")
Back to Journals » International Journal of Nanomedicine » Volume 14
Enhanced anti-tumor immunotherapy by silica-coated magnetic nanoparticles conjugated with ovalbumin
Authors Lee SJ, Kim JJ, Kang KY, Paik MJ, Lee G, Yee ST
Received 13 November 2018
Accepted for publication 27 July 2019
Published 16 October 2019 Volume 2019:14 Pages 8235—8249
DOI https://doi.org/10.2147/IJN.S194352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Sung-Ju Lee,1 Jong-Jin Kim,2 Kyung-Yun Kang,3 Man-Jeong Paik,1 Gwang Lee,4 Sung-Tae Yee1
1Department of Pharmacy, Sunchon National University, Suncheon 540-742, Republic of Korea; 2Center for Self-assembly and Complexity, Institute for Basic Science (IBS), Pohang 37673, Republic of Korea; 3Suncheon Research Center for Natural Medicines, Suncheon, Republic of Korea; 4Department of Physiology and Department of Biomedical Science, Ajou University School of Medicine, Suwon, Republic of Korea
Correspondence: Sung-Tae Yee
Department of Pharmacy, Sunchon National University, 255 Joongang-Ro, Seokhyeon-Dong, Suncheon 549-742, Republic of Korea
Tel +82 61 750 3752
Fax +82 61 750 3708
Email [email protected]
Background: The effective induction of an antigen-specific T cell immune response through dendritic cell activation is one of the key goals of tumor immunotherapy.
Methods: In this study, efficient antigen-delivery carriers using silica-coated magnetic nanoparticles were designed and, their antigen-specific T cell immune response through dendritic cell activation investigated.
Results: The results showed that the silica-coated magnetic nanoparticles with conjugated ovalbumin enhanced the production of cytokines and antigen uptake in bone marrow-derived dendritic cells. Also, this induced an antigen-specific cytotoxic T lymphocyte (CTL) immune response and activated antigen-specific Th1 cell responses, including IL-2 and IFN-γ production and proliferation. We proved that the immune-stimulatory effects of silica-coated magnetic nanoparticles with conjugated ovalbumin were efficient in inhibiting of tumor growth in EG7-OVA (mouse lymphoma-expressing ovalbumin tumor-bearing mice model).
Conclusion: Therefore, the silica-coated magnetic nanoparticles with conjugated ovalbumin are expected to be useful as efficient anti-cancer immunotherapy agents.
Keywords: antigen-delivery systems, silica-coated magnetic nanoparticles, ovalbumin, EG-7, dendritic cell, CD4+ T cell, CD8+ T cell
Introduction
Tumor-immunotherapy has emerged as an alternative and innovative therapeutic intervention that can overcome the side effects and limited efficacy of conventional chemotherapy against chemo-resistant and relapsing tumors.1–3 A major milestone in the development of tumor-immunotherapy is the development of dendritic cells (DCs)-based therapy or T-cell adoptive transfer therapy and has been validated in several clinical trials.1,4,5 Although DC-based therapy approaches have been shown to be effective in clinical trials, they are complex and require multiple ex vivo manipulations beginning from the isolation of DCs from the blood of the patients, their exposure to antigens and other maturation stimuli, and finally their reinjection into the patients.6,7 This is a personalized but expensive therapeutic approach, and these cell-based therapeutic strategies require significant cost, labor, and time for the isolation, activation, and proliferation of these immune cells before they are re-injected into the patient.1 Therefore, nanoparticle-based vaccines have attracted substantial attention for the induction of an immune response without any ex vivo manipulations to overcome these limitations.1,8,9 Nanoparticles are being studied as the next-generation platform in the pharmaceutical and biomedical fields due to their high potential for the controlled intracellular delivery of biomolecules and drugs.6 Also, in recent studies, various types of polymer nanoparticles that can target and deliver specific antigens for immunotherapy have been reported to provide protective immunity against cancer and infectious diseases.10–14
Recently, nanoparticles have attracted a great deal of attention as potential candidates for antigen delivery vehicles.6,10 Most nanoparticles-based active tumor immunotherapy studies have demonstrated the enhanced function of DCs and their antigen-specific response.10 However, the problem of the potential toxicity of the nanoparticles has not yet been solved.15–20 Therefore, we used nanoparticles coated with silica (SiO2) which are known to be biocompatible materials, on the particle surfaces to overcome these problems,15–20 and we chose ovalbumin (OVA) as a model antigen to study the function of DCs and their antigen-specific response.
DCs are professional antigen-presenting cells (APCs) involved in immune responses that regulate various types of immune cells.5,21–23 Especially, DCs trigger the activation of helper T cells or cytotoxic T cells.21–24 Therefore, DCs induce cell-mediated immune responses and have anti-tumor effects on cytotoxic T cells. Also, DCs play a major role in the production of antigen-specific cytotoxic T lymphocytes (CTLs) and CTL-mediated tumor immunotherapy. Therefore, the development of nanoparticle-based vaccine formulations that can generate strong Th1 and CTL-mediated immune responses is paramount.
In this research, we explored the effects of silica-coated magnetic nanoparticles with conjugated OVA on the cytotoxicity and activation of DCs. Also the response of the OVA-specific Th1 cells was increased by the silica-coated magnetic nanoparticles with conjugated OVA, and we showed their potent applications in cancer immunotherapies.
Materials and methods
Animals and experimental treatments in vivo
Female 8- to 12-week-old C57BL/6 mice, weighing 20–22 g each, were purchased from Orientbio (Orientbio Inc., Seongnam, Korea). The animals were housed in a controlled environment [22±2 °C and 50±5% (relative humidity)] in polycarbonate cages and fed a standard animal diet with water. All of the mice were treated in strict accordance with the guidelines issued for the care and use of laboratory animals by the Sunchon National University Institutional Animal Care and Use Committee (SCNU IACUC). All procedures were approved by the SCNU IACUC (Permit Number: SCNU IACUC-2017-07)
Reagents and antibodies
Recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (rmIL)-4 were purchased from R&D Systems (Minneapolis, MN, USA), propidium iodide (PI), and ovalbumin (OVA) were purchased from Sigma-Aldrich (Steinheim, Germany), and lipopolysaccharide (LPS) and OVA-Alexa 488 were purchased from Invitrogen (Carlsbad, CA, USA). The following FITC- or PE-conjugated monoclonal antibodies (Abs) and non-labeled Abs were purchased from BD Biosciences (San Jose, CA, USA): FITC-annexin V, CD16/32 (2.4G2), CD11c (HL3), IA[b] (AF6–120.1), IFN-γ, CD4, PE-CD8, CD4. Cytokine ELISA primary and secondary -antibodies specific for murine IL-1β, IL-6, IL-12p70, IFN-γ, IL-2, IL-10, and TNF-α were purchased from BD Biosciences (San Jose, CA, USA). 5-Bromo-2′-Deoxy-Uridine Labeling and Detection Kit III and collagenase D were purchased from Roche (Salt Lake City, UT, USA).
Cell culture
E.G7-OVA, a chicken egg OVA gene-transfected clone of EL4, which presents OVA with MHC class I molecules, was obtained from the American Type Culture Collection (Manassas, VA, USA).25
Silica-coated magnetic nanoparticles: MNPs@SiO2(RITC)
Silica nanoparticles were purchased from Biterials (Korea). MNPs@SiO2(RITC) particles containing a cobalt ferrite core (CoFe2O3) sheathed by a silica shell that is chemically bonded to rhodamine isothiocyanate dye (RITC). The silica nanoparticles are identical to the MNPs@SiO2(RITC) except that they lack a cobalt ferrite core and show similar tendencies concerning their biological effects. The MNPs@SiO2(RITC) and silica nanoparticles are 54.2 nm ±8.9%. In diameter, and the size distribution and zeta-potential of both nanoparticles have been previously reported (Figure 1A ).20,26,27
Preparation of OVA-silica-coated magnetic nanoparticles
MNPs@SiO2(RITC) with conjugated Ovalbumin were prepared by mixing 50 μg/mL of OVA with MNPs@SiO2(RITC) at a concentration of 50 μg/mL using a vortex for 60 min (Figure 1A 20). The morphology of the OVA- MNPs@SiO2(RITC) was examined using a Confocal Laser Scanning Microscope (CLSM) (Rhodamine 123: excitation558, emission; 581 FITC: excitation 488, emission 519) (Figure 1B).
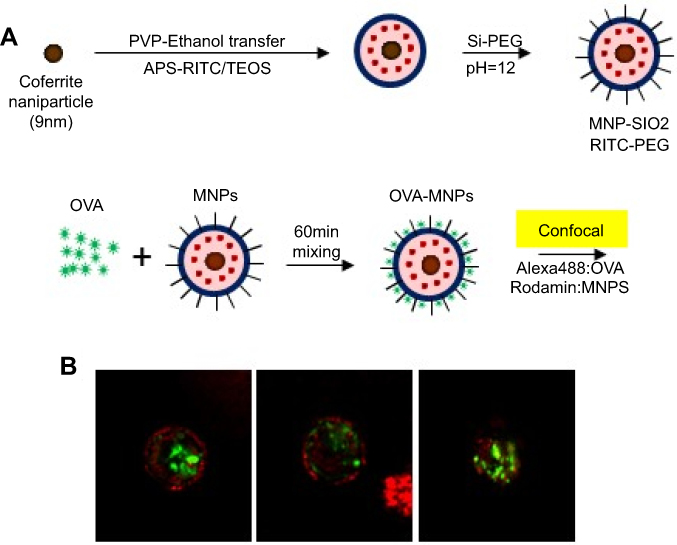 |
Figure 1 Confocal Laser Scanning Microscope (CLSM) image of OVA- MNPs@SiO2(RITC). (A) For the preparation of the nanoparticles,20 OVA (50 μg/mL) and MNPs@SiO2(RITC) (50 μg/mL) were mixed for 60 min. (B) MNPs@SiO2(RITC): Rhodamine 123: excitation558, emission; 581 OVA: FITC: excitation 488, emission 519. |
Generation and activation of bone marrow-derived DCs
BMDCs were obtained from the femurs and tibias of C57BL/6 mice, and the red cells were treated with lysis buffer solution (4.15 g of ammonium chloride per 500 mL of 0.01 M Tris–HCl buffer). The cells were washed and cultured in six-well tissue culture plates at 1×106 cells/mL in complete RPMI culture medium supplemented with 10% fetal bovine serum (FBS), 2-mercaptoethanol (50 μM/mL), IL-4 (1,000 U/mL), and GM-CSF (1,000 U/mL). The culture medium was changed on culture days 2 and 4. New medium and cytokines (ie, GM-CSF and IL-4) were added after flushing out the non-adherent cells. On day 6, the loosely adherent clustered cells were defined as immature BMDCs, treated with LPS, OVA, and OVA- MNPs@SiO2(RITC) for 24 h and then harvested.
Annexin-V/PI assay
Apoptosis induction by MNPs@SiO2(RITC) was quantified via flow cytometry using Annexin V-FITC and a propidium iodide (PI) solution, according to the manufacturer’s instructions. Briefly, BMDCs (1×106/well) were seeded onto 24-well plates and exposed to MNPs@SiO2(RITC) or for 24 h. Apoptosis was quantified by staining with Annexin V-FITC/PI. Finally, the samples were analyzed by flow cytometry, and the apoptosis percentages were calculated by counting the Annexin V-positive cells.
Antigen uptake assay
Antigen uptake by BMDCs was measured using the standard method. OVA-Alexa488 (Invitrogen) was used as the model antigen. BMDCs (2×105) were incubated for various time points at 37 °C with OVA-Alexa488 (50 µg/mL) or OVA-Alexa488-MNPs (OVA-Alexa488 50 µg/mL/MNPs 25, and 50 µg/mL) particles. Uptake was terminated by washing the cells with ice-cold fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline containing 1% FBS and 0.1% NaN3). Cells were stained with PE-conjugated anti-CD11c (BD Biosciences). Alexa488 fluorescence of CD11cpositive cells was measured using an FACS CantoII (BD Biosciences). The morphology of the BMDCs was examined using a Confocal Laser Scanning Microscope (CLSM) (PE: excitation 496, emission; 578 FITC: excitation 488, emission 519).
OVA-specific Th1 cell preparation
The mice were immunized by intraperitoneal injection (i.p.) with incomplete Freund’s adjuvant (IFA) or complete Freund’s adjuvant (CFA) (Pierce) and immunized with OVA on days 0 (OVA-IFA) and 7 (OVA-CFA). The spleens were harvested from the OVA-immunized mice on day 14. CD4+ T cells were isolated from the spleens using a CD4+ T cell Isolation Kit II and then activated with OVA (100 μg/mL) and syngeneic APCs. The OVA-specific CD4+ T cells were selected by limiting dilution, and the OVA-specific T (CD4+, IFN-γ +) cells were counted using a FACScanto II.5,21,28
Proliferation assay of OVA-specific Th1 cells
To investigate the antigen-presenting ability of the OVA or OVA-MNPs@SiO2(RITC)-pulsed BMDCs, 3×104 cells were incubated with 5×104 OVA-specific Th1 cells for 24 h or 48 h in 200 μL of culture medium in 96-well cell culture plates. The proliferation was evaluated using a 5-bromo-2′-deoxy-uridine Labeling and Detection Kit III. The cytokine levels were determined via ELISA. The results are expressed as the mean of experiments performed in triplicate.
CD8+ OT-1 cell proliferation
OVA257-264-specific CD8+ T cells were obtained from splenocyte suspensions from C57BL/6 OT-1 T-cell receptor (TCR) transgenic mice. BMDCs were incubated with or without various concentrations of OVA and OVA+MNPs@SiO2(RITC) for 24 h. Mature BMDCs co-cultured with CD8+ T cells. CD8+ T cell proliferation was determined using a 5-bromo-2′-deoxy-uridine Labeling and Detection Kit III, and cytokine secretion and intracellular cytokine concentrations were determined by ELISA and flow cytometry.
Stimulation of mouse splenocytes and draining lymph nodes
The splenocyte and lymphocyte cells obtained from the spleen and lymph nodes from the two groups of C57BL/6 mice cultured in RPMI medium, respectively, were re-stimulated with OVA at concentrations of 100 μg/mL, and 1000 μg/mL or not stimulated. After 48 h of incubation, the spleen cell and lymph node cell proliferations were determined using a 5-bromo-2-deoxy-uridine labeling and detection kit III and the cytokine production was measured with the ELISA assay. The results are expressed as the mean of triplicate experiments. The intracellular cytokine concentrations were determined by flow cytometry to analyze the cytotoxic T cell population.
Cytokine assay
The culture supernatants were analyzed by the enzyme-linked-immunosorbent assay (ELISA). The levels of the various cytokines secreted by the spleen cells and lymph node cells re-stimulated with OVA were measured by ELISA.
Isolation of tumor cell populations
The tumors were dissected using razor blades, digested with 1.5 mg/mL collagenase D (Roche) for 1 h at 37 °C, and then filtered through a 40-μm cell strainer, pooled, and treated with red blood cell lysis buffer.
Intracellular cytokine staining
The CD4+ T cells and CD8+ T cells were treated in the presence of anti-FcR (2.4G2), fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% saponin, and stained with FITC-anti-IFN-γ and PE-anti-CD8. The CD8+ T cells were then gated and analyzed using a FACScanto II (BD Biosciences).
Flow cytometry
The FcγII and FcγIII receptors on the cells were blocked by incubation with anti-mouse CD16/32 (1 μg/1×106 cells), and stained with fluorescence-labeled antibodies specific for the following markers: anti-mouse CD16/32 (2.4G2), CD11c (HL3), IA[b] (AF6–120.1), IFN- γ, CD4 -FITC and -PE on ice. After 30 min of incubation, the cells were washed and read on a BD FACScanto™ II. Data analysis was performed using BD FACS Diva software.
Anti-tumor activity
To measure the therapeutic efficacy, C57BL/6 mice were inoculated subcutaneously with 1×105 EG7 tumor cells, an OVA-expressing EL4 variant, into a flank site. After 1 day, mice were injected subcutaneously with PBS (200 μL), OVA (50 μg/200 μL), and OVA+ MNPs@SiO2(RITC) (50/50 μg/200 μL). On day 22, the mice were euthanized. Tumor growth was monitored and measured every 2 days (n=4 mice/group) by measuring the major and minor axes of the tumors using calipers. The tumor volume was calculated using the following formula: tumor volume (mm3) = major axis×minor axis2×0.5.
Cytotoxicity assay
Splenocytes were used as effector cells without in vitro stimulation, and EG7-OVA cells were used as the target cells. 1×104 cells target cells were incubated with effector cells at effector/target ratio of 25/1, 50/1, and 100/1 in 96-well round bottom plates. The CTL activity after 4 h was measured by the lactate dehydrogenase (LDH) cytotoxicity detection assay and evaluated at various ratios of effector cells to target cells (EG7-OVA) using an EZ-LDH Cell Cytotoxicity Assay Kit (Dogen) according to the manufacturer’s instructions. The cytotoxicity (%) was calculated using the following equation: (Experimental value of Effector Cell Spontaneous Control-Target Cell Spontaneous Control)/(Target Cell Maximum Control-Target Cell Spontaneous Control) × 100.29 The IFN-γ cytokine secretion caused by the cytotoxicity activity after 24 h was determined by ELISA.
Statistical analysis
The differences between the groups are presented as the mean ± SD of three replicate experiments. The significance of the differences was determined using the Student’s t-test. Probability values of <0.05 were considered significant (p-values are indicated as follows: (p-values *<0.05, **<0.01, ***<0.001)
Results
MNPs@SiO2(RITC) is low cytotoxicity to BMDCs
We investigated whether MNPs@SiO2(RITC) at different concentrations induces apoptosis in BMDCs. As shown in Figure 2, MNPs@SiO2(RITC) survived 75% of BMDC treated with MNPs@SiO2(RITC) (100 μg/mL). However, approximately 85% of BMDCs treated with MNPs@SiO2(RITC) (50 μg/mL) survived. Furthermore, other concentrations of MNPs@SiO2(RITC) did not induce apoptosis containing MNPs@SiO2(RITC) (50 μg/mL).
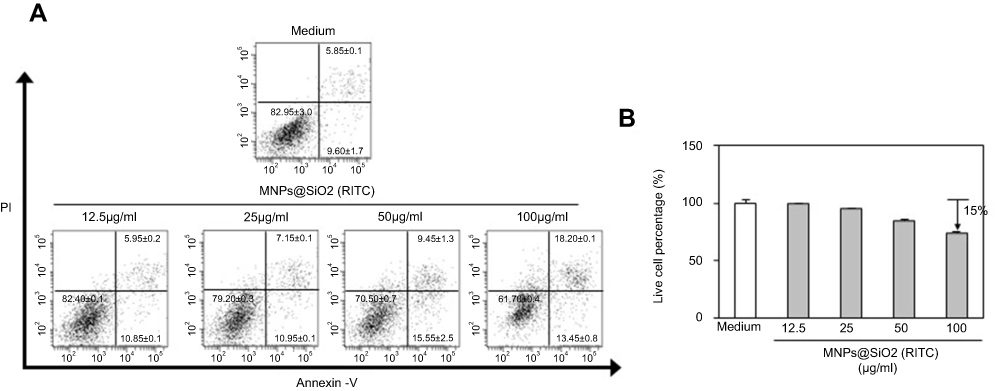 |
Figure 2 MNPs@SiO2(RITC) were low cytotoxic to BMDCs. BMDCs were treated with the indicated concentrations (12.5 μg/mL, 25 μg/mL, 50 μg/mL, and 100 μg/mL) of MNPs@SiO2(RITC) for 24 h, stained with Annexin V-FITC and PI, and analyzed by flow cytometry (A) Dot plot, (B) Live cell percentage, %. The results are representative of three experiments. |
OVA-MNPs@SiO2(RITC) enhances cellular antigen uptake to BMDCs
To further evaluate the efficiency of the uptake, BMDCs were treated with OVA-Alexa488 or OVA-Alexa488-MNPs@SiO2(RITC), and the fluorescence intensity of the cells was analyzed by confocal laser scanning microscope (CLSM) (Figure 3A) and flow cytometry (Figure 3B). To quantify the cellular uptake of the antigen, OVA-Alexa488 was incorporated into the test samples. The BMDCs treated with OVA-Alexa488-MNPs@SiO2(RITC) showed higher uptake than those treated with OVA-Alexa488. Also, as shown in Figure 3, a significant increase in fluorescence intensity was observed compared with the BMDCs treated with OVA-Alexa488. These data indicate that the OVA-MNPs@SiO2(RITC) were taken up much more efficiently by the DCs at the same concentration, and delivered the OVA antigen into the DCs efficiently.
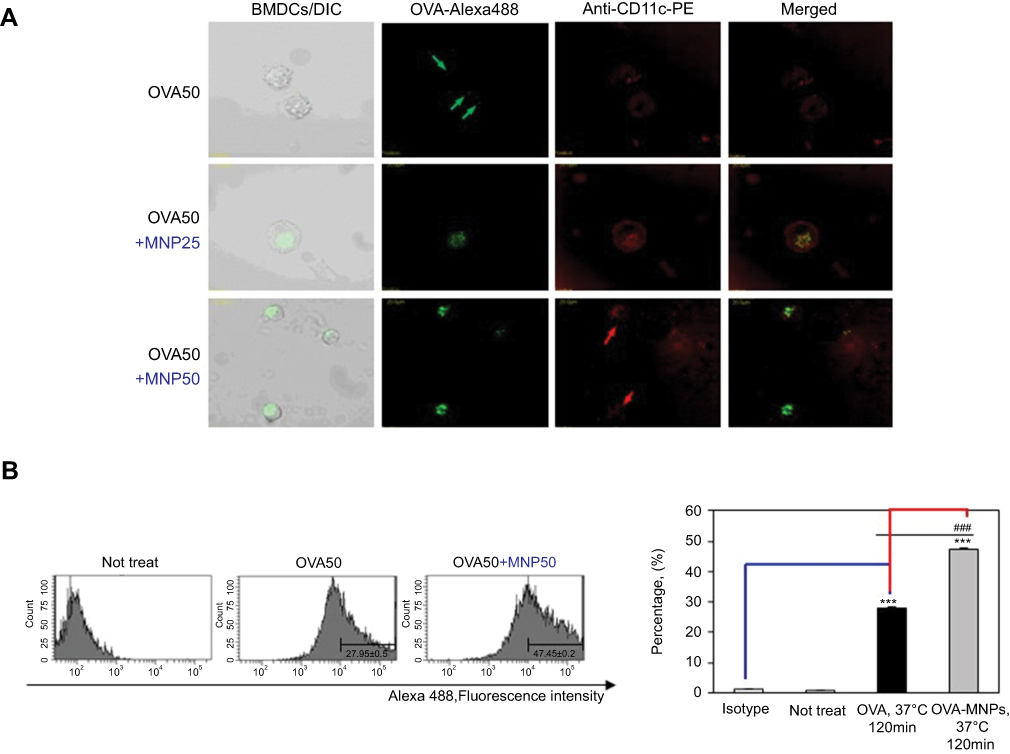 |
Figure 3 Antigen uptake of OVA-Alexa488 by BMDCs is enhanced by MNPs@SiO2(RITC). BMDCs were incubated for various times with either OVA-Alexa488 or OVA-Alexa488-MNPs@SiO2(RITC) and stained with PE-conjugated anti-CD11C. BMDCs incubated at 37 °C with OVA-Alexa488 or OVA-Alexa488-MNPs@SiO2(RITC). Intracellular antigen uptake of OVA-Alexa488 was investigated by (A) confocal microscopy (B) Flow cytometry. Data are presented as the percentage (%) of Alexa488 fluorescence of CD11C-positive cells. The results are representative of three experiments. ***p<0.001 vs untreated BMDCs. ###p<0.001 vs OVA-pulsed BMDCs. |
OVA-MNPs@SiO2(RITC) enhances cytokine production involved in BMDCs activation
To assess the role of the MNPs@SiO2(RITC) in DC activation and stimulation, BMDCs were stimulated with various concentrations of MNPs@SiO2(RITC). After 24 h, we harvested and measured the secretion of pro-inflammatory cytokines, which are known to be important markers of BMDC maturation.21–24 TNF-α, IL-6, IL-1β, and IL-12p70 play important roles in the anti-tumor activity of BMDCs.21–24 In particular, IL-10 and IL-12p70 stimulate the proliferation and differentiation of Th1 and Th2 cells, respectively.21–24 In this experiment, BMDCs treated with LPS were used as a positive control. The secretion of IL-12p70 by the BMDCs treated with MNPs@SiO2(RITC) was higher than that of the untreated BMDCs (Figure 4). The secretion of IL-1β, IL-6, and TNF-α by the BMDCs exhibited similar concentration-dependent similar patterns. However, the level of IL-10 secretion was not changed (Figure 4). In conclusion, these findings indicate that MNPs@SiO2(RITC) can be effectively used for the delivery of antigens into DCs and the activation of antigen-specific Th1 cells. Also, as shown in Figure 5, the cytokine levels of the OVA-MNPs@SiO2(RITC)-treated BMDCs was much higher that of the OVA-treated BMDCs.
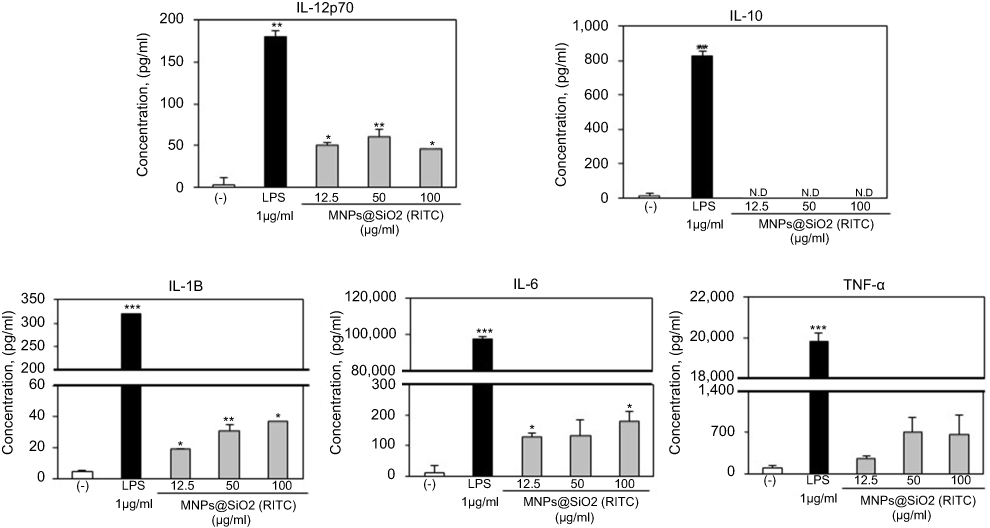 |
Figure 4 MNPs@SiO2(RITC) enhances BMDC cytokine production. Immature BMDCs (1×106 cells) derived from C57BL/6 mice were incubated for 24 h at 37 °C with LPS or MNPs@SiO2(RITC). Cytokine were analyzed by ELISA. The results are representative of three experiments. *p<0.05, **p<0.01, and ***p<0.001 vs untreated BMDCs. |
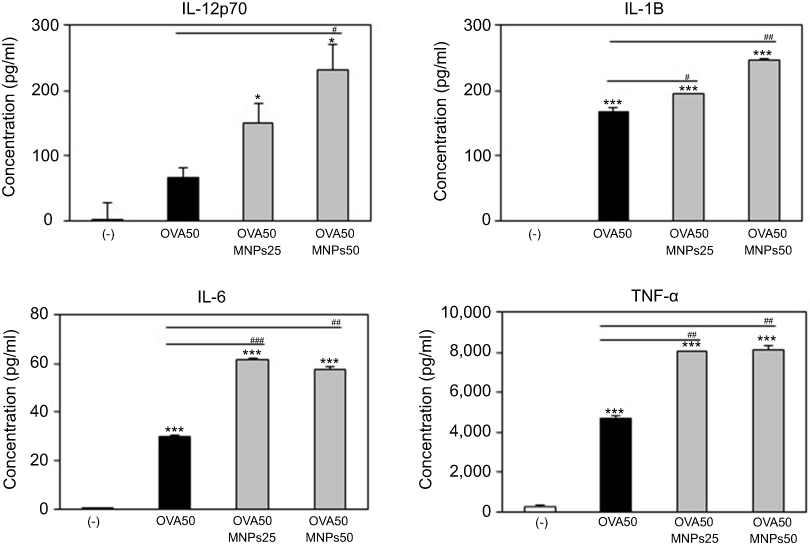 |
Figure 5 MNPs@SiO2(RITC) enhanced BMDC cytokine production. Immature BMDC derived (1×106) from C57BL/6 mouse were incubated for 24 h at 37 °C with OVA, OVA-MNPs@SiO2(RITC). Cytokines were analyzed by ELISA. The results are representative of three experiments. *p<0.05 and ***p<0.001 vs compared untreated BMDCs. #p<0.05, ##p<0.01, and ###p<0.001 vs OVA-pulsed BMDCs. |
OVA-MNPs@SiO2(RITC) enhances OVA-specific T cell responses
We investigated how to OVA-MNPs@SiO2(RITC) induce an OVA-specific response by using OVA-specific CD4+ Th1 cells. To measures the proliferation and cytokine secretion of the OVA-specific CD4+ Th1 cells, we co-cultured OVA-specific Th1 cells with or OVA-MNPs@SiO2(RITC)-pulsed BMDCs. The results showed that the proliferation of antigen-specific CD4+ Th1 cells was higher than that of the OVA-treated mBMDCs and increased with increasing concentration of OVA-MNPs@SiO2(RITC)-treated mBMDCs. Furthermore, the OVA-specific CD4+ Th1 cells co-cultured with the OVA-MNPs@SiO2(RITC)-treated mBMDCs secreted significantly more IFN-γ, and IL-2 than the cells co-cultured with OVA-treated mBMDCs, indicating that MNPs@SiO2(RITC) can be effectively used for the delivery of an antigen into DCs and the activation of antigen-specific Th1 cells (Figure 6). Also, we used S.I. to identify the incremental increase in OVA-MNPs@SiO2(RITC)-treated mBMDCs per group (Figure S1A). The presentation of captured antigens to cytotoxic CD8+ T cells is important for the induction of anti-tumor immunity.23 For this reason, we examined the ability of OVA-MNPs@SiO2(RITC)-treated BMDCs to increase antigen-presentation to CD8+ T cells using OT-1 mice. Proliferation (Figure 7A), S.I. about proliferlation (Figure S1B), and cytokine production of CD8+ T cells were measured by anti-CD8-PE staining after splenocytes from OT-1 mice were co-cultured with various BMDCs. CD8+ T cells co-cultured with OVA-MNPs@SiO2(RITC)-treated BMDCs pulsed with OVA showed greater proliferation than those co-cultured with untreated BMDCs after 48 hrs, demonstrating that MNPs@SiO2(RITC) played role as a potent antigen delivery and immune stimulator of antigen specific CD8+ T cells via DC activation. We then investigated IFN-γ production in CD8+ T cells activated with MNPs@SiO2(RITC)-treated BMDCs pulsed with OVA. CD8+ T cells primed with MNPs@SiO2(RITC)-treated BMDCs produced significantly higher levels of IFN-γ than cells primed with untreated BMDCs through intracellular and extracellular too (Figure 7). These results proved that MNPs@SiO2(RITC) effectively used for the delivery of an antigen into DCs and the activation of antigen-specific CD8+ cells.
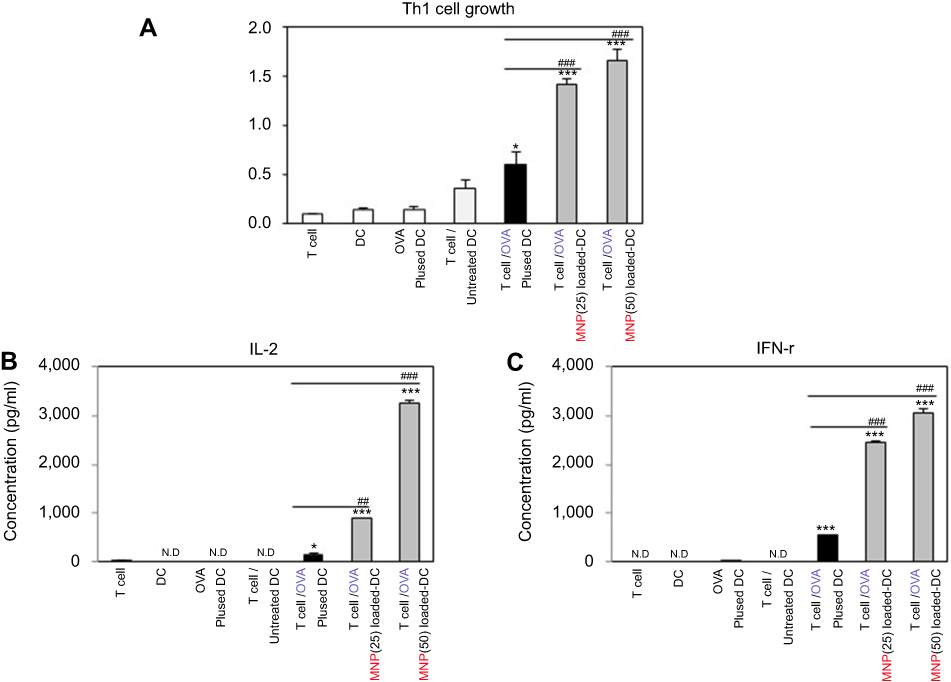 |
Figure 6 MNPs@SiO2(RITC) enhanced antigen-specific CD4+ T cell (A) proliferation and (B, C) cytokine production. CD4+ T cell proliferative and cytokine productive responses by mBMDC pretreated with OVA, OVA-MNPs@SiO2(RITC) were assessed. *p<0.05 and ***p<0.001 compared with T cell/Untreated BMDCS. ##p<0.01 and ###p<0.001 compared with T cell/OVA pulsed BMDCs. |
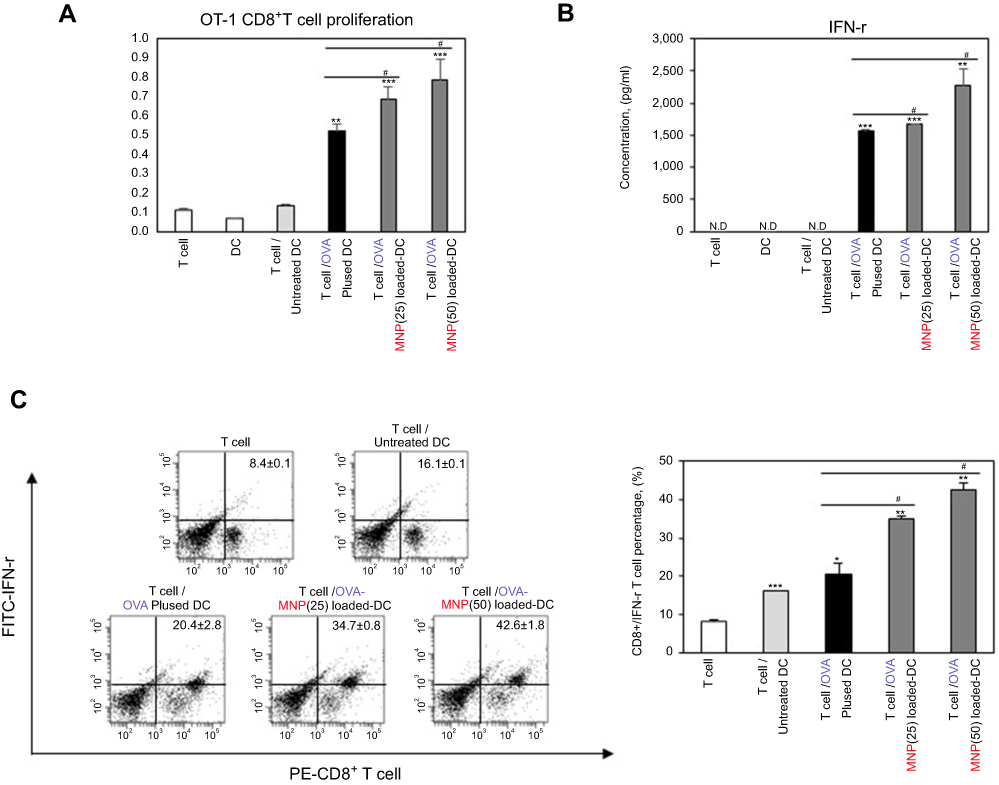 |
Figure 7 MNPs@SiO2(RITC) enhanced antigen-specific CD8+ T cell (A) proliferation and (B, C) cytokine production. CD8+ T cell proliferative and cytokine productive responses by mBMDC pretreated with OVA, OVA-MNPs@SiO2(RITC) were assessed. *p<0.05, **p<0.01, and ***p<0.001 compared with T cell/Untreated BMDCS. #p<0.05 compared with T cell/OVA pulsed BMDCs. |
In vivo anti-tumor therapeutic effect of OVA-MNPs@SiO2(RITC)
To evaluate the in vivo anti-tumor effects of the OVA-MNPs@SiO2(RITC) combination antigen delivery system, we subcutaneously injected OVA-MNPs@SiO2(RITC) into EG7-OVA tumor-bearing mice once a week for three weeks (Figure 8A). After 12 days of inoculation, the tumor volume measurement was obtained at 2-day intervals when an average tumor diameter of 4–6 mm (n=4). The OVA-MNPs@SiO2(RITC) injected groups showed a significantly inhibited tumor growth (Figure 8B). Also, the inhibition of the tumor weight exhibited a similar pattern, too (Figure 8C). We also evaluated the effect of OVA-MNPs@SiO2(RITC) on the stimulation of the immune cells ex vivo after their injection into the tumor site. Moreover, the infiltration population of in activated immune cells in the tumor was analyzed by flow cytometric analysis. The phenotype analysis further revealed that the injection with OVA-MNPs@SiO2(RITC) increased the number of helper T cells (CD4+ T cells), cytotoxic T cells (CD8+ T cells) increased in the tumor cells as compared to the OVA-treated group (Figure 8D and E). These experimental results suggest that the immune-stimulation effect of OVA-MNPs@SiO2(RITC) antigen delivery systems can successfully inhibit tumor proliferation and thus provide a significant therapeutic effects.
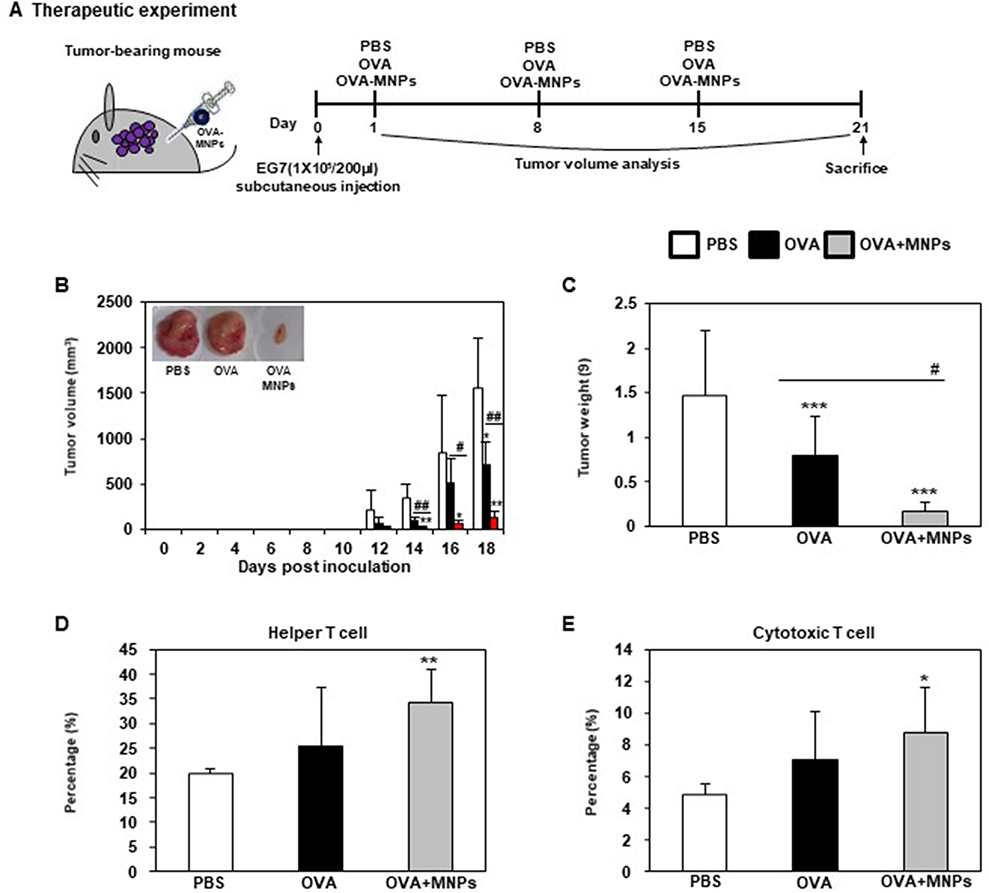 |
Figure 8 Analysis of anti-tumor immunity for therapeutic effect. EG7-OVA tumor cells (1×105 cells) were inoculated subcutaneously into mice (n=4/group) 1days before injection of OVA or OVA-MNPs@SiO2(RITC). (A) Experimental scheme for the anti-tumor immunity therapy (B) Tumor growth was monitored by measuring the tumor volume of mice. Photographs of the dissected tumor tissue from each group are also shown. (C) Tumor weight of the dissected tumor tissue from each group was also shown. (D, E) Subsets in tumor cells measured gated and analyzed using a FACScanto II (BD Biosciences). Data are represented as mean ± SD of four mice per group. *p<0.05, **p<0.01, and ***p<0.001 compared with negative PBS group. #p<0.05 and ##p<0.01 compared with OVA group. |
To further test the CTL response in vivo, splenocytes were collected and co-incubated with EG7 cells at 7 days post immunization. CTLs are CD8+ T cells that can induce an antigen-specific adaptive immune response through the presentation of antigens to MHC class I molecules and can kill cancer cells by the release of cytotoxic granules, such as perforin and granzymes. Synergistically, OVA-MNPs@SiO2(RITC) resulted in a significantly higher CTL activity than OVA alone at all target cell/effector cell ratios. These results suggest that the OVA-MNPs@SiO2(RITC) were able to induce antigen-specific CTL responses for efficient DC-based cancer therapy (Figure 9). Also, to ensure the Th1 response in the context of the cell-mediated immune response, we isolated the splenocytes from each group and measured the antigen-specific response.
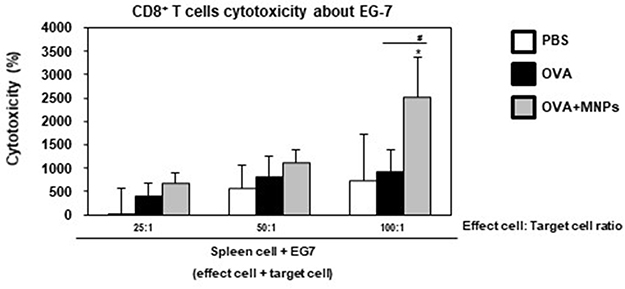 |
Figure 9 Analysis of anti-tumor immunity for therapeutic effect. EG7-OVA tumor cells (1×105 cells) were inoculated subcutaneously into mice (n=4/group) 1 days before injection of OVA or OVA-MNPs@SiO2(RITC). OVA or OVA-MNPs@SiO2(RITC) immunization induces OVA-specific CTL activity. The mice (n=4/group) were sacrificed and the splenocytes of each group were harvested for the in vitro killing assay (E/T=25, 50, and 100). Data are represented as mean ± SD of four mice per group. *p<0.05 compared with negative PBS group. #p<0.05 compared with OVA group. |
The OVA-MNPs@SiO2(RITC) vaccine induced the strong antigen-specific proliferation of splenocytes in response to OVA protein stimulation. The maximum splenocytes proliferation in response to OVA-MNPs@SiO2(RITC) was about 1.6-fold higher than that in response to OVA alone. The maximum IL-2, and IFN-γ concentrations in the supernatant were also significantly higher on treatment with OVA-MNPs@SiO2(RITC) compared with OVA (Figure 10A). Additionally, we confirmed the secretion of IFN-γ from CD8+ T cells by intracellular staining using flow cytometry. Higher secretion of IFN-γ was observed in the OVA-MNPs@SiO2(RITC) group than in the other groups. IFN-γ secretion by CD8+ T cells is important to induce specific cytotoxic responses for the clearance of infectious pathogens and cancer cells. This is consistent with the observed CTL response (Figure 10B). Interestingly, we confirmed the secretion of IFN-γ from CD4+ T cells by intracellular staining using flow cytometry too. Higher secretion of IFN-γ from CD4+ T cells was observed in the OVA-MNPs@SiO2(RITC) group than in the OVA groups (Figure 10C).
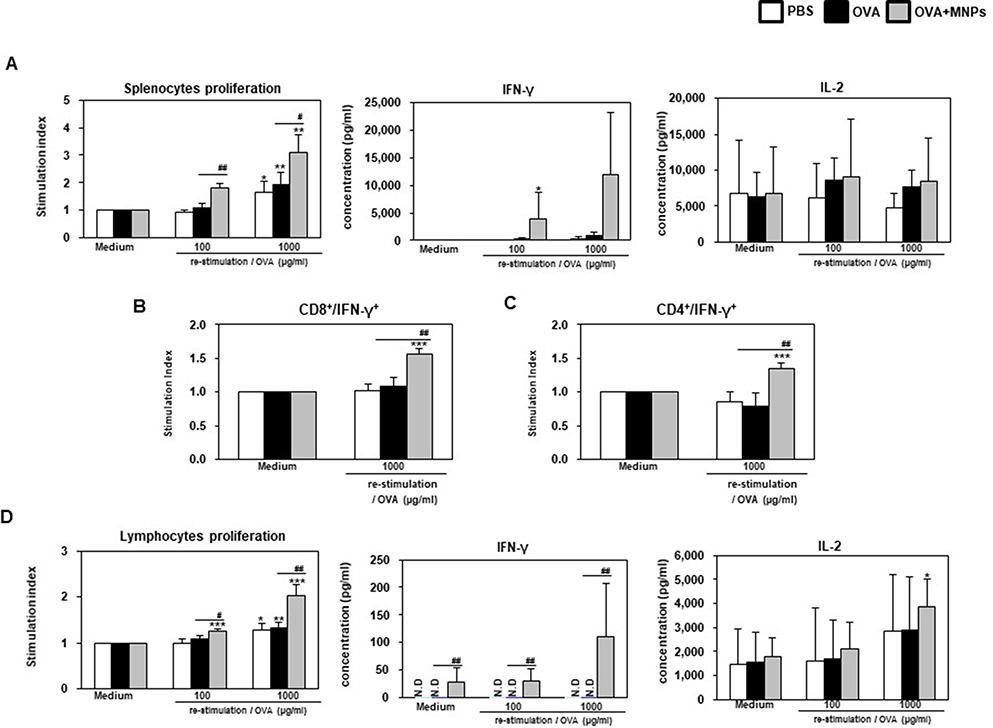 |
Figure 10 Analysis of anti-tumor immunity for therapeutic effect. EG7-OVA tumor cells (1×105 cells) were inoculated subcutaneously into mice (n=4/group) 1 days before injection of OVA or OVA-MNPs@SiO2(RITC). The splenocytes of each group were re-stimulated with OVA 7 days after the last immunization. (A, D) The proliferation of the splenocytes and lymphocytes were assessed using a Bromo-kit. The culture supernatants were harvested after 24 h, and the cytokine levels were measured by ELISA. The splenocytes of each group were re-stimulated with OVA 7 days after the last immunization and (B)CD8+, and (C)CD4+ T cells were then gated and analyzed using a FACScanto II (BD Biosciences) by staining with FITC-anti-IFN-γ and PE-anti-CD4/CD8. Data are represented as mean ± SD of four mice per group. *p<0.05, **p<0.01, and ***p<0.001 compared with negative PBS group. #p<0.05, and ##p<0.01 compared with OVA group. |
The proliferation of lymphocytes in response to OVA-MNPs@SiO2(RITC) was about 1.5-fold higher than that in response to OVA alone. The maximum IL-2, and IFN-γ concentrations in the supernatant were also significantly higher on treatment with OVA-MNPs@SiO2(RITC) compared with OVA. As shown in Figure 10D, OVA-MNPs@SiO2(RITC) vaccine induced the strong antigen-specific response of the lymphocytes in response to OVA protein stimulation. Collectively, these findings indicate that the development of OVA-MNPs@SiO2(RITC) that can deliver the antigen (OVA) efficiently lead to an enhanced CTL response and cell-mediated immune response in comparison to the antigen alone. Also, these data indicate that the immune-stimulation effect of MNPs@SiO2(RITC) based antigen delivery systems can successfully inhibit tumor proliferation and elicit potent prophylactic anti-tumor immunity, as well as a therapeutic response.
Discussion
In this research, we evaluated their performance as therapeutic vaccines for tumor immunotherapy using nanoparticles as antigen carriers. Nanoparticles have been reported to have a variety of advantages as antigen and adjuvant co-loading of immune-stimulatory adjuvants via electrostatic surface charge interactions.30 We used silica-coated magnetic nanoparticles as a platform for the efficient delivery of OVA into the dendritic cells ensure their immune-stimulatory activity. Nanoparticles in silica coated surface onto particle are known as biocompatible materials.15–20 However, in the case of magnetic nanoparticle at high concentrations, concerns regarding their toxicity and safety have been reported.26,27 Figure 2 shows that MNPs@SiO2(RITC) at a concentration 100 µg/mL induced apoptosis. However, other concentrations of MNPs@SiO2(RITC) did not induce apoptosis. In this study, we confirmed that immunization by MNPs@SiO2(RITC) delivery elicited a higher cellular and humoral immune response than soluble OVA. Also, OVA-MNPs@SiO2(RITC) delivery induces a synergistic anti-tumor effect in an animal tumor model, including the suppression of tumor proliferation.
DCs are the most potent antigen presentation cells and are a good candidate for an immune cell vaccine.5,21–23 Immature DCs capture antigens generated by pathogens and tumors and then enter a maturation cascade.5,21–23 The resulting mature DCs exhibit the elevated expression of co-stimulatory molecules and secrete more cytokines to promote immune synapse formation, antigen processing, antigen presentation to T cells, and T cell activation.5,21–23 Therefore, DCs could potentially be used in vaccines for tumor immunotherapy and to treat other diseases. DCs secrete various cytokines (ie, IL-1β, IL-6, IL-12p70, TNF-α, and IL-10) so as to elicit antigen-specific immune responses.21–23 IL-10 and IL-12p70 stimulate the proliferation and differentiation of Th1 and Th2 cells, respectively.5 Also, IL-1β, IL-6, and TNF-α of pro-inflammatory type cytokines induce a humoral immune response.21–23 When MNPs@SiO2(RITC) were applied to BMDCs, various types of cytokines were secreted at higher levels than in the case of the negative control. As shown in Figure 4, we suggest that the higher level of IL-12p70 in the BMDCs means that the naïve Th cells differentiated into Th1 cells, and then enhanced the cell- meditated immune response. Also, we showed that the level of IL-10 secretion was unchanged. And Figure 3. clearly shows that OVA-MNPs@SiO2(RITC) contributed to the enhanced antigen (OVA) uptake efficiency. The antigen uptake by DCs is very important in the antigen-specific immune response. The immature BMDCs were treated with OVA-MNPs@SiO2(RITC) after 18 h. Subsequently, the mature BMDCs were harvested and co-cultured with OVA-specific Th1 cells to confirm the maintenance of their antigenic properties in the MNPs@SiO2(RITC)
It was shown that the OVA-MNPs@SiO2(RITC) treated BMDCs more potently increased the OVA-specific Th1 cell growth and cytokine secretion than the OVA-treated BMDCs. These data suggest that the MNPs@SiO2(RITC) maintained their antigenic properties and enhanced the antigen-specific immune response.
Based on the in vitro results, we confirmed that OVA-MNPs@SiO2(RITC) could be used as a the therapeutic vaccine for tumor immunotherapy. Also, the OVA-MNPs@SiO2(RITC) also boosted the proliferation of immune cells through its immune-stimulatory effect, which is necessary in an immune-suppressive tumor microenvironment. A strategically important target of vaccination in order to induce an anti-tumor immune response is the induction of a specific immune response through the interaction of Th1 cells and cytotoxic T cells. The OVA-MNPs@SiO2(RITC) vaccine induced the strong antigen-specific proliferation of splenocytes and lymphocyte in response to OVA protein stimulation. The maximum IL-2 and IFN-γ concentrations in the supernatant were also significantly higher on treatment with OVA-MNPs@SiO2(RITC) compared with OVA. The results showed that OVA-MNPs@SiO2(RITC) increased the cell-mediated immune responses. Higher secretion of IFN-γ from CD4+/CD8+ T cells was observed in the OVA-MNPs@SiO2(RITC) group than in the other groups. IFN-γ secretion by CD8+ T cells has been reported to be important to induce specific cytotoxic responses for the clearance of infectious pathogens and cancer cells. Also, this is consistent with the observed CTL response. In addition, we proved that OVA-MNPs@SiO2(RITC) contributed to the inhibition of tumor growth and prevention of tumor formation in the context of a therapeutic and prophylactic vaccine, respectively, through the EG7 tumor mouse model.
Tumor immunotherapy is an active area of tumor research. Both medical and pharmaceutical research scientists have been exploring tumor immunotherapy.6–25,30 The most general concept for tumor-immunotherapy is the development of DCs-based therapy or T-cell adoptive transfer therapy and this has been validated in several clinical trials.1,6,28 However, it has many limitations, such as the significant investment in terms of medical personnel, money, time, and labor associated with the ex vivo manipulation of DCs.1,6,30 To overcome these limitations, the most general concept for immunotherapy is the delivery of adjuvants and antigens to DCs.1,6,21,30 Thus, we examined antigen delivery using nanoparticle carrier systems for DC-based tumor immunotherapy without the ex vivo manipulation of DCs.1,6,21,30 We directly injected the antigen conjugated MNPs@SiO2(RITC) into the tumor-bearing mice to target the DCs. Therefore, we can overcome the limitations associated with the ex vivo manipulation of DCs for DC-based tumor immunotherapy. This approach using a nanoparticle conjugated antigen has broad applicability for the active delivery of adjuvants and antigens to DCs without ex vivo manipulation, leading to strong cytotoxic CD8+ T cell-mediated immune responses and increased therapeutic efficacy.1,6,21,30 This approach leads to Th1 cell and cytotoxic T cell-mediated immune responses for therapeutic efficacy in a tumor mouse model and has broad applicability for the active delivery of antigens to DCs without ex vivo manipulation. We also demonstrated that an effective antigen-specific immune response to the targets was induced, thereby enhancing the therapeutic efficacy. The major progress for cancer immunotherapy has involved the development of dendritic cells (DCs)-based therapy or T-cell adoptive transfer therapy. However, these cell-based therapeutic strategies require significant cost, labor, and time in the isolation, activation and proliferation of these immune cells before they are re-injected into the patient.1,10,24,31,32 An alternative strategy that overcomes the limitation of ex vivo cultured immune cell-based cancer immunotherapy is to activate and proliferated immune cells in vivo using various immunomodulatory materials.1,10,24,31,32 Therefore, antigen delivery by nanoparticles is highly efficient, because of its potential to overcome the limitations of DC or T cell-based immunotherapy.
Conclusions
In this study, the data confirmed that MNPs@SiO2(RITC) based direct antigen delivery systems can enhance the antigen-specific Th1, CTL -mediated immune response and provide active anti-tumor immunotherapy without toxicity and ex vivo manipulation. Therefore, MNPs@SiO2(RITC)-based systems are expected to be useful as efficient antigen-delivery carriers for the enhancement of immune responses in active cancer immunotherapy by direct injection into tumor-bearing mice. Also, this MNPs@SiO2(RITC)-based platform may be adapted for use in other target delivery antigens and other immunological diseases. Finally, antigen delivery systems using MNPs@SiO2(RITC) may be attractive for diverse biomedical applications.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shin WJ, Noh HJ, Noh YW, Kim S, Um SH, Lim YT. Hyaluronic acid-supported combination of water insoluble immunostimulatory compounds for anti-cancer immunotherapy. Carbohydr Polym. 2017;155:1–10. doi:10.1016/j.carbpol.2016.08.040
2. Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. doi:10.1146/annurev.immunol.021908.132544
3. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi:10.1038/nature10611
4. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nature Rev Immunol. 2007;7(10):790–802. doi:10.1038/nri2173
5. Kim JJ, Hwang YH, Kang KY, et al. Enhanced dendritic cell maturation by the B-chain of Korean mistletoe lectin (KML-B), a novel TLR4 agonist. Int Immunopharmacol. 2014;21(2):309–319. doi:10.1016/j.intimp.2014.05.010
6. Han HD, Byeon Y, Jang JH, et al. In vivo stepwise immunomodulation using chitosan nanoparticles as a platform nanotechnology for cancer immunotherapy. Sci Rep. 2016;6:38348. doi:10.1038/srep38348
7. Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32(9):456–465. doi:10.1016/j.tibtech.2014.06.007
8. Kim J, Mooney DJ. In vivo modulation of dendritic cells by engineeredmaterials: towards new cancer vaccines. Nano Today. 2011;6(5):466–477. doi:10.1016/j.nantod.2011.08.005
9. Li WA, Mooney DJ. Materials based tumor immunotherapy vaccines. Curr Opin Immunol. 2013;25(2):238–245. doi:10.1016/j.coi.2012.12.008
10. Song C, Noh YW, Lim YT. Polymer nanoparticles for cross-presentation of exogenous antigens and enhanced cytotoxic T-lymphocyte immune response. Int J Nanomed. 2016;11:3753–3764. doi:10.2147/IJN.S110796
11. Friede M, Aguado MT. Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems. Adv Drug Deliv Rev. 2005;57(3):325–331. doi:10.1016/j.addr.2004.10.001
12. Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35(1):590–600. doi:10.1016/j.biomaterials.2014.01.026
13. Noh YW, Hong JH, Shim SM, et al. Polymer nanomicelles for efficient mucus delivery and antigen-specific high mucosal immunity. Angew Chem Int Ed Engl. 2013;52(30):7684–7689. doi:10.1002/anie.201302881
14. Serda RE. Particle platforms for cancer immunotherapy. Int J Nanomed. 2013;8:1683–1696. doi:10.2147/IJN.S37465
15. Kim BS, Chun SY, Lee JK, et al. Human amniotic fluid stem cell injection therapy for urethral sphincter regeneration in an animal model. BMC Med. 2012;10:94. doi:10.1186/1741-7015-10-94
16. Park JS, Na K, Woo DG, et al. Non-viral gene delivery of DNA polyplexed with nanoparticles transfected into human mesenchymal stem cells. Biomaterials. 2010;31(1):124–132. doi:10.1016/j.biomaterials.2009.09.023
17. Kim JS, Yoon TJ, Yu KN, et al. Toxicity and tissue distribution of magnetic nanoparticles in mice. Toxicol Sci. 2006;89(1):338–347. doi:10.1093/toxsci/kfj027
18. Park KS, Tae J, Choi B, et al. Characterization, in vitro cytotoxicity assessment, and in vivo visualization of multimodal, RITC-labeled, silica-coated magnetic nanoparticles for labeling human cord blood-derived mesenchymal stem cells. Nanomedicine. 2010;6(2):263–276. doi:10.1016/j.nano.2009.07.005
19. Lee JK, Chun SY, Im JY, Jin HK, Kwon TG, Bae JS. Specific labeling of neurogenic, endothelial, and myogenic differentiated cells derived from human amniotic fluid stem cells with silica-coated magnetic nanoparticles. J Vet Med Sci. 2012;74(8):969–975. doi:10.1292/jvms.12-0016
20. Yoon TJ, Kim JS, Kim BG, Yu KN, Cho MH, Lee JK. Multifunctional nanoparticles possessing a “magnetic motor effect” for drug or gene delivery. Angew Chem Int Ed Engl. 2005;44(7):1068–1071. doi:10.1002/anie.200461910
21. Kim JJ, Nam JP, Nah JW, Jang MK, Yee ST. Immunoadjuvant efficacy of N-carboxymethyl chitosan for vaccination via dendritic cell activation. J Med Food. 2014;17(2):268–277. doi:10.1089/jmf.2013.2921
22. Kim JJ, Hwang YH, Kang KY, et al. Antitumor effect of KML-B-treated dendritic cells via induction of lymphocyte activation. J Immunol Res. 2017;2017:2471627. doi:10.1155/2017/5974574
23. Lee SJ, Kim JJ, Kang KY, et al. Herbal preparation (HemoHIM) enhanced functional maturation of bone marrow-derived dendritic cells mediated toll-like receptor 4. BMC Complement Altern Med. 2016;16:67. doi:10.1186/s12906-016-1045-9
24. Kim SY, Phuengkham H, Noh YW, Lee HG, Um SH, Lim YT. Immune complexes mimicking synthetic vaccine nanoparticles for enhanced migration and cross-presentation of dendritic cells. Adv Funct Mater. 2016;26(44):8072–8082. doi:10.1002/adfm.201603651
25. Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54:777–785. doi:10.1016/s0092-8674(88)91043-4
26. Shim W, Paik MJ, Nguyen DT, et al. Analysis of changes in gene expression and metabolic profiles induced by silica-coated magnetic nanoparticles. ACS Nano. 2012;6(9):7665–7680. doi:10.1021/nn301113f
27. Phukan G, Shin TH, Shim JS, et al. Silica-coated magnetic nanoparticles impair proteasome activity and increase the formation of cytoplasmic inclusion bodies in vitro. Sci Rep. 2016;6:290095. doi:10.1038/srep29095
28. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi:10.1038/nri1001
29. Ko BK, Lee SY, Lee YH, et al. Combination of novel HER2-targeting antibody 1E11 with trastuzumab shows synergistic antitumor activity in HER2-positive gastric cancer. Mol Oncol. 2015;9:398–408. doi:10.1016/j.molonc.2014.09.007
30. An M, Li M, Xi J, Liu H, Liu H. Silica nanoparticle as a lymph node targeting platform for vaccine delivery. ACS Appl Mater Interfaces. 2017;9(28):23466–23475. doi:10.1021/acsami.7b06024
31. Yang HL, Zhao JL, Wu CY, Ye CQ, Zou DW, Wang SG. Facile synthesis of colloidal stable MoS2 nanoparticles for combined tumor therapy. Chem Eng J. 2018;351:548–558. doi:10.1016/j.cej.2018.06.100
32. Zhao J, Xie P, Ye C, et al. Outside-in synthesis of mesoporous silica/molybdenum disulfide nanoparticles for antitumor application. Chem Eng J. 2018;351(1):157–168. doi:10.1016/j.cej.2018.06.101
Supplementary material
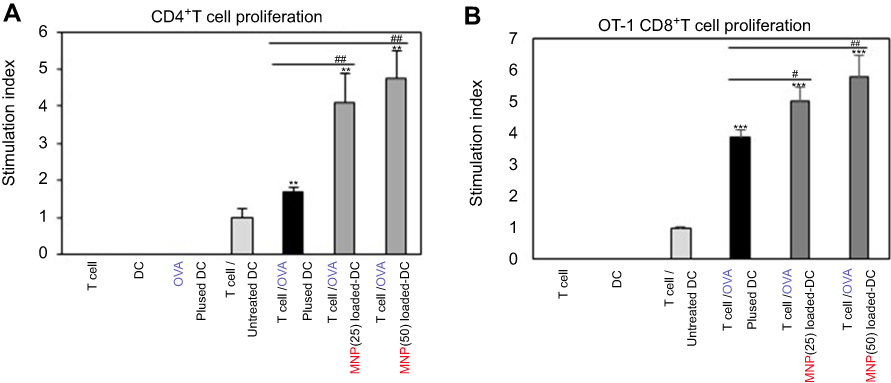 |
Figure S1 Proliferation of CD4+ T cells and CD8+ T cells. MNPs@SiO2(RITC) enhanced (A) antigen-specific CD4+ T cell proliferation stimulation index. (B) antigen-specific CD8+ T cell proliferation. CD4+ and CD8+ T cell proliferative responses by mBMDC pretreated with OVA, OVA-MNPs@SiO2(RITC) were assessed. *p<0.05, **p<0.01 or ***p<0.001 compared with T cell/Untreated BMDCS. #p<0.05 and ##p<0.01 compared with T cell/OVA pulsed BMDCs. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.