Back to Journals » Drug Design, Development and Therapy » Volume 14
Engineering of Naproxen Loaded Polymer Hybrid Enteric Microspheres for Modified Release Tablets: Development, Characterization, in silico Modelling and in vivo Evaluation
Authors Hameed HA, Khan S ![]() , Shahid M
, Shahid M ![]() , Ullah R, Bari A
, Ullah R, Bari A ![]() , Ali SS, Hussain Z
, Ali SS, Hussain Z ![]() , Sohail M
, Sohail M ![]() , Khan SU
, Khan SU ![]() , Htar TT
, Htar TT ![]()
Received 23 September 2019
Accepted for publication 13 December 2019
Published 7 January 2020 Volume 2020:14 Pages 27—41
DOI https://doi.org/10.2147/DDDT.S232111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Hajra Afeera Hameed,1 Shahzeb Khan,1–3 Muhammad Shahid,4 Riaz Ullah,5 Ahmed Bari,6 Syed Saeed Ali,6 Zahid Hussain,7 Muhammad Sohail,8 Shafi Ullah Khan,9 Thet Thet Htar9
1Department of Pharmacy, University of Malakand, Chakdara, Khyber Pakhtunkhwa 18800, Pakistan; 2Discipline of Pharmaceutical Sciences, School of Health Sciences, University of KwaZulu-Natal, Durban 4000, South Africa; 3Division of Molecular Pharmaceutics and Drug Delivery, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA; 4Department of Pharmacy, Sarhad University of Science and Information Technology, Peshawar, Khyber Pakhtunkhwa, Pakistan; 5Medicinal, Aromatic & Poisonous Plants Research Center (MAPPRC), College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia; 6Central Laboratory, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia; 7Department of Pharmaceutics and Pharmaceutical Technology, College of Pharmacy, University of Sharjah, Sharjah 27272, United Arab Emirates; 8Department of Pharmacy, COMSATS University Islamabad, Abbottabad 22060, Pakistan; 9School of Pharmacy, Monash University Malaysia, Bandar Sunway, Subang Jaya 47500, Malaysia
Correspondence: Shahzeb Khan
Department of Pharmacy, University of Malakand, Dir Lower KPK, Pakistan
Email [email protected]
Background: Naproxen (NP) is a non-steroidal anti-inflammatory drug with poor aqueous solubility and low oral bioavailability, which may lead to therapeutic failure. NP causes crucial GIT irritation, bleeding, and peptic and duodenal ulcers.
Purpose of the study: This study aimed to engineer and characterize polymer hybrid enteric microspheres using an integrated (experimental and molecular modelling) approach with further development to solid dosage form with modified drug release kinetics and improved bioavailability.
Materials and methods: NP loaded polymer hybrid enteric microspheres (PHE-Ms) were fabricated by using a modified solvent evaporation technique coupled with molecular modelling (MM) approach. The PHE-Ms were characterized by particle size, distribution, morphology, crystallinity, EE, drug-polymer compatibility, and DSC. The optimized NP loaded PHE-Ms were further subjected to downstream procedures including tablet dosage form development, stability studies and comparative in vitro-in vivo evaluation.
Results: The hydrophobic polymer EUD-L100 and hydrophilic polymer HPMC-E5 delayed and modified drug release at intestinal pH while imparting retardation of NP release at gastric pH to diminish the gastric side effects. The crystallinity of the NP loaded PHE-Ms was established through DSC and P (XRD). The particle size for the developed formulations of PEH-Ms (M1-M5) was in the range from 29.06 ±7.3–74.31 ± 17.7 μm with Span index values of 0.491–0.69, respectively. The produced NP hybrid microspheres demonstrated retarded drug release at pH 1.2 and improved dissolution at pH 6.8. The in vitro drug release patterns were fitted to various release kinetic models and the best-followed model was the Higuchi model with a release exponent “n” value > 0.5. Stability studies at different storage conditions confirmed stability of the NP loaded PHE-Ms based tablets (P<0.05). The molecular modelling (MM) study resulted in adequate binding energy of co-polymer complex SLS-Eudragit-HPMC-Naproxen (−3.9 kcal/mol). In contrast to the NP (unprocessed) and marketed formulations, a significant increase in the Cmax of PHE-MT1 (44.41±4.43) was observed.
Conclusion: The current study concludes that developing NP loaded PHE-Ms based tablets could effectively reduce GIT consequences with restored therapeutic effects. The modified release pattern could improve the dissolution rate and enhancement of oral bioavailability. The MM study strengthens the polymer-drug relationship in microspheres.
Keywords: naproxen, hybrid microspheres, molecular modelling, dissolution, bioavailability, modified-release tablets
Introduction
Smart drug delivery approaches have encouraged the design and development of new nonsteroidal anti-inflammatory drugs (NSAIDs) products with improved physiochemical, biopharmaceutical and therapeutic attributes.1–3 The advancements in novel drug delivery strategies include microspheres, microcapsules, micro-sponges, nanoparticles, nanocrystals, controlled drug delivery, and modified drug delivery systems. This advancement in NSAIDs pharmaceutics has led to providing clinical benefits such as improvement in adverse event profile with reduced dosing frequency and addressed serious dose-dependent gastrointestinal (GI) adverse effects (AEs). Owing to poor aqueous solubility, most of the compounds have become a great challenge during drug delivery for scientists to effectively develop compounds into the dosage form.4,5
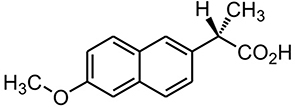 |
Figure 1 Chemical structure of naproxen. |
The BCS II (Biopharmaceutical classification system) class drug compounds are considered as having great potential as drug molecules.6 However, the poor dissolution which is the rate-limiting step in absorption of these APIs has led to poor drug development. The slow and limited rate of drug release in GIT fluids mostly results in low oral bioavailability. Consequently, the key challenge in the development of new APIs is to implement such approaches that advance the solubility and dissolution of poor solubilized drugs but also to improve and boost oral bioavailability with more therapeutic effect.
Conventional techniques utilized for the development of drug-polymer systems have been reported with noticeable challenges and drawbacks including the unnecessary use of solvent, chemical and thermal solute degradation, structural variations, high residual solvent concentrations and additionally difficulty in control of the particle size (PS) and particle size distribution (PSD).7,8 The majority of pharmaceuticals cause irritations in the gastric tract due to an increased concentration of the drug after dose administration. Others have observed a therapeutic phase for a short period of time, after which a rapid decline inside the drug concentration was observed until new dose management achieved.9 It is a prerequisite to sort out appropriate dosing to the patient, how to maximize patient compliance, and to understand the novelty that will modify the proprietary position of the final product prior to designing a functional and effective delivery system that exploits the latest technologies.8,10
The pH-responsive polymers have been found very useful for the enteric coating and development of acid-labile drug molecules.8 The commonly used pH-responsive polymers for enteric coatings mostly contain carboxylic groups.11 In the stomach at low pH, these polymers remain un-ionized. However, at higher pH conditions like the small intestine, these polymers become ionized permitting the dissolution of the enteric coating and drug release. Eudragit-L100 is methacrylic acid and methyl methacrylic acid copolymer of carboxylic acid.12 Eudragit L-100 is a pH-responsive polymer that can be used at gastric or lower pH levels to minimize drug release and increase drug release at higher pH levels like those in the intestine.
Naproxen [(S)-2-(6-methoxynaphthalen-2-yl) propionic acid], is a non-selective COX inhibitor (Figure 1).13 Naproxen is a potent non-steroidal anti-inflammatory drug widely used in the therapeutic relief of pain, rheumatoid arthritis, osteoarthritis, acute gout, and as an antipyretic agent.14 However, the use of Naproxen can lead to severe GIT side effects. Naproxen belongs to BCS II class of drug compounds with poor water solubility, leading to limited dissolution and erratic drug absorption.15–17
To address the above challenges associated with NP, there several formulation approaches that have been previously used including, complexation with cyclodextrins, self-emulsifying drug delivery system enteric coating, sustain released, matrix system, floating drug delivery, micellar solubilization solid dispersion, hot-melt extrusion, and lipid carrier system.11,17–19 However most of these formulations have not focused simultaneously on the three aspects including safe transit to the intestine, significant improvement in solubility and microsphere behavior in the tablet dosage form. In addition, the molecular-level understanding of these selected polymers/surfactants and NP in microsphere formulations has not yet been uncovered.20,21
One of the potential advantages of utilizing microparticles is their ability to entrap the active drug in the amorphous state, potentially improving the dissolution and in some instances drug delivery in a controlled manner.22 The combination approach of bio-compatible and non-biodegradable biopolymers and surfactants to incorporate gastric-irritant drugs into pH-responsive microspheres has been anticipated as an alternative approach. This approach is also used to prevent drug release in the stomach and to target the small intestine.23 Therefore, it would be imperative to develop NP loaded polymer hybrid microspheres to address the issues of gastric irritation, gastric mucosa damaging, poor aqueous solubility and consequently enhanced bioavailability.
This study aimed to develop stable polymer hybrid enteric microspheres with subsequent conversion to modified-release tablets. Furthermore, the molecular modelling (MM) approach was designed to identify the binding affinity of NP with individuals and with combinations of polymers and surfactant molecules. The MM and simulations study uncovered and clarified the molecular-level of the encapsulation of NP within the chosen pair of polymer and surfactants with the subsequent linkage to entrapment efficiency (EE) and the release kinetics of drug. The produced microspheres were subjected to solid-state characterization and conversion to tablet formulations. Comparative in vivo evaluation and stability studies of the developed tablet formulations were conducted to correlate with their in vitro dissolution assessment.
Materials and Methods
Materials
Naproxen (Batch No: 210711) was provided by Medicraft Pharmaceutical (Pvt.Ltd Peshawar, Pakistan). Chloroform was purchased from Ali Baba Chemical Lahore, Pakistan. Eudragit L100 (B.No.150313) was obtained from Anhui Sunhere Pharmaceuticals Excipients Co Ltd, China. Ethanol, Methanol and Ortho phosphorous acid were procured from Sigma-Aldrich, USA. HPMC (Grade E-5) was purchased from Anhui Sunhere Pharmaceuticals Excipients. Co Ltd Economical Technological Developing Zone Huainan, Anhui China). Citric acid was obtained from Alchemy, Lahore Pakistan. Avicel (pH-102) and Kollidon® (B.No 30-4129) was purchased from Anhui Sunhere Pharmaceuticals Excipients Co Ltd, China. Talcum powder and Mg. Stearate (B.No.2291) was obtained from Aries Pharma Peshawar.
Methods
Preparation of NP Loaded PHE-Ms
NP was fabricated in the form of Polymeric Hybrid microspheres using a modified “Emulsion solvent evaporation (ESE) technique”.24,25 Briefly, 3% (w/v) EUD- L100 solution was prepared in methanol and added to 3% (w/v) ethanol solution of NP and sonicated for 02 mins. The EUD-L100 ratios in different formulations were increased with the fixed ratio of NP and the required volumes were made up with chloroform, followed by continuous stirring until the uniform drug-polymer solutions were obtained. The copolymer and surfactant solutions in water that were composed of HPMC-E5 (0.5, 1, 1.5, 2, and 2.5% (w/v)) and SLS (0.2%) were gently added and stirred vigorously at 1200 rpm with a magnetic stirrer at 35°C. The stirring was continued for 08hrs in order to completely evaporate the solvents. The remaining solution after evaporation was subjected to low-speed centrifugation to harvest microspheres. The microspheres were washed with n-hexane and subsequently, assembled through filtration (vacuum filtration). Furthermore, the filtered microspheres were dried properly and stored. The formulation variables and process parameters are as mentioned in Table 1.
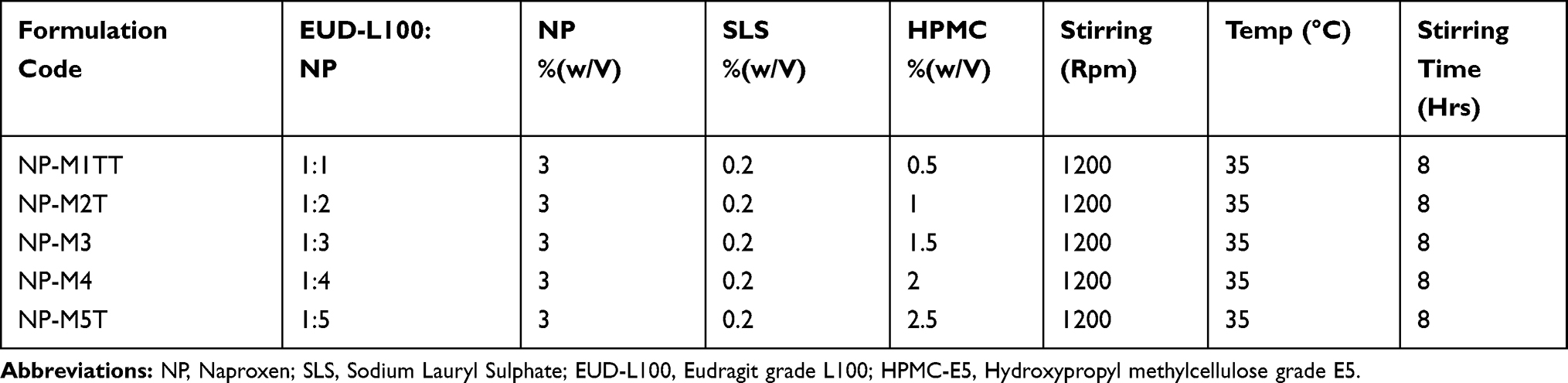 |
Table 1 Process Parameter and Compositions of NP Loaded PHE-Ms |
Characterization of Particle Size and Analysis
The particle size measurement and distribution of the engineered microspheres were carried out by laser diffraction using Mastersizer laser diffraction analyzer (Model Hydro 2000, Malvern Instruments, UK). The method as reported by Rahman et al22 was employed for correct particle size analysis of the produced microspheres. To avoid agglomeration in microspheres, 50 mg from each formulation (NP-M1-NP-M5) were acquired and dispersed in 5mL of water (distilled) comprising 2% w/v of Tween 80. In the water bath, the dispersions were sonicated for 3 min. Microspheres’ average particle size was designated as the volume mean diameter D [4, 3]. The span index value was calculated by the interpretation of the following equation:
(1)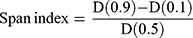
Where, D (0.1), D (0.5) and D (0.1) particle diameters were observed at 90th, 50th and 10th percentile of particles respectively.
FTIR Spectral Analysis
The FTIR spectral analysis of unprocessed NP, and developed NP loaded PHE-Ms (NP-M1) was carried out to identify and assess the interaction between components of microspheres. The NP-M1 powder (5mg) was well mixed with 250 mg of pure dry powdered potassium bromide. Finally, the mixture was pressed into a disc by means of a hydraulic press (100 kg/cm2) for 10 min. The subsequent mixtures were employed in a sampler of diffuse reflectance and scanned across the wavelength region of 400 to 4000 cm−1 at a resolution of 4 cm−1 with a scan speed of 1 cm/s through FTIR Spectrophotometer (IR Prestige 21 Shimadzu, Japan).
Differential Scanning Calorimetry (DSC)
DSC study of the unprocessed and processed NP samples was carried out using DSC (Model 404 F1/F3 Pegasus, Germany). The instrument was standardized with Indium under the stream of nitrogen gas. The samples were heated at a heating rate of 20°C/min from 20°C to 180°C. DSC thermograms of all the samples were recorded in triplicate.
Powder X-Ray Diffraction
The crystallography patterns of the produced microspheres, individual polymers, unprocessed NP and Physical mixture of NP with HPMC-EUD and SLS were investigated by X-ray diffraction (JDX-3532, JEOL Japan). The samples were loaded into silicon wells and scanned at 2-theta range from 5–40°C.
Scanning Electron Microscopy (SEM) Analysis
Scanning electron micrographs of the NP and produced NP loaded Polymer Hybrid Enteric Microspheres were obtained using SEM (Hitachi (S-570), Japan). The samples were coated with a double-sided sticking tape, sealed under reduced pressure and coated with gold using an ion sputtering device for 15 mins and scanned through a scanning electron microscope. Images of the unprocessed and processed samples were captured at magnification levels including 500X and 2000X respectively.
Drug Loading Entrapment Efficiency and Percent Yield
The entrapment efficiency of produced NP loaded PHEMs was analyzed and evaluated using the previously reported method with a slight modification.26 For determination of drug loading and entrapment efficiency, 50mg sample of microspheres was precisely weighed and dissolved in phosphate buffer solution (pH 6.8) and sonicated for 45 min. The content of NP was quantified in all formulations using a UV spectrophotometer (IRMECO Model U2020, Germany) at the ʎ of 332nm. The percent drug entrapment efficiency (DEE) and drug loading (DL) were calculated by the following formulas:
(2) (3)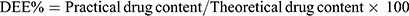
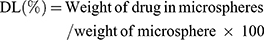
The microspheres were accurately weighed followed by calculation of the percentage yield using the following formula:
(4)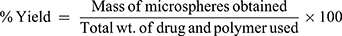
Molecular Modelling Study
The Structures of Naproxen, HPMC, and SLS were downloaded from PubChem while Eudragit was build-up using Chemdraw Professional v15.0 (Figure 2). Energy minimization of all the generated structures was carried out using YASARA-Structure software.27 The structures of polymers and surfactants including HPMC, Eudragit, and SLS were considered as alternative receptors (host) and ligand (guest) to obtain the stable complex of co-polymeric structure, while NP was used as only ligand (guest) structure for the molecular docking simulations. AutoDock Vina was used for molecular docking calculation in PyRx,28 in which the grid box was set to cover the entire polymer to ensure that all possible interactions with the drug were explored.29 The best-docked complex between co-polymer and drug was then subjected to molecular dynamics (MD) to divulge its stability in time and under the influence of explicit solvent molecules. MD simulations were carried out in the YASARA-Structure program using the YASARA force field with knowledge-based components.27 Chimera was used for the visualization and graphical representations of all co-polymer and drug complex.30
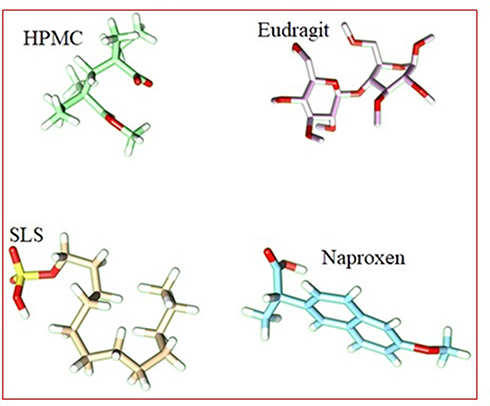 |
Figure 2 Minimized structures of polymers, surfactants, and NP. |
Preparation of Tablets from NP Loaded PHE-Ms
The NP loaded PHE-Ms equivalent to 250mg of the NP were mixed with different ratios of the chosen excipients for all batches (NP-M1-NP-M5) and compressed into tablets by using the Direct Compression machine (ZP.17). The compression force employed was in the range of 250 PSI for 40 seconds with a die size of 16mm. Oblong biconvex tablets of each ratio were obtained and assigned with formulation codes including NP-MT1, NP-MT2, NP-MT3, and NP-MT4and NP-MT5. The NP loaded PHE-Ms based tablets composition are shown in (Table 2).
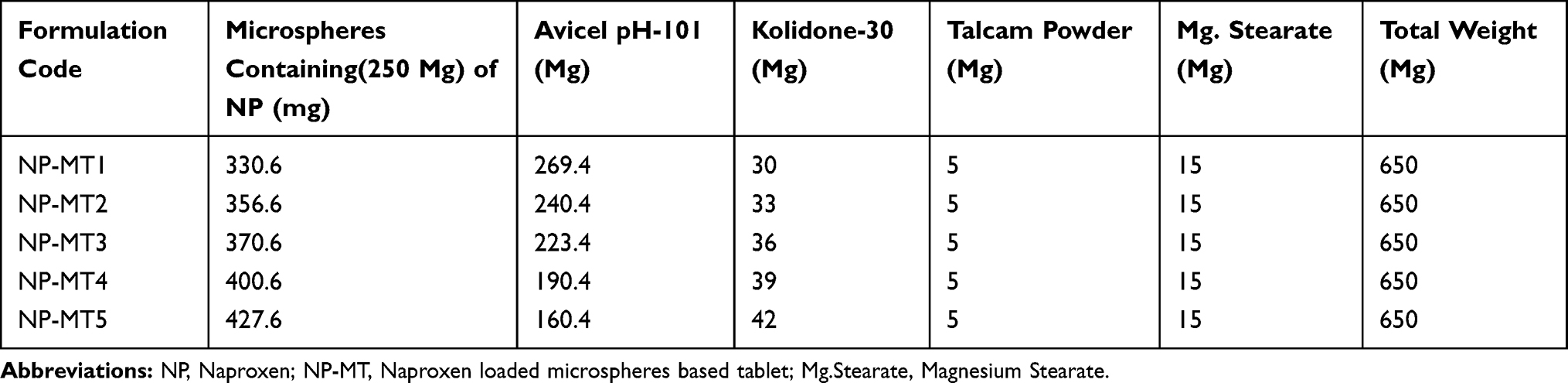 |
Table 2 Composition of NP Loaded PHE-Ms Based Tablets Formulations (MT1-MT5) |
Evaluation of PHE-Ms Tablets
The hardness of the microspheres based tablets was evaluated by Monsanto hardness tester (MH-1/Galvano scientific). A calibrated Vernier caliper (Y128 In) was used to determine the diameter and thickness of tablets. For the friability test, the tablets were positioned in a friabilator (Roche friabilator) and were revolved at a speed of 25 rpm for 4 mins. The tablets from each batch were exposed to weight variation and drug content uniformity tests. For drug content uniformity evaluation 03 tablets from each batch were crushed into powder and weighed (equivalent to 250 mg of NP). The powder form of tableted microspheres was suspended in 50 mL solution of phosphate buffer (PBS, pH 6.8).26 The obtained mixture was then filtered, diluted suitably and analyzed through UV- spectrophotometer at 332 nm for drug content. The dissolution study was conducted for drug release from the developed NP loaded polymer hybrid enteric microspheres (PHE-Ms) based on tableted formulations (NP-MT1-NP-MT5). The USP paddle-type II dissolution apparatus (DL-0601/Curio/Paddle Type II) was used with dissolution media of 900 mL. The pH of the dissolution medium was kept at pH 1.2 and at pH 6.8 correspondingly. The dissolution analysis at pH 1.2 and pH 6.8 was carried out for 120 mins and 6 hrs respectively. The drug contents in the respective collected samples (5mL) were analyzed using a UV spectrophotometer. Comparative dissolution study of optimized polymer hybrid enteric microspheres based tablets (NP-MT1) were executed with the marketed drug and unprocessed NP powders. The pH of the dissolution medium, for the first 2 hrs was kept at pH of 1.2 followed by pH 6.8 and continued dissolution for a further 6 hrs.
Modelling of Drug Release Kinetics
The NP loaded PHE-Ms based tablets were subjected to drug release kinetic study to identify the mechanism of the drug release from the developed tablets. Nicholas Peppas was the first who presented an equation, which defined the mechanism of drug release from the polymeric system.
(5)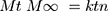
Where: Mt and M∞ are the absolute cumulative amount of drug released at time t and infinite time, respectively; k is a constant including the structural and geometric characteristic of the system, and n is a release exponent
In vivo Pharmacokinetic Study
For the pharmacokinetic experiment, male Sprague-Dawley rats weighing (200 ± 15 gm) were used. The rats were fasted for 10 h before the experiment and randomly allocated to various groups, each group contained six animals (n = 6). The research was carried out in accordance with the Animal Care Policy Manual and Use of Laboratory Animals as approved and promulgated through the NIH, Pak. The experimental procedures on animals were approved officially by the departmental research ethical committee of the University of Malakand and applicable Bye-Laws 2008 (Scientific Procedure Issue-1).
The NP (Unprocessed), commercialized NP tablets and the optimized formulation (NP-MT1) were orally administered to the animals at a dose equivalent to 40 mg/kg. The blood samples from retro-orbital plexus were collected at specific intervals of 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h. The blood samples were centrifuged at 7000 rpm at 4°C for 20 min to isolate plasma. The plasma samples were analyzed by means of a high-performance liquid chromatography method as Patino, el al previously reported. The analysis was performed on C18 with particle size of (5 mm), length (150 mm) and diameter symmetry (3.9), using a mixture of sodium acetate buffer adjusted at pH 3.14 (0.05M), with acetonitrile (60:40, v/v) as the mobile phase, and a flow rate of 1 mL/min. From the column, the effluent was detected at an absorbance of 235 nm (Millipore HPLC 501pump water/484 tunable UV absorbance Detector model#M.45).31 The pharmacokinetic parameters Cmax, Tmax, (t1/2), and AUC were determined by a trapezoidal rule.
Stability Study
The stability study of the optimized PHEMs based Tablets (NP-M1) was carried out at refrigerator (5±2), room (25±2°C) and accelerated temperature (40±2°C, RH of 75±5%) under the standard protocols of International conference on Harmonization (ICH) for zones (III &IV). The tablets were packed in screw-capped high-density polyethylene (HDPE) containers, subjected to a stability chamber for 6 months. The tablets were evaluated for several parameters, including visual examination, (%) drug content, hardness, friability, % moisture content through Karl Fischer titration (METTLER TOLEDO Moisture Analyser), and dissolution.
Statistical Analysis
The generated data was reported as a grand mean of triplicate of samples ± standard deviation and standard error mean. In addition, the generated data were subjected to one-way analysis of variance (ANOVA) and two-tailed t-tests with significant values <0.05. In plasma, the pharmacokinetics parameters were determined by means of pharmacokinetic software WinNonLin (v 4.0; Pharsight Software, Mountain View, CA, USA).
Results and Discussion
Development of Microspheres and Impact of Process Conditions
The impact of the process conditions and proper control of all the considerations for the optimization of NP loaded PHE-M were significant which resulted in microspheres with more spherical and discrete morphology. The EUD-L100 concentration in the organic phase stimulated solvent diffusion and emulsification at a constant stirring rate (1200 rpm). The mean diameters and particle size of NP loaded PHEM were dependent on polymer concentration distributed in the organic phase (Table 3). The stirring rate (shearing force) became a key factor in dispersing the organic phase into the subsequent aqueous phase, attributing to an increase in small droplet size of the emulsion. The stirring speed higher than 1500rpm would lead to the rupturing of the dispersion droplet and resulted in the formation of fewer microspheres.32 The SLS (0.2% w/v) as a surfactant (HBL value 40) ensured its superiority in emulsion solvent evaporation (ESE), influencing the size of the emulsion droplet and reducing surface tension.33 In addition, the recovered quantity of total microspheres established the appropriateness of process variables during solvent evaporation.
 |
Table 3 Particle Size (μm) and Span Index of NP Loaded PHE-MS of Formulations (M1–M5) |
The particle sizes for the formulations (M1-M5) were found to be in the range of 30.5 ± 0.2–74.31 ± 0.26 μm. The lowest span value 0.49 for NP-M1 formulation was observed compared to the rest of the formulations which was indicative for monodispersed of the microspheres.22 Alhalaweh et al previously reported,34 the value of D (50th) represented the particle size at which 50% of NP-M were smaller and 50% larger than this size. D (10th) was the size of particles for which 10% of the NP-Ms lowered this size. D (90th) specified particle size for which 90% of the NP-M was below this size. By increasing the polymer concentration, the particle size of the NP-M increased with high span values.22 PS and PSD of NP loaded Polymeric Hybrid Enteric Microspheres (PHE-Ms) are represented in Table 3.
FTIR Analysis
FTIR spectroscopy was used to explore the interaction of the chosen polymers/surfactant with NP in polymer hybrid microspheres. However, the FTIR data for the hybrid system demonstrated the compatibility of the NP with the excipients and did not show any type of chemical interaction. To characterize the microspheres, the FTIR spectra for NP (Unprocessed) and NP-M1 were compared in terms of functional groups. The FTIR spectrophotometer analysis exposed characteristic peaks bend at Carboxyl Group (-COOH and O-H stretch) a broad absorption peak at 3443cm−1 and peak of (C=O stretch) was observed at 1721 cm−1. The Aromatic Ring (C=C-C stretch) was perceived at 1608 cm−1. The peaks of (Aryl-O stretch) were detected at 1264 cm−1.35 Lacking the additional peaks in the FTIR spectrum of microspheres exposed homogeneous dispersion of NP in the polymer matrix without shifting of the functional group (Figure 3A and B).9
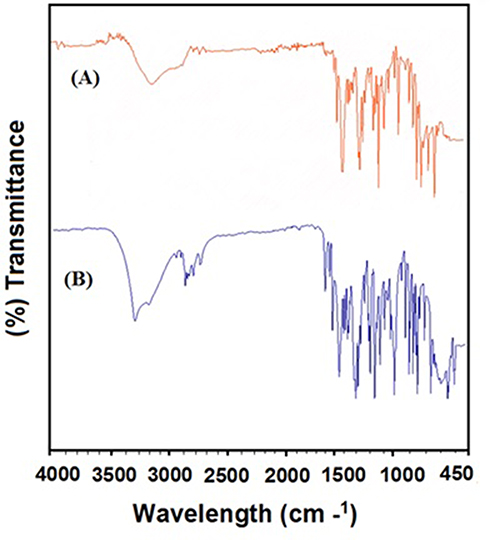 |
Figure 3 (A) FTIR spectra for NP (Unprocessed) and (B) NP-M1 optimized formulation. |
Morphology Study
The SEM photomicrographs of the developed NP-loaded PHE-Ms indicated that the microspheres were uniform, spherical and interlinked to each other with a distinct symmetry in Figure 4B. The particles of the unprocessed NP were found with prism, plates and elongated morphologies in the size range from 60 to 80 micron in Figure 4A. The surface texture of the optimized microspheres (NP-M1) was much smoother, no crystals were present on the surface indicating that NP was successfully enclosed in enteric-coated EUD-L100/HPMC. The solvents chloroform and EtOH allowed complete dissolution of the enteric EUD-L100, even though retaining the integrity of the inner microspheres reported by Dan Zhous et al, formerly.36
 |
Figure 4 (A) Unprocessed NP at magnification of 500× and (B) NP-M1 optimized formulation (2000×). |
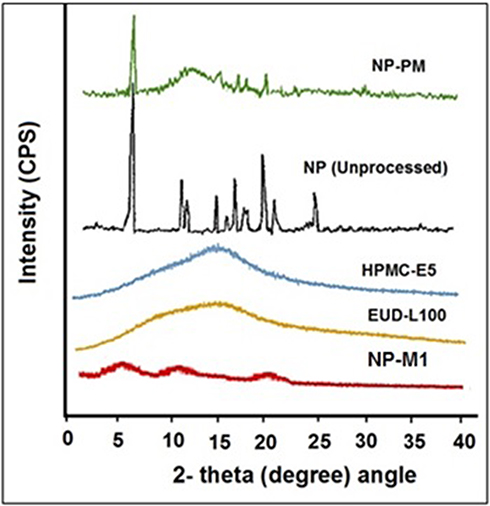 |
Figure 5 P-XRD pattern of NP (Unprocessed), NP-PM, NP-M1, EUD, and HPMC. |
X-Ray Diffraction
The crystallinity of Naproxen (Unprocessed) was found to be more than NP-M1 and NP-PM. The relative intensity of all the peaks of NP (Unprocessed) was more than that of the peaks at the corresponding position in the diffractograms from NP-M1 and NP-PM. Though the NP-M1 lost most of the XRD peaks and the intensities of the rest of the peaks were lower associated with those of the NP (Unprocessed). The P-XRD studies of the polymers (EUD-L100 and HPMC-E5) were also performed to assess their influence on the crystallinity of the developed PHE-Ms. However, on diffractogram, there were no dominant consequences of the polymers on the developed microspheres. This decrease and absence of the major peaks in NP microspheres revealed a reduction in the degree of crystallinity or particle size reduction, which led to an amorphous state. The NP-M1 in an amorphous state has been signified in Figure 5.37
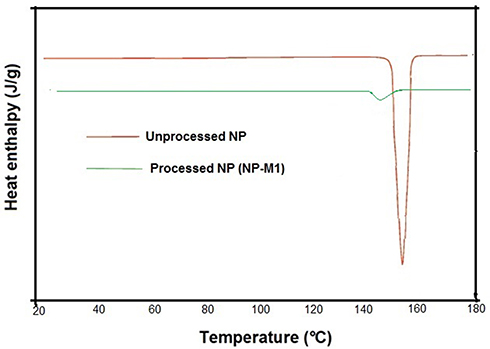 |
Figure 6 DSC Thermograms of NP (Unprocessed) and NP-M1. |
Differential Scanning Calorimetry (DSC)
The melting point of NP (Unprocessed) showed a single sharp melting peak at about 157.50°C (Figure 6). However, we observed a very small and broader peak for the processed NP. The reduction in peak intensity of the processed NP and a broader appearance indicated size reduction, amorphous transition and shifting towards lower melting point. The PXRD data for the processed hybrid NP also showed a few small peaks which exhibited traces of crystalline NP. The sharp peak was a characteristic of naproxen’s crystalline nature which has been reported previously by Dixit et al38 Varshosaz et al, reported that the reduction in micro-particle size and the presence of a stabilizer (polymer, surfactant) affected the enthalpy and the precipitation of a drug was sterically stabilized against crystal growth on the polymer surface. Therefore, it resulted in the lowering of the surface energy and enthalpy of the system with smaller particle sizes.19,39
Determination of % Entrapment Efficiency (EE) and % Drug Loading (DL)
The percent entrapment efficiency of PHE-MS formulations was found to be increased with a high ratio of the polymers, whilst the percent DL decreased irrespective of the percent EE. The (%) Yield of formulations NP (M1-M5) was obtained in a minimum to the maximum range of (73.12±0.31–91.73±0.72). Consistent with the higher (%) yield for NP-M5, higher (%) EE was also observed for the same formulation. The drug/Polymer ratio during microspheres preparation was a critical factor and by increasing polymer concentration there was a significant increment in drug entrapment.40 Drug–polymer composition affected the (%) EE of the drug inside polymer molecules and the adsorption of drug molecules on the surface of the polymeric matrix because of electrostatic adhesions.41 The impact of formulation variable in Table 4.
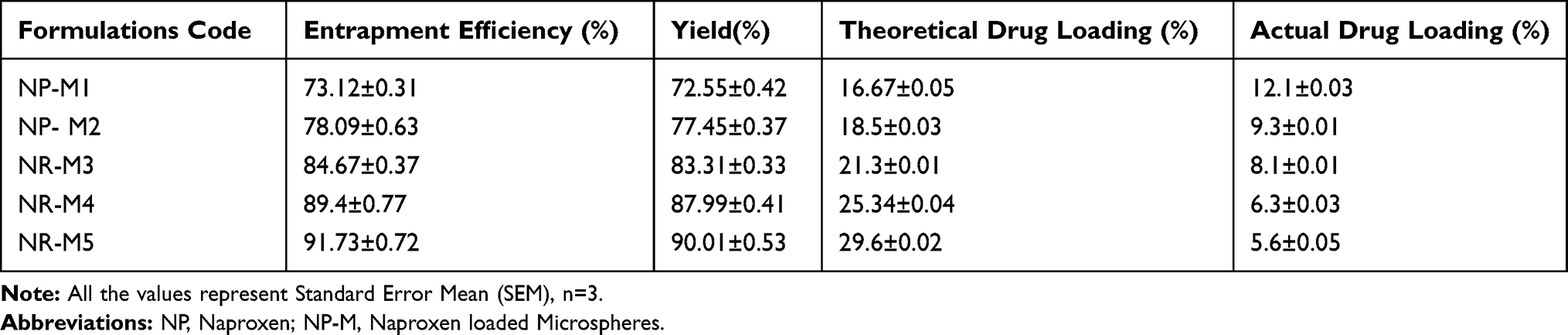 |
Table 4 Impact of Formulation Variables on (%) Yield, Drug Loading, and Encapsulation Efficiency on Formulations (M1-M5) |
In silico Modelling Study
Molecular docking is an important modelling approach that gives an idea about the interactions between receptor (host) and ligand (guest). This in-silico method allowed us to depict the ligand-binding sites and conformations within a host. Mostly, the molecular docking simulation gives insight about the alignment of the drug in a binding site (termed as “pose”) and also gives an estimation of the binding affinity of the identified pose in the form of scoring value.29 The AutoDock-VINA algorithm utilizes a “machine-learning” method that merges the advantages of knowledge-based potentials and empirical scoring-functions to calculate the binding energy of a given ligand pose. The relative binding free energies between the naproxen and three different polymer molecules as well as co-polymeric molecules were calculated using Autodock Vina as indicated in Table 5. The binding free energies calculations between the polymers and surfactants including HPMC, EUD, SLS (host) and NP (Naproxen) molecule (guest) will estimate the strength of the interactions between them. Tighter interactions between the drug molecules and polymer might lead to a stable drug-polymer complex and may result in a more sustained drug release profile and effective encapsulations compared to the looser interaction/binding.42–44
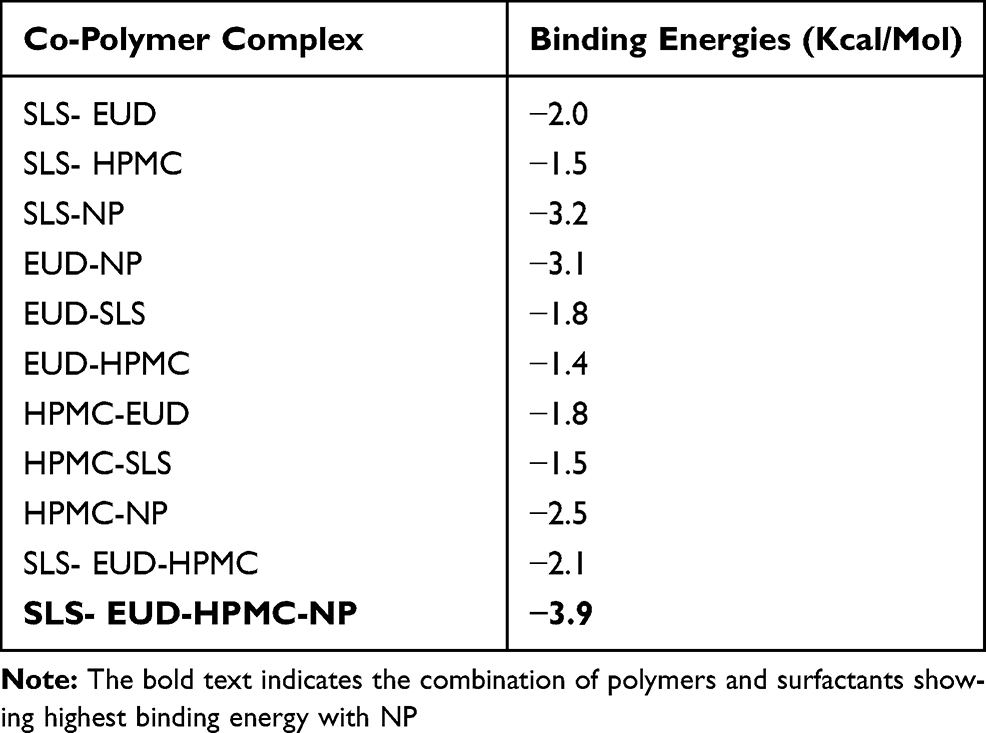 |
Table 5 Binding Energies Calculations for Different Co-Polymeric Systems of SLS, Eudragit (EUD), HPMC and Naproxen (NP) |
It has become apparent from the binding free energies table that the mono-polymeric form has a lower binding affinity than the co-polymeric form. In the case of mono-polymeric complexes, HPMC and Naproxen (−2.5 kcal/mol) complex were found to have the least binding affinity compared to Eudragit-Naproxen (−3.1 kcal/mol)) than the SLS-Naproxen complex (−3.2 kcal/mol). The bare co-polymeric complex between SLS- Eudragit-HPMC (−2.1 kcal/mol) was found to have a lower binding affinity compared to their complex with naproxen. The best binding affinity and binding energy were shown by co-polymer complex SLS- Eudragit-HPMC-Naproxen which was −3.9 kcal/mol as shown in Table 5. This has substantiated our experimental results, where we have chosen HPMC-EUD-SLS as the best combination to effectively encapsulate and accommodate the NP with subsequent high controlled release kinetics.
In order to comprehend the binding mode, interaction mechanism, and complex stability, between the NP and co-polymers complex, MD simulations were also performed. The stability of the simulated systems was evaluated by computing the differences in the root mean square deviation (RMSD) in relation to the minimized structures. MD simulation also revealed the stability of co-polymeric complex with NP. The binding orientation of Naproxen within the complex structure of SLS-Eudragit-HPMC is shown in Figure 7.
 |
Figure 7 3D surface and structure representation of SLS and Eudragit complex (A). 3D surface and structure representation of SLS-Eudragit (host) and HPMC (guest) complex structure (B). 3D surface and structure representation of SLS-Eudragit-HPMC host) and Naproxen (guest) complex structure (C). |
Development and Evaluation of NP Loaded Hybrid Microspheres Based Tablets
The NP-loaded Polymeric Hybrid Enteric Microspheres (PHE-Ms) based tablets subjected to recommended quality-control tests resulted in acceptable limits (Supplementary data). All the results were found statistically significant (P<0.05). The tablet thickness ranged from 4.11 ±0.02 to 4.12±0.01mm and average weight ranged from 653.0±5.23 to 659.0±3.52 mg. The friability was noted less than 1% friability, from 0.76% to 0.78%, signifying appropriate mechanical strength.45 The (%) Drug content (DC) of the formulations (MT1-MT5) closely varied between 96.2±2.64 to 98.7±4.12% suggesting uniform drug distribution. The formulations (MT1-MT5) exhibited appropriate hardness ranged from 4.41±0.02 to 6.23±0.13 kg/cm2. The hardness of the tablets varied, depending upon the amount of MCC (Avicil 101), coating polymer HPMC-E5 and polymer used as a core material EUD-L100. The coat of HPMC-E5 contributing elasticity to withstand the compaction force and protected the enteric microspheres from rupturing and any other mechanical shock. The Microcrystalline cellulose (MCC) was used to assist the method of compaction of the microspheres formulations relative to avoid the rupturing and damaging of the microspheres. Matrix monolithic polymeric microspheres were stated to be more resistant and more robust to rupture on tableting. The tableted microspheres specified that in the tablets, microspheres were present as a single and discrete particle without getting compressed during the process of compression. During compression, the polymers retained their integrity, withstood the compaction force, and remained intact without cracks.46,47
In-vitro Drug Release
The drug release profile of NP from various PHEMs based tablet formulations is shown in Figure 8. The comparative dissolution studies demonstrated that the hybrid microspheres effectively retarded the drug release at pH 1.2(0.1N HCl) in the first 120 mins and showed a <5% drug release (Figure 8B). It has become evident from Figure 8B, that at pH (1.2), the PHE-MT slightly swelled then remained intact. The polymers were dissolved in a controlled manner and the microspheres resulted in more than 85% drug release at pH 6.8 (Figure 8A. Eudragit L100 has no solubility in aqueous media but has shown a remarkable release of the drug at alkaline media (>6 pH).48 Pinto, et al reported, HPMC was estimated to illustrate pH-independent dissolution behavior, as it contained no ionizable groups. However, the HPMC dissolution and release rate were affected and controlled by their kinetics of swelling. Furthermore, HPMC-E5 as a component of the enteric EUD-L100 microspheres is apparently dependent not only upon the dissolution of the polymeric layer but also on the permeability and mechanical characteristics of the resulting PHE-MS. The augmented pH-responsiveness of the EUD-L100 layer was related to the swelling index of the HPMC-E5.49
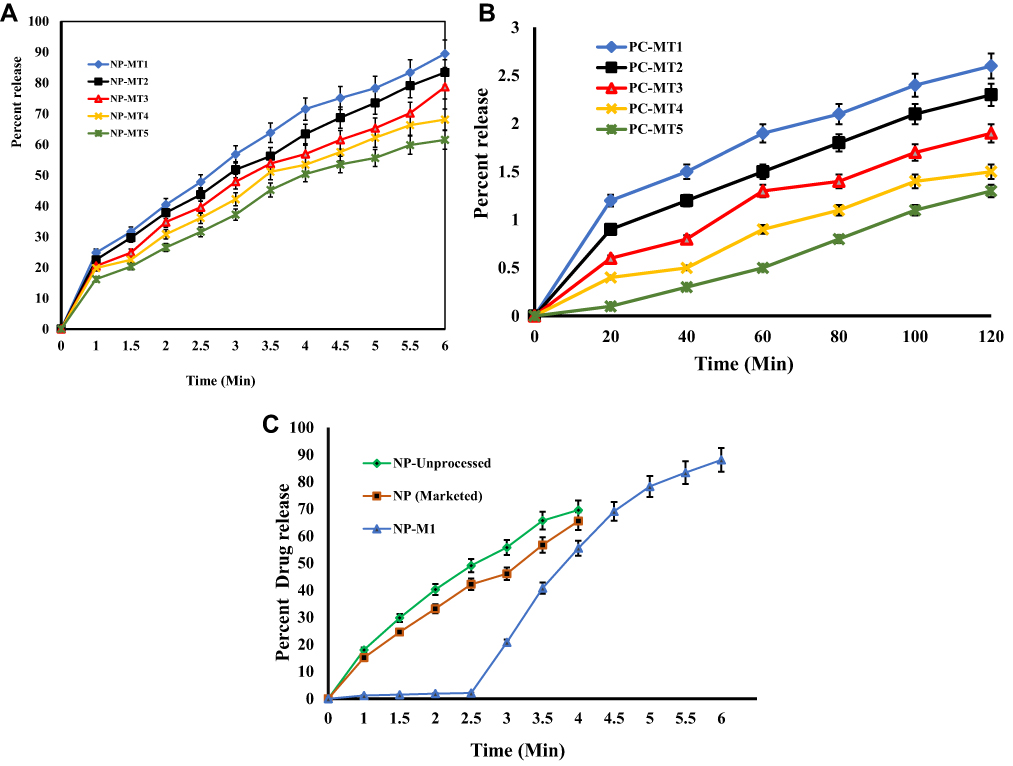 |
Figure 8 (A) Drug release from formulations of NP (MT1-MT5) at pH 6.8. (B) Drug release from formulations of NP (MT1-MT5) at pH 1.2. (C) Comparative drug release profile of NP (Marketed Drug), NP (Unprocessed) and NP-MT1 at pH1.2 and pH 6.8. |
The HPMC-E5 exhibited unique release-modifying properties, primary rate-controlling contributor swelled and a gel layer formation on the surface of the system as delaying lag time. Diffusion and erosion of the PHE-Ms based tablets followed a change in the diffusion pathway due to polymer erosion, modified and delayed-release system. The hydrophobic EUD-L100 core system, contained NP in disperse form throughout the matrix, comprises negligibly and an essential increase on system surface or impart change in dimension during drug release, followed by dissolution and formerly diffusion of the drug in alkaline media.50,51 The NP pKa value was 4.2 and showed a unionized and ionized form conditional to pH change. Furthermore, the pH environment of NP was affected with the involvement of the Hybrid system of hydrophobic and hydrophilic polymer composite presented pH-regulated NP release.
The PHE-Ms based tablet of 1:1 ratio showed more significant drug release and in a more controllable manner.48 When considering variation in the surface area we found NP loading and particle size have intermingled effects on dissolution. The comparative study of the optimized PHE-Ms based tablets (NP-MT1), NP (Unprocessed) and Proxen (Marketed drug) showed the dissolution characteristics represented in Figure 8C. The NP-MT1 showed retardation of NP release at pH 1.2 and significant and enhanced release pattern at pH 6.8 in contrast to NP (Unprocessed) and Proxen (Marketed Naproxen). The NP release from formulations (MT1-MT5) was considered p < 0.05 as significant value.
Drug Release Kinetics Study
The NP loaded Polymeric Hybrid Enteric Microspheres (PHE-Ms) based tablets were analyzed for the NP modified-release kinetics. The time versus cumulative release (%) data was tailored to the Higuchi, First- Order, Zero-Order and the Korsmeyer–Peppas equations. The equation for drug release was designated by the Higuchi model for microspheres.
(6)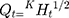
In the equation, Qt represented the % cumulative drug released at time t while kH was a constant characteristic of the process. The (%) cumulative release from the NP loaded PHE-Ms based tablets was proportional to the square root of time signifying diffusion-controlled release from the microspheres based tablets. The values of the rate constants kH were greatly influenced by the process parameters (stirring speed, surfactant nature, and concentration, the number of polymers and excipients used in tableting). The superlative fit model with the uppermost correlation coefficient r2 was practically observed in the Higuchi model (Table 6). Additionally, the mechanism of drug release was determined by fitting the release versus time data to the Korsmeyer and Peppas semi-empirical equation as follows:
(7)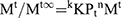
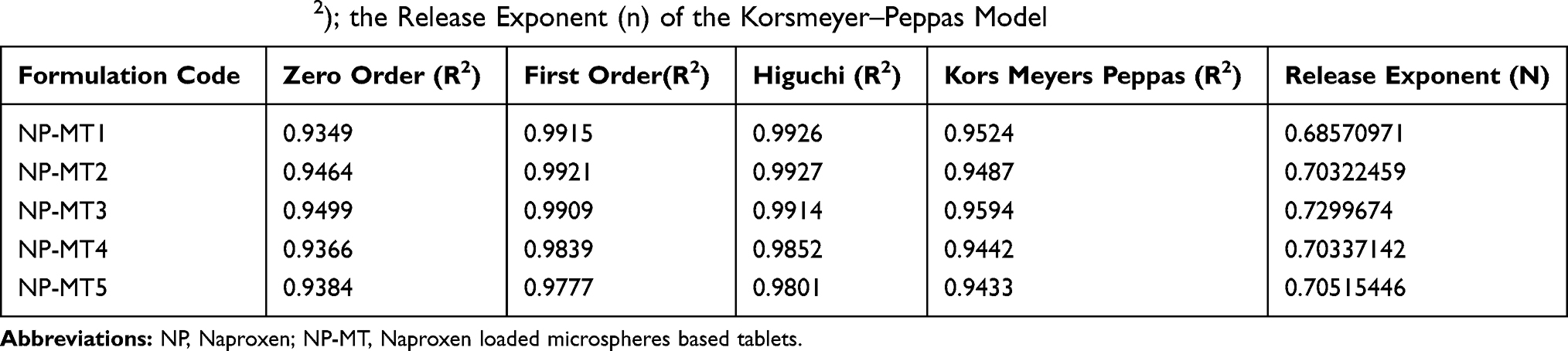 |
Table 6 Modulation and the Model Equations Applied to the in- Vitro Release Kinetics of PHE-Ms Based Tablets Respectively; the Correlation Coefficient (R2); the Release Exponent (n) of the Korsmeyer–Peppas Model |
Where Mt/Mt∞ was the drug released fraction at time t, Mt and Mt∞ described the masses of drug release at time t and t∞, correspondingly. The kKP was the rate constant and n represented as an observed parameter illustrating the mechanism of drug release. The values of n for cylinders and spheres were described by Ritger and Peppas.52 For spherical geometry, intermediate values ranging between 0.43 and 0.85 were accredited to anomalous or non-Fickian transport of drug release with mix function of swelling, erosion, and diffusion. Mouffok, Meryem, et al reported,40 for the Korsmeyer–Peppas model, the obtained values of rate constants were dependent on n value.
The non-Fickian release mechanism of the drug was termed by two mechanisms: one was the coupling of the drug diffusion and the second was the relaxation of the polymer. The first step in diffusion was the wetting of NP-Ms by water, followed by its dissolution to ensure the availability of its molecular form to diffuse out NP from the hydrophobic core of the matrix. Hence, the polymer and the excipient in tablet form significantly influenced the diffusion process and the rate of NP release from PHE-M.53
Oral Bioavailability Study
The pharmacokinetic profile of NP in plasma after oral administration of NP (Unprocessed), NP (Marketed drug) and NP loaded Polymeric Hybrid Enteric Microspheres based Tablet formulation were shown in Figure 9. The pharmacokinetic parameters, including AUC, Cmax, and biological half-life have been shown in Table 7.
 |
Table 7 Pharmacokinetics Parameters for NP (Unprocessed), NP (Marketed Drug), and NP MT1 Formulation in Rats |
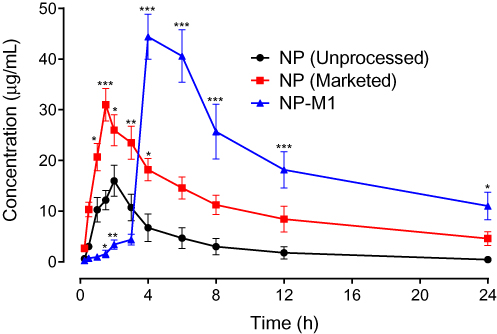 |
Figure 9 Pharmacokinetic profile of NP (Unprocessed), NP (Marketed drug) and NP-MT1 in rats. The plot of plasma concentration (µg/mL) versus time (h). Data represented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 in contrast to NP (Unprocessed) treated animals group at respective time-period; two-way repeated-measures ANOVA followed by post hoc Bonferroni’s analysis was used. |
The Administration of 40mg/kg dose of NP (Unprocessed) exhibited a mean elimination phase from 12 to 24 h with an elimination half-life of 6.38 h and a clearance of 640 mL/h. The distribution phase (DP) was observed from 6 to 8 h with an acquired volume of 6263mL of distribution. The absorption phase occupied a range from 0.3 to 4 h. The Cmax was in the range of 16.01 µg/mL at 2.39 h. The AUC was 81.01 µg-h/mL from time zero to 24 h. For the NP (Marketed), a significant increase in the plasma concentration was observed, which was first prominent after 1 h (P < 0.05) and remained significant for the subsequent time duration of 1.5 h and 2–4 h (P < 0.05, P < 0.01) as compared to the NP (Unprocessed) as shown in Figure 9. The pharmacokinetic parameters for the NP (Marketed drug) were observed as an elimination half-life of 13.97 h, maximal plasma concentration of 31.01 µg/mL (P < 0.05 as compared to NP (Unprocessed), time to reach maximal plasma concentration as 1.65 h, AUC of 259.1 µg-h/mL, and a volume of distribution of 3318 mL. The optimized–formulation of PHE-Ms (NP-MT1) showed an enteric and delayed onset as a substantial increase in the plasma concentration was first apparent at 4 h (P < 0.001) and this increased proclivity of plasma concentration was significant for the subsequent experimental time duration i.e. 6–24 h (P < 0.05, P < 0.001), as compared to the NP (Unprocessed) in Figure 9. A significant increase (P < 0.01) in the maximum plasma concentration of 44.41 µg/mL was observed with a significant increase in the time (4.31 h, P < 0.05) to reach maximum plasma concentration. The volume of distribution was noted as 2329 mL (distribution phase: 6 to 8 h). A significant decrease (P < 0.05) in the clearance was also observed i.e. 94.90 mL/h. Moreover, the microspheres significantly increased (P < 0.01) the plasma exposure of NP as revealed from the AUC from time zero to 24 h (444.9 µg h/mL).31
Stability Study
The stability studies demonstrated that the produced PHE-Ms based tablets (NP-MT1) were stable at different stability conditions. The results of assays characterizing hardness, friability, % drug release, physical appearance, and moisture content %(w/w) demonstrated that the produced microspheres based tablets were stable at different temperatures and humidity levels (Supplementary data). At storage conditions, no significant changes were found in the sample monitored with several parameters after 180 days suggesting its reasonable strength to withstand the accelerated conditions. The key physicochemical attributes of the % drug release and assay level were maintained at acceptable limits. No statistically significant variances in the % release profiles were detected among the NP-MT1 stored at various conditions.54 All the results were found statistically significant with a paired t-test, one-way ANOVA, exhibited P<0.05.
Conclusion
The Polymeric Hybrid delivery system was used to develop NP loaded enteric Microspheres through the “Solvent Evaporation Technique” using biocompatible pH-responsive EUD-L100 in conjunction with HPMC and SLS. The enteric microspheres of NP potentially retarded release at pH1.2 (stomach pH) and enhanced drug release at pH 6.8 (small intestine pH). NP loaded microspheres based tablets exhibited a modified release pattern with retardation of NP release in acid pH while, modified-release at alkaline pH in a more controlled manner. The molecular modelling (MM) and simulations studies substantiated the experimental results and rationalized EUD-HPMC-SLS as an effective hybrid system with the highest binding energy to improve the encapsulation of NP with a modified release profile. The stability studies demonstrated no loss of integral properties of the Polymeric Hybrid enteric microspheres based tablet at storage conditions. In addition, the in vivo studies ensured the target objectives of the developed hybrid microspheres with improved dissolution and bioavailability. In the future perspective, this technology can potentially be extended towards other drug molecules to be developed as novel Polymeric Hybrid microspheres based systems to diminish poor aqueous solubility and gastro-intestinal tract related issues.
Acknowledgments
The authors extend their appreciation to the research supporting Project (No: RSP-2019/110) at King Saud University for funding and supporting this research work The support of Navegal Laboratories (Pvt) Industrial state Hattar Haripur Pakistan is also appreciated and acknowledged for providing the facility for tablet manufacturing and characterizations. We are also very grateful to Heather C. Radford (University of Texas at Austin, USA) for her support to thoroughly check this manuscript for grammatical mistakes.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Auriemma G, Cerciello A, Aquino RP. NSAIDS: design and development of innovative oral delivery systems. Nonsteroidal Anti-Inflammatory Drugs. 2017;33.
2. Altman R, Bosch B, Brune K. Advances in NSAID development: evolution of diclofenac products using pharmaceutical technology. Drugs. 2015;75(8):859–877. doi:10.1007/s40265-015-0392-z
3. Mrsny RJ. Oral drug delivery research in Europe. J Control Rele. 2012;161(2):247–253. doi:10.1016/j.jconrel.2012.01.017
4. Tsume Y, Mudie DM, Langguth P. The Biopharmaceutics Classification System: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci. 2014;57:152–163. doi:10.1016/j.ejps.2014.01.009
5. Boyd BJ, Bergström CA, Vinarov Z. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur J Pharm Sci. 2019;137:104967. doi:10.1016/j.ejps.2019.104967
6. Kansara H, Panola R, Mishra A. Techniques used to enhance bioavailability of BCS class II drugs: a review. Int J Drug Dev Res. 2015;7(1):82–93.
7. Zhao X, Song K, Wang S. Micronization of the pharmaceutically active agent genipin by an antisolvent precipitation process. Chem Eng Technol. 2013;36(1):33–42. doi:10.1002/ceat.201200036
8. Montes A, Kin N, Gordillo M. Polymer–naproxen precipitation by supercritical antisolvent (SAS) process. J Supercritc Fluids. 2014;89:58–67. doi:10.1016/j.supflu.2014.02.004
9. Alhnan MA, Basit AW. In-process crystallization of acidic drugs in acrylic microparticle systems: influence of physical factors and drug–polymer interactions. J Pharm Sci. 2011;100(8):3284–3293. doi:10.1002/jps.22572
10. Wise DL. Handbook of Pharmaceutical Controlled Release Technology. CRC Press; 2000.
11. Liu F, Lizio R, Meier C. A novel concept in enteric coating: a double-coating system providing rapid drug release in the proximal small intestine. J Control Rel. 2009;133(2):119–124. doi:10.1016/j.jconrel.2008.09.083
12. Moustafine R, Bukhovets A, Sitenkov A. Eudragit E PO as a complementary material for designing oral drug delivery systems with controlled release properties: comparative evaluation of new interpolyelectrolyte complexes with countercharged eudragit L100 copolymers. Mol Pharm. 2013;10(7):2630–2641. doi:10.1021/mp4000635
13. McGettigan P, Henry D. Current problems with non-specific COX inhibitors. Current Pharm Des. 2000;6(17):1693–1724. doi:10.2174/1381612003398690
14. Arroyo‐Lira AG, Rodríguez‐Ramos F, Ortiz MI. Supra‐additive interaction of docosahexaenoic acid and naproxen and gastric safety on the formalin test in rats. Drug Deve Res. 2017;78(7):332–339. doi:10.1002/ddr.21396
15. Correa MA, Scarpa M, Franzini M. On the incorporation of the non-steroidal anti-inflammatory naproxen into cationic O/W microemulsions. Colloids Sur Biointerfaces. 2005;43(2):108–114. doi:10.1016/j.colsurfb.2005.04.005
16. Guo Z, Liu X-M ML. Effects of particle morphology, pore size and surface coating of mesoporous silica on Naproxen dissolution rate enhancement. Colloids Sur Biointerfaces. 2013;101:228–235. doi:10.1016/j.colsurfb.2012.06.026
17. Maghsoodi M, Taghizadeh O, Martin GP. Particle design of naproxen-disintegrant agglomerates for direct compression by a crystallo-co-agglomeration technique. Int J Pharm. 2008;351(1–2):45–54. doi:10.1016/j.ijpharm.2007.09.033
18. Maheswari P, Rambhau D, Narasu M. Micellar solubilization in the formulation development of poorly soluble naproxen. Pharm Reg Affairs. 2013;2(108):2.
19. Aabakken L, Ugstad M, Gamst O. Naproxen-associated gastroduodenal toxicity: enteric coated granules versus plain tablets. Eur J Rheum Inflam. 1992;12(2):43–48.
20. Adibkia K, Barzegar-Jalali M, Maheri-Esfanjani H. Physicochemical characterization of naproxen solid dispersions prepared via spray drying technology. Powder Technol. 2013;246:448–455. doi:10.1016/j.powtec.2013.05.044
21. Liu X, Zhou L, Zhang F. Reactive melt extrusion to improve the dissolution performance and physical stability of naproxen amorphous solid dispersions. Mol Pharm. 2017;14(3):658–673. doi:10.1021/acs.molpharmaceut.6b00960
22. Rahman MA, Ahmed N, Hasan I. Formulation and in vitro assessment of Eudragit L 100 and Eudragit S 100 based naproxen microspheres. Dhaka Univ J Pharm Sci. 2016;15(1):47–55. doi:10.3329/dujps.v15i1.29192
23. McConnell EL, Short MD, Basit AW. An in vivo comparison of intestinal pH and bacteria as physiological trigger mechanisms for colonic targeting in man. J Controll Rele. 2008;130(2):154–160. doi:10.1016/j.jconrel.2008.05.022
24. Obeidat WM, Price JC. Preparation and evaluation of Eudragit S 100 microspheres as pH-sensitive release preparations for piroxicam and theophylline using the emulsion-solvent evaporation method. J Microencapsul. 2006;23(2):195–202. doi:10.1080/02652040500435337
25. Al-Nasi AA, Al-Tahami KA. Preparation, characterization, and in vitro release of ketoprofen loaded polymeric microspheres. Int J PharmTech Res. 2016;9:313–321.
26. Zaghloul A, Faltinek J, Vaithiyalingam S. Naproxen-Eudragit microspheres: screening of process and formulation variables for the preparation of extended release tablets. Pharmazie. 2001;56(4):321–324.
27. Krieger E, Vriend G, Spronk C. YASARA–yet another scientific artificial reality application. YASARA Org. 2013;993.
28. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Bio. 2015;1263:243–250.
29. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
30. Pettersen EF, Goddard TD, Huang CC. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
31. Patiño‐Camacho SI, Moreno MGL, Flores‐Murrieta FJ. The pharmacokinetic profile of the combination of naproxen and tizanidine in rat. Drug Deve Res. 2013;74(1):31–37. doi:10.1002/ddr.21053
32. Freitas S, Merkle HP, Gander B. Microencapsulation by solvent extraction/evaporation: reviewing the state of the art of microsphere preparation process technology. J Control Rele. 2005;102(2):313–332. doi:10.1016/j.jconrel.2004.10.015
33. Li M, Rouaud O, Poncelet D. Microencapsulation by solvent evaporation: state of the art for process engineering approaches. Int J Pharm. 2008;363(1–2):26–39. doi:10.1016/j.ijpharm.2008.07.018
34. Alhalaweh A, Andersson S, Velaga SP. Preparation of zolmitriptan–chitosan microparticles by spray drying for nasal delivery. Eur J Pharm Sci. 2009;38(3):206–214. doi:10.1016/j.ejps.2009.07.003
35. Sharmin Akhter SP, Hasan I, Ayon NJ. Preparation, characterization and compatibility studies of naproxen loaded microspheres of cellulosic and polymethacrylic polymeric blend. Dhaka Univ J Phar Sci. 2013;12:11–21. doi:10.3329/dujps.v12i1.16295
36. Zhou D, Zhu X, Wang Y. Preparation and characterization of a novel pH-sensitive coated microsphere for duodenum-specific drug delivery. Arch Pharm Res. 2012;35(5):839–850. doi:10.1007/s12272-012-0509-9
37. Montes A, Bendel A, Kürti R. Processing naproxen with supercritical CO2. J Supercritic Fluids. 2013;75:21–29. doi:10.1016/j.supflu.2012.12.016
38. Dixit M, Kulkarni P, Charyulu R. Enhancing solubility and dissolution of naproxen by spray drying technique. World J Pharm Pharma Sci. 2015;4:715–725.
39. Varshosaz J, Khajavinia A, Ghasemlu M. Enhancement in dissolution rate of piroxicam by two micronization techniques. Dissolution Technol. 2013;20(3):15–23. doi:10.14227/DT200313P15
40. Mouffok M, Mesli A, Abdelmalek I. Effect of the formulation parameters on the encapsulation efficiency and release behavior of p-aminobenzoic acid-loaded ethylcellulose microspheres. J Serb Chem Soc. 2016;81(10):1183–1198. doi:10.2298/JSC160308068M
41. Jyothi NVN, Prasanna PM, Sakarkar SN. Microencapsulation techniques, factors influencing encapsulation efficiency. J Microencapsul. 2010;27(3):187–197. doi:10.3109/02652040903131301
42. Ahmed S, Govender T, Khan I. Experimental and molecular modeling approach to optimize suitable polymers for fabrication of stable fluticasone nanoparticles with enhanced dissolution and antimicrobial activity. Drug Des Dev Ther. 2018;12:255. doi:10.2147/DDDT.S148912
43. Ndlovu ST, Ullah N, Khan S. Domperidone nanocrystals with boosted oral bioavailability: fabrication, evaluation and molecular insight into the polymer-domperidone nanocrystal interaction. Drug Deliv Trans Res. 2019;9(1):284–297. doi:10.1007/s13346-018-00596-w
44. Seedat N, Kalhapure RS, Mocktar C. Co-encapsulation of multi-lipids and polymers enhances the performance of vancomycin in lipid–polymer hybrid nanoparticles: in vitro and in silico studies. Mater Sci Eng. 2016;61:616–630. doi:10.1016/j.msec.2015.12.053
45. Nandhakumar S, Alekya C. Formulation and optimization of delayed release MUPS (Multiple Unit Particulate System) tablets of omeprazole. Mater Sci. 2017.
46. Mundargi RC, Shelke NB, Rokhade AP. Formulation and in-vitro evaluation of novel starch-based tableted microspheres for controlled release of ampicillin. Carbohydr Polym. 2008;71(1):42–53. doi:10.1016/j.carbpol.2007.05.013
47. Al-Hashimi N, Begg N, Alany R. Oral modified release multiple-unit particulate systems: compressed pellets, microparticles and nanoparticles. Pharm. 2018;10(4):176.
48. Maghsoodi M. Physicomechanical properties of naproxen-loaded microparticles prepared from Eudragit L100. AAPS Pharm Sci Tech. 2009;10(1):120. doi:10.1208/s12249-009-9186-5
49. Pinto JF. Site-specific drug delivery systems within the gastro-intestinal tract: from the mouth to the colon. Int J Pharm. 2010;395(1–2):44–52. doi:10.1016/j.ijpharm.2010.05.003
50. Qiu Y, Zhou D. Understanding design and development of modified release solid oral dosage forms. J Validation Technol. 2011;17(2):23.
51. Sangalli M, Maroni A, Foppoli A. Different HPMC viscosity grades as coating agents for an oral time and/or site-controlled delivery system: a study on process parameters and in vitro performances. Eur J Pharm Sci. 2004;22(5):469–476. doi:10.1016/j.ejps.2004.05.002
52. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Rele. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
53. Peppas N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helvetiae. 1985;60(4):110–111.
54. Shah S, Madan S, Agrawal S. Formulation and evaluation of microsphere based oro dispersible tablets of itopride hcl. DARU J Pharm Sci. 2012;20(1):24. doi:10.1186/2008-2231-20-24
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.