | ||
Usha AN, Mutalik S, Reddy MS, Ranjith AK, Kushtagi P, Udupa N. Preparation and, in vitro, preclinical and clinical studies of aceclofenac spherical agglomerates. Eur J Pharm Biopharm. 2008;70(2): 674–683. | ||
Kawabata Y, Yamamoto K, Debari K, Onoue S, Yamada S. Novel crystalline solid dispersion of tranilast with high photostability and improved oral bioavailability. Eur J Pharm Biopharm. 2010;39(4):256–262. | ||
Molina-Berríos A, Campos-Estrada C, Lapier M, et al. Benznidazole prevents endothelial damage in an experimental model of Chagas disease. Acta Trop. 2013;127(1):6–13. | ||
Matadamas-Martínez F, Nogueda-Torres B, Hernández-Campos A, et al. Analysis of the effect of a 2-(trifluoromethyl)-1H-benzimidazole derivative on Trichinella spiralis muscle larvae. Vet Parasitol. 2013; 194(2–4):193–207. | ||
Daniel-Mwambete K, Torrado S, Cuesta-Bandera C, Ponce-Gordo F, Torrado JJ. The effect of solubilization on the oral bioavailability of three benzimidazole carbamate drugs. Int J Pharm. 2004;272(1–2): 29–36. | ||
Swanepoel E, Liebenberg W, Villiers MM. Quality evaluation of generic drugs by dissolution test: changing the USP dissolution medium to distinguish between active and non-active mebendazole polymorphs. Eur J Pharm Biopharm. 2003;55(3):345–349. | ||
Cirri M, Maestrelli F, Mennini N, Mura P. Influence of the preparation method on the physical-chemical properties of ketoprofen-cyclodextrin-phosphatidylcholine ternary Systems. J Pharm Biomed Anal. 2009; 50(5):690–694. | ||
Cirri M, Maestrelli F, Mennini N, Mura P. Physical-chemical characterization of binary and ternary systems of ketoprofen with cyclodextrins and phospholipids. J Pharm Biomed Anal. 2009;50(5):683–689. | ||
Vo C L, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3 Pt B):799–813. | ||
García-Rodríguez JJ, Torre-Iglesias, PM, Vegas-Sánchez MC, Torrado-Durán S, Bolás-Fernández F, Torrado-Santiago S. Changed crystallinity of mebendazole solid dispersion: Improved anthelmintic activity. Int J Pharm. 2011;403(1–2):23–28. | ||
Torrado S, López M L, Torrado G, Bolás F, Torrado S, Cadórniga R. A novel formulation of albendazole solution: oral bioavailability and efficacy evaluation. Int J Pharm. 1997;156:181–187. | ||
Beinborn NA, Dua J, Wiederhold NP, Smyth HD, Williams RO, 3rd. Dry powder insufflation of crystalline and amorphous voriconazole formulations produced by thin film freezing to mice. Eur J Pharm Biopharm. 2012;81(3):600–608. | ||
García JJ, Bolás F, Torrado JJ. Bioavailability and efficacy characteristics of two different oral liquid formulations of albendazole. Int J Pharm. 2003;250(2):351–358. | ||
Wakelin D, Lloyd M. Accelerated expulsion of adult Trichinella spiralis in mice given lymphoid cells and serum from infected donors. Parasitology. 1976;72(3):307–315. | ||
Cerdeira AM, Mazzotti M, Gander B. Formulation and drying of miconazole and itraconazole nanosuspensions. Int J Pharm. 2013; 443(1–2):209–220. | ||
Karavas E, Georgarakis E, Sigalas MP, Avgoustakis K, Bikiaris D. Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug–polymer interactions. Eur J Pharm Biopharm. 2007;66(3):334–347. | ||
Jana S, Maji N, Nayak AK, Sen KK, Basu SK. Development of chitosan-based nanoparticles through inter-polymeric complexation for oral drug delivery. Carbohydr Polym. 2013;98(1):870–876. | ||
De Villiers MM, Terblanche RJ, Liebenberg W, Swanepoel E, Dekker TG, Song M. Variable-temperature X-ray powder diffraction analysis of the crystal transformation of the pharmaceutically preferred polymorph C of mebendazole. J Pharm Biomed Anal. 2005;38(3): 435–441. | ||
Mura P, Manderioli A, Bramanti G, Furlanetto S, Pinzauti S. Utilization of differential scanning calorimetry as a screening technique to determine the compatibility of ketoprofen with excipients. Int J Pharm. 1995;119:71–79. | ||
Pani NR, Nath LK, Acharya S, Bhuniya B. Application of DSC, IST, and FTIR study in the compatibility testing of nateglinide with different pharmaceutical excipients. J Therm Anal Calorim. 2012;108(1): 219–226. | ||
Damian F, Blaton N, Naessens L, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10(4):311–322. | ||
Froehlich PE, Gasparotto FS. Mebendazol: identificação das formas polimórficas em diferentes matérias-primas e medicamentos (referência e genéricos) disponíveis no mercado nacional. [Mebendazole: identification of polymorphic forms in different raw materials and drugs (reference and generics) available domestically]. Rev Ciênc Farm Básica Apl. 2005;26(3):205–210. Portuguese. | ||
Moes J, Koolen S, Huitema A, Schellens J, Beijnen J, Nuijen B. Development of an oral solid dispersion formulation for use in low-dose metronomic chemotherapy of paclitaxel. Eur J Pharm Biopharm. 2013;83(1):87–94. | ||
Karmwar P, Graeser K, Gordon KC, Strachan CJ, Rades T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int J Pharm. 2011;417(1–2): 94–100. | ||
Mura P, Bettinetti GP, Cirri M, Maestrelli F, Sorrenti M, Catenacci L. Solid-state characterization and dissolution properties of Naproxen–Arginine–Hydroxypropyl-β-cyclodextrin ternary system. Eur J Pharm Biopharm. 2005;59(1):99–106. | ||
Chadha R, Bala M, Arora P, Jain DV, Pissurlenkar RRS, Coutinho EC. Valsartan inclusion by methyl-β-cyclodextrin: Thermodynamics, molecular modelling, Tween 80 effect and evaluation. Carbohydr Polym. 2014;103(15):300–309. | ||
Krishnaiah YS, Veer RP, Dinesh KB, Satyanarayana V, Karthiekeyan RS, Bhaskar P. Pharmacokinetic evaluation of guar gum-based colon-targeted drug delivery systems of mebendazole in healthy volunteers. J Control Release. 2003;88(1):95–103. | ||
Zhang M, Li H, Lang B, et al. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur J Pharm Biopharm. 2012;82(3):534–544. | ||
Yang W, Johnston KP, Williams RO, 3rd. Comparison of bioavailability of amorphous versus crystalline itraconazole nanoparticles via pulmonary administration in rats. Eur J Pharm Biopharm. 2010; 75(1):33–41. | ||
Hess JA, Chandrasekar PH, Mortiere M, Molinari JA. Comparative Efficacy of Ketoconazole and Mebendazole in Experimental Trichinosis. Antimicrob Agents Chemother. 1986;30(6):953–954. | ||
de-la-Rosa JL, Alvarez N, Gomez-Priego A. Study of the reproductive capacity of Trichinella spiralis recovered from experimentally infected mice under-dosed with albendazole or mebendazole. Trop Biomed. 2007;24(2):93–97. | ||
Maki J, Yanagisawa T. Comparative efficacy of flubendazole and mebendazole on encysted larvae of Trichinella spiralis (USA strain) in the diaphragm of mice and rats. J Helminthol. 1988;62(1):35–39. | ||
Zardi EM, Petrarca V, Pozio E, Uwechie V, Tamburrini A, Laghi V. A case from India of neurocysticercosis responsive to albendazole treatment. Parasite. 2000;7(1):54–56. | ||
Yépez-Mulia L, Morales-Hurtado R, Viveros-Guzmán N, et al. Evaluation of albendazole prodrugs in experimental trichinellosis. Arch Med Res. 1999;30(5):368–374. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.