Back to Journals » Neuropsychiatric Disease and Treatment » Volume 19
Endo-Lysosomal and Autophagy Pathway and Ubiquitin-Proteasome System in Mood Disorders: A Review Article
Authors Matutino Santos P ![]() , Pereira Campos G
, Pereira Campos G ![]() , Nascimento C
, Nascimento C ![]()
Received 9 August 2022
Accepted for publication 8 December 2022
Published 14 January 2023 Volume 2023:19 Pages 133—151
DOI https://doi.org/10.2147/NDT.S376380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Petala Matutino Santos,1 Giovanna Pereira Campos,1 Camila Nascimento2
1Center for Mathematics, Computing and Cognition (CMCC), Federal University of ABC (UFABC), São Paulo, Brazil; 2Department of Psychiatry, University of São Paulo Medical School, São Paulo, Brazil
Correspondence: Camila Nascimento, Department of Psychiatry, University of São Paulo Medical School, 785, Doutor Ovídio Pires de Campos Street, São Paulo, SP, Brazil, Tel +55 11 2661 7928, Email [email protected]
Abstract: Mood disorders are disabling conditions that cause significant functional impairment. Due to the clinical heterogeneity and complex nature of these disorders, diagnostic and treatment strategies face challenges. The etiology of mood disorders is multifactorial, involving genetic and environmental aspects that are associated with specific biological pathways including inflammation, oxidative stress, and neuroprotection. Alterations in these pathways may reduce the cell’s ability to recover from stress conditions occurring during mood episodes. The endo-lysosomal and autophagy pathway (ELAP) and the ubiquitin-proteasome system (UPS) play critical roles in protein homeostasis, impacting neuroplasticity and neurodevelopment. Thus, emerging evidence has suggested a role for these pathways in mental disorders. In the case of neurodegenerative diseases (NDDs), a deeper understanding in the role of ELAP and UPS has been critical to discover new treatment targets. Since it is suggested that NDDs and mood disorders share clinical symptomatology and risk factors, it has been hypothesized that there might be common underlying molecular pathways. Here, we review the importance of the ELAP and UPS for the central nervous system and for mood disorders. Finally, we discuss potential translational strategies for the diagnosis and treatment of major depressive disorder and bipolar disorder associated with these pathways.
Keywords: mood disorders, endo-lysosomal pathway, ubiquitin-proteasome system, autophagy, endo-lysosome, neuroplasticity, novel treatments, treatment target, translational science
Introduction
The term mood disorders is used to describe psychiatric conditions presenting with changes in emotional states. Bipolar disorder (BD) and major depression disorder (MDD) are among the most studied mood disorders that cause significant functional impairment.1,2 Although the etiology of mood disorders has not been completely elucidated, it has been proposed that both environmental and genetic factors may underlie their pathophysiology.1,2 Regarding the contribution of genetic factors, the role of multiple genes showing small size effects is the most acceptable model. For the environmental conditions, several risk factors have been associated with increased risk of developing MDD and BD including, early adverse stress, psychological stressors, and drug abuse.3–5 The combination of environmental and genetic factors may lead to intermediate phenotypes, such as altered brain gene expression and circuitry dysfunction, that affect mood disturbances.6 These alterations may be associated with changes in different cellular pathways that culminate in inflammation,7,8 decreased neuroplasticity,9 increased oxidative stress,10 and cell death.11 The endo-lysosomal and autophagy pathway (ELAP) and the ubiquitin-proteasome system (UPS) play critical roles in maintaining cellular homeostasis by controlling the quality and trafficking of the proteins. Since protein quality is crucial for the pathways involved in mood disorders, recent discoveries have shed light on the role of ELAP and UPS in these disorders.
The presence of misfolded proteins in specific brain regions are pathological hallmarks of neurodegenerative diseases (NDDs) including Alzheimer’s disease (AD).12 For this reason, the role of ELAP and UPS in NDDs is well recognized13,14 and has offered a starting point for new possibilities for the treatment of these diseases.15 Clinically, NDDs patients show psychiatric and behavioral alterations in addition to cognitive impairment.16 Furthermore, a recent follow-up study showed that having a mental disorder (especially MDD and BD) can increase the risk of developing dementia later in life.17 Shared clinical symptoms and the fact that mood disorders may increase the risk for NDDs suggest that common molecular mechanisms may underlie mental disorders.16 Although the contribution of specific molecular pathways has been addressed, a subset of MDD patients does not respond to current pharmacological treatments available.1 Furthermore, the diagnosis and management of MDD and BD suggest that patients may benefit from new treatment strategies. A better understanding of the molecular pathways involved in mood disorders can bring insights into novel translational strategies for their diagnostic and treatment. Here, we review the importance of ELAP and UPS to the brain and the findings on their role in MDD and BD. Ultimately, we discuss potential translational strategies for mood disorders involving these cellular pathways.
Endo-Lysosomal and Autophagy Pathway in Mood Disorders
The ELAP is a network for protein trafficking and degradation (Figure 1). Its goal is homeostasis maintenance, and this is especially important in neurons.13,18 This system is composed of mitochondria, lysosomes, endosomes, and exosomes. Several studies have found that failure in one of these components is associated with mental illness due to aberrant protein sorting. In the following topics, we detail the findings related to the ELAP in mood disorders for each of its components.
 |
Figure 1 The endo-lysosomal and autophagy pathway. The process of endocytosis is regulated by a Rab GTPase called rab5, which helps with protein internalization and endosome maturation, trafficking, and signaling. Once the early endosome is formed, the process of maturation continues, the rab5-to-rab7 conversion underlies the cargo transition from early to late endosomes/ MVBs. This process is also mediated by mitochondria, which send MDVs for the formation of MVBs. The endocytic pathway has two main routes: either the MVBs can fusion with autophagosomes, together with lysosomes leading to the autophagic process; or merge to the plasma membrane and release their vesicles into the extracellular space, which will later be called exosomes. Abbreviations: MVBs, Multivesicular bodies; MDVs, Mitochondria-derived vesicles. |
Mitochondria and Mitophagy
It is well known that mitochondria are responsible for maintaining cells’ integrity and functioning through ATP production.19 Additionally, mitochondria play a role in the quality control of cellular molecules. Under stress conditions, the first response of this organelle is the formation of vesicles containing mitochondrial DNA and proteins that are targeted to the endo-lysosomal system.20 This reaction is an alternative stress response, functioning as a quality control mechanism. Furthermore, stress conditions can lead to mitochondrial dysfunction, which has been related to lysosomal impairment,21 inflammatory responses,22 and accumulation of autophagy substrates.23 The following response under persistent stress conditions is the clearance of the damaged mitochondria (mitophagy).24,25 Mitophagy failure is detrimental to neurons and brain function.26 Its dysfunction can disturb mitochondrial functioning leading to the accumulation of defective organelles.27
Because of the sensitivity of the mitochondria to stress conditions, mitochondrial impairment has been largely studied in mood disorders.28,29 Recent studies have hypothesized the involvement of the mitochondria in the progression and severity of MDD and BD.30 A study focusing on the dorsolateral prefrontal cortex (dlPFC) from MDD patients found differentially expressed mitochondrial genes (Table 1) and several of these genes participate in regulatory processes of the nervous system.31 In BD, mitochondrial alterations were associated with calcium signaling, a decrease in energy production, production of reactive oxygen species (ROS), and impairment of mitochondrial dynamics.32–34 Mitochondria-related changes have also been found in the peripheral tissue of BD and MDD patients. Copy number variations in mitochondrial DNA are observed in patients with BD35,36 and MDD.37 Additionally, a study found changes in the levels of mitochondria proteins associated with the severity of MDD including MFN2, FIS-1, LC3B, Pink, and parkin.38 A question remains whether these findings could be used to predict new episodes or function as biomarkers to measure disease severity.
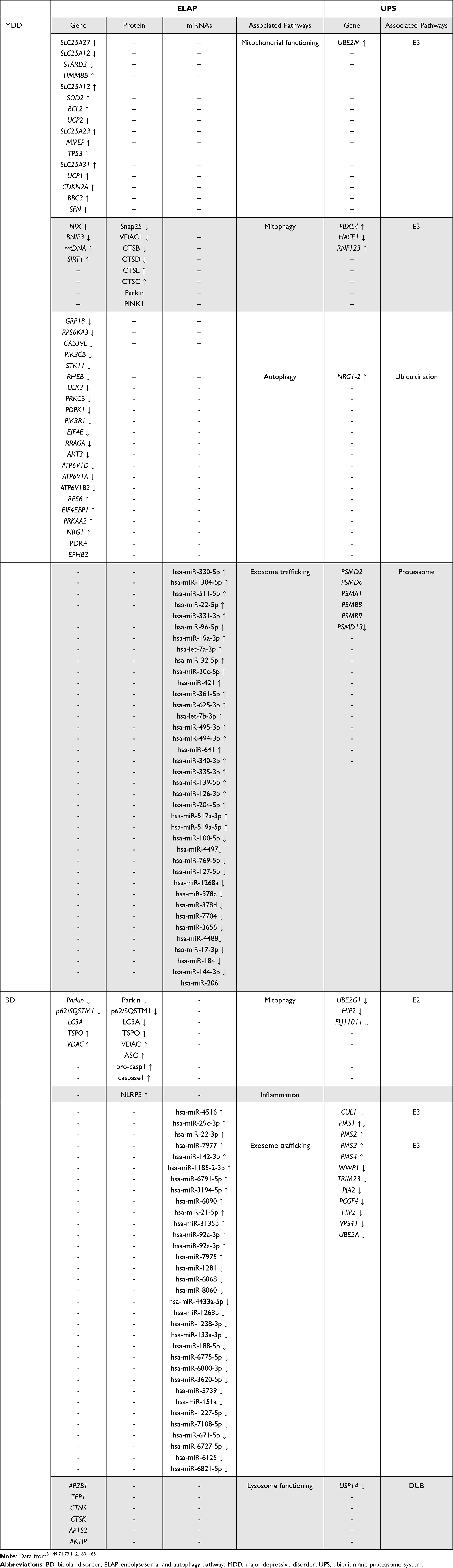 |
Table 1 Molecules Involved on the Endolysosomal and Autophagy Pathway and the Ubiquitin and Proteasome System in Mood Disorders |
Another way to address mitochondrial changes in the nervous system is using induced pluripotent cells (iPSCs) differentiated into neural cells. This approach gives us the opportunity of understanding the mechanisms that may be occurring in the brain of psychiatric patients. In BD, studies using iPSCs suggests alterations in mitochondria-related calcium signaling and alterations in mitochondrial size, function, and overexpression of mitochondrial-genes.39,40
Regarding mitophagy, one study showed that damaged mitochondria located in axons can trigger this pathway within tens of minutes.41 This suggests that mitophagy dysregulation is associated with synaptic failure and consequently the development/progression of mental disorders.24,42 In BD patients, findings show an upregulation of gene expression and protein levels of TSPO and VDAC (Table 1).43 Interestingly, higher levels of TSPO reduce mitophagy.43 These proteins, in addition to Pink1 and Beclin1, were decreased in a MDD rat model.44 Interestingly, while levels of Pink1 were elevated, parkin levels were lower in the peripheral blood of MDD subjects.38 It is possible that mitophagy is dysregulated in mood disorders, which may lead to mitochondrial accumulation and damage.
Autophagy
Whereas mitophagy is a clearance pathway for mitochondria, the degradation of the whole cell is called autophagy. Autophagy is a lysosome-dependent cell death system that degrades and recycles damaged organelles and unfolded proteins.45 Interestingly, treatment with silibinin inhibits autophagy and protects AD animal models from exhibiting anxiety/depression-like behaviors.46
Recent findings have suggested a role of autophagy in MDD pathophysiology and treatment.47,48 Analysis of gene expression data from different databases showed an association of autophagy-related genes with MDD (Table 1).49 Furthermore, these genes were associated with the immune microenvironment in MDD, which is a well-known pathway related to this disorder.50 Additionally, glucocorticoids can negatively regulate autophagy pathway.48
In BD, alterations of the signaling pathway mammalian target of rapamycin (mTOR) have been addressed.51,52 A transcriptome study of the prefrontal cortex (PFC) showed that genes related to the mechanistic target of the mTOR pathway were downregulated in patients while genes interrelated with the mTOR were upregulated (Table 1).53 Additionally, gene co-expression network analyses showed an enrichment of mTOR signaling and mTOR-related signaling pathways in endothelial and neuronal cells.53 This suggests the participation of different cell types in mTOR pathways in the PFC of BD subjects. Additional findings show the decreased activity of mTOR through the reduction of ULK1 kinase protein phosphorylation, which caused increased autophagy in cortical neurons of the PFC in BD patients.54
A recent review study presented several findings showing autophagic alterations in mood disorders.55 In animal models, increased expression of the mTOR pathway and decrease of ULK1 protein causes depressive, anhedonia, and anxiety-like behaviors. Moreover, the mTOR is downregulated in both the peripheral blood of depressive BD patients and the PFC of MDD patients. Taken together, these findings suggest that the mTOR pathway could be playing a role in the pathophysiology of mood disorders by regulating the autophagy of cortical neurons.
Endosomes and Exosomes
Endosomes are sorting compartments that transport proteins throughout the trans-Golgi network.56 Its functions include recycling and reusing proteins, as well as delivering them to lysosomes for hydrolysis.56,57 Endosomes are the major vesicles/organelles of the endocytic pathway and are classified as early, recycling, and late endosomes. If the cargo is retained in early endosomes and matures into late endosomes the process is called multivesicular bodies (MVBs).58 When late endosomes fusions with plasma membranes, vesicles are released into the extracellular environment, these are called exosomes.59 Studies focusing on endosomes and exosomes in mood disorders are only beginning to emerge, and recent findings require more investigation. Most studies have analyzed either the membrane proteins or the cargo of these vesicles.
A GWAS study60 of MDD and AD patients found 40 genes that were associated with both disorders, nine of them had already been published as AD risk genes,61 and out of these nine genes, four are involved in endocytosis (BIN1, PICALM, SORL1, and PTK2B). This suggests different mental disorders may share the contribution of endocytic pathways in their pathophysiology. Since depressive symptoms can precede clinical manifestations of AD, understanding factors associated with molecules involved in endocytosis might even help in preventing AD later in life.
Cathepsin D is a protease involved in the ELAP as a hydrolase.62 A recent study conducted in mice model deficient to cathepsin D showed that the animals exhibited anxiety-like, depressive-like behaviors, and mixed manic/depressive-like behavior.63 This suggests that cathepsin D may be associated with affective symptoms and potentially be an underlying molecular mechanism of mood disorders.
Besides their role in signaling and trafficking of molecules through vesicles, exosomes are also important for quality control of toxic or damaged proteins, lipids, RNAs, and microRNAs (miRNAs),64 being crucial to cell homeostasis and communication.65,66 Interestingly, certain molecules belonging to the autophagic machinery were found to contribute to exosome biogenesis.67
Once it is challenging to investigate specific molecular alterations in the brains of patients in vivo, the study of brain-derived exosomes allows us to explore alterations in these organelles in the peripheral blood.68 This is because exosomes exhibit specific proteins in their membrane that end up functioning as markers of the tissue from which they originated. Additionally, exosomes are suggested to be the primary vehicle for miRNA transportation throughout the body.69 Interestingly, miRNA level alterations are frequently seen in mental disorders.70
A recent study focusing on brain-derived exosomes found that several of their miRNAs were differentially expressed in MDD (Table 1).71 The top differentially expressed exosomal miRNA was the miR-139-5p, which was also altered in brain-derived exosomes of MDD in another study.72 Interestingly, injection of brain-derived human exosomal miR-139-5p caused depressive-like behaviors in mice models for MDD, while exosomes from healthy patients had antidepressant-like effects.71 Regarding BD, analysis of plasma exosomes showed thirty-three differentially expressed exosomal miRNAs (Table 1).73 Among them was miR-29c-3p, a miRNA that had been previously associated with BD.74 All these findings suggest that peripheral blood and brain exosomes can be biomarkers of mood disorders.75 Brain-derived exosomes may better correspond to clinical characteristics, since exosomes can cross the blood-brain barrier, which could be reflecting changes in cell communication and targeted cell behavior occurring in the brain.
Lysosomes
Lysosomes are digestive organelles belonging to the endocytic degradation pathway responsible for finalizing the autophagic process. Lysosomes have also been implicated in mitochondrial stress and mitophagy. Together with mitochondrial stress, the increase of lysosomal biogenesis leads to an increase in mitophagy.21 There are few studies investigating the role of lysosomes in mood disorders. Since lysosomal storage diseases show psychiatric and behavioral manifestations seen in MDD and BD,76 studying the role of lysosomal pathway deserves better attention.
Transcriptome analysis of the anterior cingulate showed that among the differentially expressed genes in BD the most enriched pathway was the lysosomal system (genes shown in Table 1).77 Moreover, depressive-like behaviors are observed in mice models overexpressing acid sphingomyelinase (ASM), a glycoprotein present in lysosomes that act in the catalyzation of hydrolysis.78 Interestingly, antidepressants seem to downregulate the expression of ASM both in mice models and non-treated MDD patients.79 Cathepsins are also involved in mood disorders in the context of lysosomal degradation. Downregulation of cathepsins is frequently seen in BD while their upregulation is associated with depressive-like behavior.80 These findings suggest further investigation into the role of cathepsins in mood states.
Ubiquitin and Proteasome System Dysfunction in Mood Disorders
The endoplasmic reticulum (ER) is responsible for the production and folding of proteins, an essential mechanism for proteins to function correctly. However, the formation of poorly folded proteins occurs constitutively in the organism. About 30% of the synthesized proteins do not achieve the appropriate final conformation.81 Therefore, the protein quality control machinery is present in the ER, comprising the Unfolded Protein Response (UPR), ER-associated degradation (ERAD), and the ubiquitin-proteasome system (UPS)82,83 (Figure 2).
 |
Figure 2 Protein quality control systems are activated upon the occurrence of unfolded proteins. The UPR acts in three ways: (A) increasing the transcription of chaperones; (B) inhibiting translation; (C) positively regulating ERAD components. Abbreviations: ERAD, ER-associated degradation; UPR, Unfolded protein response; UPS, Ubiquitin–proteasome system; aUb, Ubiquitin activated; Ub, Ubiquitin. |
Unfolded Protein Response – UPR
The UPR is activated when there is an insufficient capacity to fold proteins, and it acts by increasing the transcription of chaperones (Figure 2A), inhibiting translation (Figure 2B), and positively regulating ERAD components (Figure 2C).84 Protein synthesis and folding processes depend on appropriate environmental, genetic, and metabolic conditions. When there is a failure in restoring homeostasis, chronic UPR activation occurs, which can induce an apoptotic response.
Oxygen consumption in the brain is high and consequently, ROS production and oxidative stress are increased when compared to other organs. Oxidative stress is one of the factors responsible for protein malformation and requires the activation of UPR.85 Interestingly, evidence has suggested that UPR dysfunction may be involved in MDD, and BD.86 Several studies showed the implication of UPR in mood disorders (Figure 3).
 |
Figure 3 UPR’s mechanisms of action. In stress situations, PERK and IRE1α proteins dissociate from the GRP78 chaperone, autophosphorylates, form dimers, and phosphorylate other proteins. PERK phosphorylates eIF2-α, causing attenuation of protein translation. When the levels of eIF2-α are limited, ATF4 mRNA is translated and up-regulates CHOP, which responds to induce apoptosis. IRE1α activates XBP1 mRNA splicing, thus inducing the expression of chaperones. Abbreviations: PERK, protein kinase-like endoplasmic reticulum kinase; IRE1α, inositol requiring enzyme 1α; GRP78, glucose-regulated protein 78; eIF2-α, eukaryotic initiation factor 2 alpha; eIF2α-P, phosphorylation of eukaryotic initiation factor 2 alpha; XBP1, x-box binding protein 1; XBP1s, spliced form of XBP1; CHOP, C/EBP homologous protein; ATF4, activating transcription factor 4. Note: Flores-Santibáñez F, Medel B, Bernales JI, Osorio F. Understanding the Role of the Unfolded Protein Response Sensor IRE1 in the Biology of Antigen Presenting Cells. Cells. 2019 Dec 4;8(12):1563. doi: 10.3390/cells8121563.166 http://creativecommons.org/licenses/by/4.0/. |
Lymphocytes from individuals with BD subjected to stress did not show variation in the levels of GRP78 protein nor phosphorylation of eIF2α initiation factor or CHOP protein, which suggests that they present inherent a priori dysfunction in stress-response.87 Furthermore, lymphocytes in the BD group demonstrated significant cell death suggesting that UPR-induced apoptosis may reestablish homeostasis. Interestingly, when patients were divided considering disease chronicity, only patients in early stages submitted to prolonged stress and with advanced-stage BD did not present UPR modulation, showing that UPR dysfunction may be associated with decreased resilience and, consequently, the progression of the disease.87 Genetic studies corroborate the hypothesis that UPR is involved in the pathophysiology of BD since an association was observed between this disorder and polymorphisms in the promoter regions of the genes GRP7888 and XBP1.89 Furthermore, the expression of XBP1 was significantly decreased in the peripheral blood of BD.90
In MDD, mRNA levels of GRP78, GRP79, and ATF4C transcription factors were elevated in individuals with the disorder who died from suicide.91 This suggests that UPR mediation may be a critical factor for suicide among subjects with MDD.91 Taken together, we hypothesize that UPR alterations could be a molecular mediator of stress response caused by environmental modifications, often experienced by mood disorder patients.
Ubiquitin–Proteasome System – UPS
The UPS is the main pathway of protein degradation in eukaryotes. Through this pathway, ubiquitin marks misfolded or damaged proteins in a process named ubiquitination.92 This process is performed by ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3)93 (Figure 4) and serves as a signal for degradation. Ubiquitinated proteins will be targeted to the proteasome for proteolysis. The ubiquitination process is reversible and regulated by deubiquitinating enzymes (DUB), which remove ubiquitin from the substrate before it is degraded (Figure 4).
 |
Figure 4 The ubiquitin-proteasome pathway. The ubiquitin (Ub) binds to E1, and it is activated (aUb). Then it is transferred to E2. The complex (E2 + aUb) binds to E3 and then aUb connects to the target protein. Other monomers bind to aUb, forming a polyubiquitinated chain, which serves as a signal for degradation. If the linkage involves lysine-63, degradation occurs through the lysosome, whereas if the linkage involves lysine-48, degradation is dependent on the 26S proteasome. The deubiquitination enzymes (DUBs) remove the substrate from the protein, before being degraded. |
UPS is involved in the development and neuronal function and in cell signaling during stress, inflammation, and apoptosis.94,95 For this reason, many studies have associated UPS and mood disorders.96,97
The E3 play a critical role in the selectivity of substrates, which acts in maintenance of synaptic plasticity and transmission, remodeling of the dendritic column, as well as in the regulation of neurotransmitters receptors.98–100 In an animal study, both mRNA and protein levels of the E3 Nedd41 were increased on the medial PFC (mPFC) of mice susceptible to stress as well as the ubiquitination of the neurotrophin neuregulin (NRG1).101 This suggests that NRG1 is ubiquitinated by Nedd41 and that this ubiquitination regulates its levels. These findings are relevant in the context of mood disorders since NRG1 regulates processes of neuronal development, such as synaptic plasticity.102 Brain-derived neurotrophic factor (BDNF), which is another neurotrophin involved in MDD,103 has its expression mediated by an E3 substrate (Pdcd4). Exposure to chronic restriction stress decreases the ubiquitination of Pdcd4, resulting on its overexpression causing a decrease in BDNF levels in the hippocampus of mice.104 Despite the well-known changes in BDNF expression in MDD, its regulation by UPS is a new perspective.
These findings suggest that environmental stressors could promote alterations in the functioning of the UPS pathway, which could result in dysfunctional behavioral phenotypes. Aberrant regulation of neuronal plasticity may result in structural changes that contribute to the pathophysiology of MDD.105 Neuroimaging studies have shown that depressed patients have a reduced volume of the mPFC and hippocampus106 and postmortem studies have revealed a significant decrease in the number of synapses in the PFC.107 It is known that exposure to stress leads to a decrease in neurotrophic factors, atrophy, and cell loss in the hippocampus.105 We hypothesize that changes in UPS components could be participating in stress-induced neurochemical and structural changes in mood disorders.
Different studies that sought to understand the antidepressant response and find biomarkers for clinical outcomes demonstrated that ubiquitin ligases are associated with therapeutic efficiency.108–110 The intergenic variant rs6916777, mapped to the RNF217 gene/protein (an E3), was associated with a better response to venlafaxine in a study of GWAS in depressive elderly patients.109 Another gene expression evaluation was performed in rats submitted to maternal separation and three proteins involved in ubiquitination (Herc6, Asb5, Rnf7) were associated with the antidepressant response.108 Investigation of peripheral blood mononuclear cell methylome from depressed patients undergoing electroconvulsive therapy (ECT) revealed that, out of five genes implicated in the ECT response, two (RNF175 and RNF213) encode ubiquitin ligases.110 Considering that a significant proportion of patients with MDD do not achieve remission with pharmacotherapy or ECT,111 the findings presented here are relevant, since they can help in the identification of new treatment approaches.
In addition to the important role of E3 in mood disorder, a meta-analysis of risk genes for MDD identified UBE2M (coding for an E2) as one of the genes with the most consistent differential expression (Table 1).112 In BD, studies have shown that E2 and E3 may be involved in the pathophysiology of the disorder.113,114 UBE3A and CUL1 genes (coding for E3) are negatively expressed in the frontal cortex of BD subjects.114 Valproic acid, frequently used as an antimanic agent in BD can induce differential expression of ANAPC4, CDC23, FBXO2, FBXO4, NEDD4, TRAF6, UBE2D1, UBE2E1, UBE2E3 and UBE2G2, genes encoding E2 and E3113 in human neuroblastoma cells.
Genes encoding proteasome subunits (PSMD2, PSMD6, PSMA1, PSMB8, and PSMB9) are differentially expressed in the umbilical cord of newborns of mothers exposed to MDD.115 A proteomic analysis of the PFC of rats found that chronic social isolation negatively regulated Psmb7, Psmb3, Psmb2, Psmb6, Psma6 and Psmb4, which are cytosolic proteasomal proteins.116 The authors suggested that these proteins have been proposed as potential markers of depression induced by environmental stressors. Interestingly, a GWAS study reported an upregulation of several proteasome-related genes in MDD as one of the most robust results.117 Taken together the results indicate the hypothesis that dysregulation of proteasomal subunits contributes to the expression of the depressive phenotype. Finally, the PSMD13 rs3817629 G allele (a subunit of the proteasome) was associated with an increased risk of developing treatment-resistant depression.118 Furthermore, expression of this genotype was associated with reduced transcription of PSMD13 in fibroblasts from MDD patients.118
The function of DUBs as ubiquitination antagonists is equally important. These specific enzymes, which regulate multiple cellular processes, have been implicated in MDD and BD.119,120 The USP46 gene (DUB encoding) was identified as responsible for depressive-like behavior in mice.119 The lack of expression of USP46 showed antidepressant effects.121 A study in humans demonstrated significant differences in USP46 single nucleotide polymorphisms between MDD patients and controls.122 Recently a study found that the CC genotype of USP46 rs346005 is related to a higher depressive temperament score in males,123 which are risk factors for suicide in mental disorders.124
One of the most recent large-scale GWAS studies included 41,917 confirmed ANK3 as the main locus of risk for BD.125 ANK3 encodes ankyrin-G, which acts in processes such as synaptogenesis, synaptic plasticity, action potential generation and transmission, and ion channel regulation.126–128 Recently, it was found that ankyrin-G is controlled by USP9X (a DUB) and that this process is essential for the development of the dendritic column.120 USP9X knockout mice show reduced levels of ankyrin-G, which are recovered 12 weeks after birth.120 However, these mice had a reduction in cortical spine density, which persisted into adulthood.120 This reveals that although protein levels are transiently affected, behavioral and clinical dysfunctions are persistent. To date, there are no studies investigating the functioning of USP9X in BD, despite the ANK3 gene variants being among the risk factors most strongly associated with this disorder. Whereas a high expression of the ANK3 isoform in BD129 and a significant reduction in the density of the dendritic column in the dlPFC of individuals with BD130 were observed, investigating the expression/function of USP9X in BD patients may provide relevant insights into the mechanisms underlying the disorder and possible alternative therapeutic approaches.
Targeting Endo-Lysosomal/Autophagy and Ubiquitin/Proteasome Pathways: Translating Basic Findings to Clinical Practice
Endo-Lysosomal and Autophagy Pathways
Synthetic biology is an emerging technology that can be applied for therapeutic purposes using gene circuits to change cellular behavior.131 There are several potential ways of using synthetic biology for therapeutic capabilities, one of them is building models to sense the expression of endogenous miRNAs as biomarkers.132,133 Additionally, miRNAs can also be used as inputs to gene circuits triggering changes in cellular responses. miRNA has been used as biomarkers in synthetic circuitry to induce apoptosis in cancer cells.134,135 Since alterations in the expression of specific miRNAs are observed in BD (miR-29c-3p) and MDD (miR-139-5p), this strategy could be used to regulate cellular behavior in mood disorders.71,73 Given that miRNAs are present in exosomes of BD and MDD, validating these molecules as biomarkers for these disorders could be a starting point for the use of synthetic biology in mood disorders. Once robust findings on miRNAs as biomarkers of mood disorders have been established, these molecules could be used to activate gene circuitry (ie, TSPO) to promote specific cellular responses (ie, mitophagy) in BD, for example. This idea could be applied to other target molecules within the ELAP or UPS (for example the ones cited in Table 1). Preclinical strategies, such as the use of synthetic biology in IPSC of mood disorder patients may bring insights to further clinical applications.
Recently, targeted degradation has emerged as a promising therapeutic strategy.136 Lysosomal-targeting chimeras (LYTACS) and autophagy-targeting chimeras (AUTACS) use targeted degradation and explore ELAP and autophagic degradation pathways.136 AUTACS are heterobifunctional compounds, which contain guanine derivatives, which function as a tag for autophagic degradation, connected to a specific ligand of an intracellular target.137 On the other hand, LYTACS are intended for membrane-bound and extracellular proteins. In this sense, LYTACS are composed of two domains, one of which is an extracellular/membrane-bound protein binding element and the other a chemically synthesized glycopeptide whose function is to transport this target substrate to the lysosome.138
In addition to targeting proteins to autophagy, AUTACs have also been shown to target mitochondria to mitophagy. Mitochondrial dysfunctions have been associated with the severity and progression of BD and MDD,24,42 which causes cellular toxicity, and vulnerability to future episodes.139 Therefore, ensuring the maintenance of cellular resilience is a strategy to increase neuroprotection. In this context, treatment with mitochondria-targeted AUTACs (mito-AUTAC) may bring a possibility of novel adjunctive therapies for maintaining cellular resilience in mood disorders. In Down´s syndrome, a cell culture study showed that treatment with mito-AUTAC accelerated the removal of dysfunctional fragmented mitochondria, as well as the biogenesis of fully functioning mitochondria.137
Ubiquitin Proteasome System
Stimulation of neuronal plasticity via the UPS could also be a way of promoting neuroprotection in mood disorders as an adjunctive therapy. Neuronal plasticity allows for the readaptation of brain networks under stress conditions.140 BDNF-mediated plasticity is one of the mechanisms responsible for the clinical effects of antidepressants.140 Modulating BDNF levels by increasing the ubiquitination of Pdcd4 may be one considerable strategy. Studies have shown that other types of E3 ligase, such as TRIM27141 and βTRCP142,143 can regulate Pdcd4 expression by inducing its degradation via UPS. Therefore, studies investigating the specificity of these E3 and whether their expression in the hippocampus can promote antidepressant effects are necessary.
Stimulation of plasticity through UPS could also be mediated by the BDNF receptor TRKB. Recent studies have found that antidepressants bind directly to TRKB facilitating its location on the synaptic surface and signaling.144 In addition to being phosphorylated, TRKB can also be ubiquitinated and deubiquitination altering its biological functions.145,146 Recently it was shown that USP8 (a DUB) binds to the TRKB receptor and upregulates its levels and activation.146 Post-mortem studies have demonstrated the decreased expression of TRKB in different brain areas of suicidal individuals.147,148 Therefore, regulating the expression and activation of TRKB through DUBs could be a promising treatment strategy, and studies evaluating the feasibility of such an application are required.
Besides the reduced neuroprotection that underlies the pathophysiology of mood disorders, inflammation is also a biological substrate of these disorders.149 Lithium, the drug most recommended for the treatment of BD, is a GSK-3β inhibitor.150 GSK-3β promotes the production of inflammatory molecules, which can cause neuronal loss.151 The effectiveness of lithium for the treatment of BD is fully recognized.152 Likewise, potential side effects and risks associated with lithium long-term use are also well known,152 but often overlooked. Studies that investigate new therapies aiming not only for a better understanding of the efficacy but also the tolerability of the pharmacological treatments are necessary. In this context, proteolysis targeting chimeras (PROTACs) emerge as a promising alternative. PROTACs are a new technology that utilizes UPS to achieve target protein degradation.153 These are micromolecules, hetero-bifunctional, composed of two interconnected active domains. One of the domains binds to the target protein and the other to the E3 ligase. In this way, PROTACs bring the ubiquitination machinery closer to the target protein and initiate the degradation cascade, finally directing the proteins to the proteasome for proteolysis.153 Recently, the first PROTAC capable of degrading the GSK-3β protein (PG21) has been developed.150 Its synthesis was based on an E3 ubiquitin, ligase cereblon.150 Considering that lithium and PG21 exert the same activity (inhibit GSK-3β), we suggest that this PROTAC could be an alternative drug for BD therapy. In addition, PROTACS have high specificity, which favors the treatment of chronic diseases such as BD, since it makes it possible to reduce the frequency of administration.153 Therefore, studies aiming to better understand the potential therapeutic effects of PG21 in mood disorders (especially BD) are needed.
On the other hand, diseases can also be caused by excessive protein ubiquitination and degradation.154 In this sense, the use of deubiquitinase targeting chimeras (DUBTACS) could be an alternative. DUBTACS are also heterobifunctional molecules composed of two interconnected domains, but one of the domains corresponds to a DUB recruiter and the other to a ligand directed to target proteins.154 In this way, a DUBTAC brings a DUB closer to a target protein, which results in the removal of ubiquitin chains, that is, in deubiquitination, which rescues the protein from degradation, leading to an increase in protein levels and stability.154
Currently, several PROTACs have been used in clinical trials.155 In these trials, two oral PROTACs (ARV-110 and ARV-471) were shown to be effective in the treatment of prostate and breast cancer.155 There are still no clinical trials involving DUBTACs. This technology is very recent, the first DUBTAC was developed in 2022.154 DUBTACs have been suggested to stabilize tumor suppressors, which are ubiquitinated and degraded in disease, maintaining cancer cell proliferation.154 In summary, both PROTACs and DUBTACs have cancer-oriented applications that extensively foster research and development in academia and pharmaceuticals. Given the fact that several studies have shown the impact of UPS in mood disorders, these new technologies (PROTACs and DUBTACs) can be applied to BD and MDD as well. In the case of DUBTACs, they could maintain cellular resilience by inducing apoptosis of dysfunctional mitochondria, through the stabilization of BAX levels (pro-apoptotic).156 Furthermore, this technology could be considered to recruit USP8 to stabilize TRKB, which is decreased in MDD patients.147
Conclusions and Future Directions
We reviewed the findings on the role of different molecules belonging to the ELAP and UPS in mood disorders. Current findings on this field suggest further investigation on the role of TSPO in regulating mitophagy in BD and mTOR in autophagy in BD and MDD. Concerning the UPS attention should be given to the potential of E3 and DUBs in mediated neuroprotection in both BD and MDD. The fact that most of the molecular alterations in these pathways were observed in the frontal cortex and that this brain region is involved in the pathophysiology of BD and MDD suggests that these pathways may be underlying the brain circuitry dysfunction seen in these disorders.157,158
Based on the literature review, we sought to bring into discussion potential translational strategies that could improve the diagnosis and treatment of mood disorders. Further characterizing miRNA contained in the brain-derived exosomes may inform more robust biomarkers with clinical meaning. Additionally, a better characterization of the miRNAs related to BD and MDD may open the possibility of applying synthetic biology to translational purposes in mood disorders. Once synthetic biology acts on specific biomarkers and gene circuits to change cellular behavior, this technology could be applied in the context of personalized medicine in subsets of patients showing common clinical phenotypes in the future. Furthermore, AUTACs, mito-AUTACS, LYTACS, PROTACS, and DUBTACS achieve targeted and selective degradation of proteins, including targets previously considered “non-druggable”.159 These strategies could act on molecules underlying the ELAP and UPS found in BD and MDD and bring new therapeutic perspectives to these mood disorders.
Abbreviations
AD, Alzheimer’s disease; ASM, acid sphingomyelinase; AUTACS, autophagy-targeting chimeras; BD, bipolar disorder; BDNF, brain-derived neurotrophic factor; dlPFC, dorsolateral prefrontral cortex; DUB, deubiquitinating enzymes; DUBTACS, deubiquitinase-targeting chimeras; ELAP, endolysosomal and autophagy pathway; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ETC, electroconvulsive therapy; E1, ubiquitin-activating enzymes; E2, ubiquitin-conjugating enzymes; E3, ubiquitin ligases; GWAS, genome-wide association studies; iPSCs, induced pluripotent cells; LYTACS, lysosomal-targeting chimeras; MDD, major depressive disorder; mito-AUTAC, mitochondria-targeted AUTACs; mPFC, medial prefrontal cortex; miRNAs, microRNAs; mTOR, mammalian target of rapamycin; MVBs, multivesicular bodies; NDDs, neurodegenerative diseases; NRG1, neuregulin; PFC, prefrontal cortex; PROTACS, proteolysis-targeting chimeras; ROS, reactive oxygen species; SS, susceptible to stress; UPR, unfolded protein response; UPS, ubiquitin-proteasome system.
Acknowledgments
CN was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo [grant number, 2017/07089-8].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Otte C, Gold SM, Penninx BW, et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2:16065. doi:10.1038/nrdp.2016.65
2. Vieta E, Berk M, Schulze TG, et al. Bipolar disorders. Nat Rev Dis Primers. 2018;4:18008. doi:10.1038/nrdp.2018.8
3. Gloger S, Vohringer PA, Martinez P, et al. The contribution of early adverse stress to complex and severe depression in depressed outpatients. Depress Anxiety. 2021;38(4):431–438. doi:10.1002/da.23144
4. Rowland TA, Marwaha S. Epidemiology and risk factors for bipolar disorder. Ther Adv Psychopharmacol. 2018;8(9):251–269. doi:10.1177/2045125318769235
5. Shadrina M, Bondarenko EA, Slominsky PA. Genetics factors in major depression disease. Front Psychiatry. 2018;9:334. doi:10.3389/fpsyt.2018.00334
6. Gandal MJ, Zhang P, Hadjimichael E, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362(6420). doi:10.1126/science.aat8127
7. Muneer A. Bipolar disorder: role of inflammation and the development of disease biomarkers. Psychiatry Investig. 2016;13(1):18–33. doi:10.4306/pi.2016.13.1.18
8. Kim Y-K, Na K-S, Myint A-M, Leonard BE. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:277–284. doi:10.1016/j.pnpbp.2015.06.008
9. Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. 2018;98(3):466–481.
10. Kim Y, Vadodaria KC, Lenkei Z, et al. Mitochondria, metabolism, and redox mechanisms in psychiatric disorders. Antioxid Redox Signal. 2019;31(4):275–317. doi:10.1089/ars.2018.7606
11. Hroudova J, Fisar Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin Neurosci. 2011;65(2):130–141. doi:10.1111/j.1440-1819.2010.02178.x
12. Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035. doi:10.1101/cshperspect.a028035
13. Cao J, Zhong MB, Toro CA, Zhang L, Cai D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci Lett. 2019;703:68–78. doi:10.1016/j.neulet.2019.03.016
14. Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi:10.1016/j.pneurobio.2013.03.001
15. Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021;7(1):e12179.
16. Nascimento C, Nunes VP, Diehl Rodriguez R, et al. A review on shared clinical and molecular mechanisms between bipolar disorder and frontotemporal dementia. Prog Neuropsychopharmacol Biol Psychiatry. 2019;93:269–283. doi:10.1016/j.pnpbp.2019.04.008
17. Richmond-Rakerd LS, D’Souza S, Milne BJ, Caspi A, Moffitt TE. Longitudinal associations of mental disorders with dementia: 30-year analysis of 1.7 million New Zealand citizens. JAMA Psychiatry. 2022;79(4):333–340. doi:10.1001/jamapsychiatry.2021.4377
18. Whyte LS, Lau AA, Hemsley KM, Hopwood JJ, Sargeant TJ. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer’s disease? J Neurochem. 2017;140(5):703–717. doi:10.1111/jnc.13935
19. Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156(4):825–835. doi:10.1016/j.cell.2013.12.042
20. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33(4):282–295. doi:10.1002/embj.201385902
21. Raimundo N, Fernandez-Mosquera L, Yambire KF, Diogo CV. Mechanisms of communication between mitochondria and lysosomes. Int J Biochem Cell Biol. 2016;79:345–349. doi:10.1016/j.biocel.2016.08.020
22. Baixauli F, Acin-Perez R, Villarroya-Beltri C, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22(3):485–498. doi:10.1016/j.cmet.2015.07.020
23. Demers-Lamarche J, Guillebaud G, Tlili M, et al. Loss of mitochondrial function impairs lysosomes. J Biol Chem. 2016;291(19):10263–10276. doi:10.1074/jbc.M115.695825
24. Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and neuroprotection. Trends Mol Med. 2020;26(1):8–20. doi:10.1016/j.molmed.2019.07.002
25. Palikaras K, Tavernarakis N. Regulation and roles of mitophagy at synapses. Mech Ageing Dev. 2020;187:111216. doi:10.1016/j.mad.2020.111216
26. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–166. doi:10.1016/j.tins.2017.01.002
27. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. doi:10.1038/s41556-018-0176-2
28. Andreazza AC, Young LT. The neurobiology of bipolar disorder: identifying targets for specific agents and synergies for combination treatment. Int J Neuropsychopharmacol. 2014;17(7):1039–1052. doi:10.1017/S1461145713000096
29. Fattal O, Budur K, Vaughan AJ, Franco K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics. 2006;47(1):1–7. doi:10.1176/appi.psy.47.1.1
30. Gimenez-Palomo A, Dodd S, Anmella G, et al. The role of mitochondria in mood disorders: from physiology to pathophysiology and to treatment. Front Psychiatry. 2021;12:546801. doi:10.3389/fpsyt.2021.546801
31. Wang Q, Dwivedi Y. Transcriptional profiling of mitochondria associated genes in prefrontal cortex of subjects with major depressive disorder. World J Biol Psychiatry. 2017;18(8):592–603. doi:10.1080/15622975.2016.1197423
32. Cyrino LAR, Delwing-de lima D, Ullmann OM, Maia TP. Concepts of neuroinflammation and their relationship with impaired mitochondrial functions in bipolar disorder. Front Behav Neurosci. 2021;15:609487. doi:10.3389/fnbeh.2021.609487
33. Kuperberg M, Greenebaum SLA, Nierenberg AA. Targeting mitochondrial dysfunction for bipolar disorder. Curr Top Behav Neurosci. 2021;48:61–99.
34. Scaini G, Andrews T, Lima CNC, Benevenuto D, Streck EL, Quevedo J. Mitochondrial dysfunction as a critical event in the pathophysiology of bipolar disorder. Mitochondrion. 2021;57:23–36. doi:10.1016/j.mito.2020.12.002
35. Scaini G, Fries GR, Valvassori SS, et al. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl Psychiatry. 2017;7(5):e1111. doi:10.1038/tp.2017.83
36. Yamaki N, Otsuka I, Numata S, et al. Mitochondrial DNA copy number of peripheral blood in bipolar disorder: the present study and a meta-analysis. Psychiatry Res. 2018;269:115–117. doi:10.1016/j.psychres.2018.08.014
37. Chung JK, Lee SY, Park M, Joo EJ, Kim SA. Investigation of mitochondrial DNA copy number in patients with major depressive disorder. Psychiatry Res. 2019;282:112616. doi:10.1016/j.psychres.2019.112616
38. Scaini G, Mason BL, Diaz AP, et al. Dysregulation of mitochondrial dynamics, mitophagy and apoptosis in major depressive disorder: does inflammation play a role? Mol Psychiatry. 2022;27(2):1095–1102. doi:10.1038/s41380-021-01312-w
39. Hahn CG, Gomez G, Restrepo D, et al. Aberrant intracellular calcium signaling in olfactory neurons from patients with bipolar disorder. Am J Psychiatry. 2005;162(3):616–618. doi:10.1176/appi.ajp.162.3.616
40. Mertens J, Wang QW, Kim Y, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527(7576):95–99. doi:10.1038/nature15526
41. Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and parkin. J Cell Biol. 2014;206(5):655–670. doi:10.1083/jcb.201401070
42. Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiol Dis. 2019;122:35–40. doi:10.1016/j.nbd.2018.07.005
43. Scaini G, Barichello T, Fries GR, et al. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacology. 2019;44(7):1291–1299. doi:10.1038/s41386-018-0293-4
44. Li D, Zheng J, Wang M, et al. Changes of TSPO-mediated mitophagy signaling pathway in learned helplessness mice. Psychiatry Res. 2016;245:141–147. doi:10.1016/j.psychres.2016.02.068
45. Uddin MS, Stachowiak A, Mamun AA, et al. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:4. doi:10.3389/fnagi.2018.00004
46. Song X, Liu B, Cui L, et al. Silibinin ameliorates anxiety/depression-like behaviors in amyloid beta-treated rats by upregulating BDNF/TrkB pathway and attenuating autophagy in hippocampus. Physiol Behav. 2017;179:487–493. doi:10.1016/j.physbeh.2017.07.023
47. Jia J, Le W. Molecular network of neuronal autophagy in the pathophysiology and treatment of depression. Neurosci Bull. 2015;31(4):427–434. doi:10.1007/s12264-015-1548-2
48. Sato M, Ueda E, Konno A, et al. Glucocorticoids negatively regulates chaperone mediated autophagy and microautophagy. Biochem Biophys Res Commun. 2020;528(1):199–205. doi:10.1016/j.bbrc.2020.04.132
49. He S, Deng Z, Li Z, et al. Signatures of 4 autophagy-related genes as diagnostic markers of MDD and their correlation with immune infiltration. J Affect Disord. 2021;295:11–20. doi:10.1016/j.jad.2021.08.005
50. Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107(2):234–256. doi:10.1016/j.neuron.2020.06.002
51. Kim YM, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57(2):207–218. doi:10.1016/j.molcel.2014.11.013
52. Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. doi:10.3389/fnmol.2014.00028
53. Park SW, Seo MK, Webster MJ, Lee JG, Kim S. Differential expression of gene co-expression networks related to the mTOR signaling pathway in bipolar disorder. Transl Psychiatry. 2022;12(1):184. doi:10.1038/s41398-022-01944-8
54. Vanderplow AM, Eagle AL, Kermath BA, Bjornson KJ, Robison AJ, Cahill ME. Akt-mTOR hypoactivity in bipolar disorder gives rise to cognitive impairments associated with altered neuronal structure and function. Neuron. 2021;109(9):1479–1496 e1476. doi:10.1016/j.neuron.2021.03.008
55. Pierone BC, Pereira CA, Garcez ML, Kaster MP. Stress and signaling pathways regulating autophagy: from behavioral models to psychiatric disorders. Exp Neurol. 2020;334:113485. doi:10.1016/j.expneurol.2020.113485
56. Schreij AM, Fon EA, McPherson PS. Endocytic membrane trafficking and neurodegenerative disease. Cell Mol Life Sci. 2016;73(8):1529–1545. doi:10.1007/s00018-015-2105-x
57. Vagnozzi AN, Pratico D. Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol Psychiatry. 2019;24(6):857–868. doi:10.1038/s41380-018-0221-3
58. Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10. doi:10.1016/j.semcdb.2014.03.034
59. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. doi:10.1007/s00018-017-2595-9
60. Lutz MW, Sprague D, Barrera J, Chiba-Falek O. Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Transl Psychiatry. 2020;10(1):88. doi:10.1038/s41398-020-0769-y
61. Pimenova AA, Raj T, Goate AM. Untangling genetic risk for Alzheimer’s disease. Biol Psychiatry. 2018;83(4):300–310. doi:10.1016/j.biopsych.2017.05.014
62. Cataldo AM, Barnett JL, Berman SA, et al. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14(3):671–680. doi:10.1016/0896-6273(95)90324-0
63. Zhou R, Lu Y, Han Y, et al. Mice heterozygous for cathepsin D deficiency exhibit mania-related behavior and stress-induced depression. Prog Neuropsychopharmacol Biol Psychiatry. 2015;63:110–118. doi:10.1016/j.pnpbp.2015.06.007
64. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514. doi:10.1146/annurev-biochem-013118-111902
65. Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403. doi:10.3389/fimmu.2014.00403
66. Desdin-Mico G, Mittelbrunn M. Role of exosomes in the protection of cellular homeostasis. Cell Adh Migr. 2017;11(2):127–134. doi:10.1080/19336918.2016.1251000
67. Xu J, Camfield R, Gorski SM. The interplay between exosomes and autophagy - partners in crime. J Cell Sci. 2018;131(15). doi:10.1242/jcs.215210
68. Saeedi S, Israel S, Nagy C, Turecki G. The emerging role of exosomes in mental disorders. Transl Psychiatry. 2019;9(1):122. doi:10.1038/s41398-019-0459-9
69. Sohel MH. Extracellular/circulating MicroRNAs: release mechanisms, functions and challenges. Achieve Life Sci. 2016;10(2):175–186. doi:10.1016/j.als.2016.11.007
70. Gruzdev SK, Yakovlev AA, Druzhkova TA, Guekht AB, Gulyaeva NV. The missing link: how exosomes and miRNAs can help in bridging psychiatry and molecular biology in the context of depression, bipolar disorder and schizophrenia. Cell Mol Neurobiol. 2019;39(6):729–750. doi:10.1007/s10571-019-00684-6
71. Wei ZX, Xie GJ, Mao X, et al. Exosomes from patients with major depression cause depressive-like behaviors in mice with involvement of miR-139-5p-regulated neurogenesis. Neuropsychopharmacology. 2020;45(6):1050–1058. doi:10.1038/s41386-020-0622-2
72. Liang JQ, Liao HR, Xu CX, et al. Serum exosome-derived miR-139-5p as a potential biomarker for major depressive disorder. Neuropsychiatr Dis Treat. 2020;16:2689–2693. doi:10.2147/NDT.S277392
73. Fries GR, Lima CNC, Valvassori SS, Zunta-Soares G, Soares JC, Quevedo J. Preliminary investigation of peripheral extracellular vesicles’ microRNAs in bipolar disorder. J Affect Disord. 2019;255:10–14. doi:10.1016/j.jad.2019.05.020
74. Banigan MG, Kao PF, Kozubek JA, et al. Differential expression of exosomal microRNAs in prefrontal cortices of schizophrenia and bipolar disorder patients. PLoS One. 2013;8(1):e48814. doi:10.1371/journal.pone.0048814
75. Fries GR, Quevedo J. Exosomal MicroRNAs as potential biomarkers in neuropsychiatric disorders. Methods Mol Biol. 2018;1733:79–85.
76. Staretz-Chacham O, Choi JH, Wakabayashi K, Lopez G, Sidransky E. Psychiatric and behavioral manifestations of lysosomal storage disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(7):1253–1265. doi:10.1002/ajmg.b.31097
77. Zhao Z, Xu J, Chen J, et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry. 2015;20(5):563–572. doi:10.1038/mp.2014.82
78. Zoicas I, Reichel M, Gulbins E, Kornhuber J. Role of acid sphingomyelinase in the regulation of social behavior and memory. PLoS One. 2016;11(9):e0162498. doi:10.1371/journal.pone.0162498
79. Rhein C, Zoicas I, Marx LM, et al. mRNA expression of SMPD1 encoding acid sphingomyelinase decreases upon antidepressant treatment. Int J Mol Sci. 2021;22(11):5700. doi:10.3390/ijms22115700
80. Niemeyer C, Matosin N, Kaul D, Philipsen A, Gassen NC. The role of cathepsins in memory functions and the pathophysiology of psychiatric disorders. Front Psychiatry. 2020;11:718.
81. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404(6779):770–774.
82. Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23(4):464–475.
83. Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84(1):25–33.
84. Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology. 2007;22:193–201.
85. Andreazza AC, Kauer-Sant’anna M, Frey BN, et al. Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord. 2008;111(2–3):135–144.
86. Muneer A, Shamsher Khan RM. Endoplasmic reticulum stress: implications for neuropsychiatric disorders. Chonnam Med J. 2019;55(1):8–19.
87. Pfaffenseller B, Wollenhaupt-Aguiar B, Fries GR, et al. Impaired endoplasmic reticulum stress response in bipolar disorder: cellular evidence of illness progression. Int J Neuropsychopharmacol. 2014;17(9):1453–1463.
88. Kakiuchi C, Ishiwata M, Nanko S, et al. Functional polymorphisms of HSPA5: possible association with bipolar disorder. Biochem Biophys Res Commun. 2005;336(4):1136–1143.
89. Kakiuchi C, Iwamoto K, Ishiwata M, et al. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet. 2003;35(2):171–175.
90. Bengesser SA, Reininghaus EZ, Dalkner N, et al. Endoplasmic reticulum stress in bipolar disorder? - BiP and CHOP gene expression- and XBP1 splicing analysis in peripheral blood. Psychoneuroendocrinology. 2018;95:113–119.
91. Yoshino Y, Dwivedi Y. Elevated expression of unfolded protein response genes in the prefrontal cortex of depressed subjects: effect of suicide. J Affect Disord. 2020;262:229–236.
92. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479.
93. Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513.
94. Hegde AN, Upadhya SC. The ubiquitin-proteasome pathway in health and disease of the nervous system. Trends Neurosci. 2007;30(11):587–595.
95. Kawabe H, Brose N. The role of ubiquitylation in nerve cell development. Nat Rev Neurosci. 2011;12(5):251–268.
96. Cheon S, Dean M, Chahrour M. The ubiquitin proteasome pathway in neuropsychiatric disorders. Neurobiol Learn Mem. 2019;165:106791.
97. Spindola L, Santoro M, Pan P, et al. SU73 - GENE EXPRESSION IN BLOOD OF ADOLESCENTS WITH PSYCHIATRIC DISORDERS. Eur Neuropsychopharmacol. 2019;29:S927–S928.
98. Kawabe H, Stegmuller J. The role of E3 ubiquitin ligases in synapse function in the healthy and diseased brain. Mol Cell Neurosci. 2021;112:103602.
99. Lee S, Park S, Lee H, et al. Nedd4 E3 ligase and beta-arrestins regulate ubiquitination, trafficking, and stability of the mGlu7 receptor. Elife. 2019;8:1.
100. Ma P, Mao B. The many faces of the E3 ubiquitin ligase, RNF220, in neural development and beyond. Dev Growth Differ. 2022;64(2):98–105. doi:10.1111/dgd.12756
101. Xu J, Guo C, Liu Y, et al. Nedd4l downregulation of NRG1 in the mPFC induces depression-like behaviour in CSDS mice. Transl Psychiatry. 2020;10(1):249. doi:10.1038/s41398-020-00935-x
102. Levchenko A, Vyalova NM, Nurgaliev T, et al. NRG1, PIP4K2A, and HTR2C as potential candidate biomarker genes for several clinical subphenotypes of depression and bipolar disorder. Front Genet. 2020;11:936. doi:10.3389/fgene.2020.00936
103. Dwivedi Y. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat. 2009;5:433–449. doi:10.2147/NDT.S5700
104. Li Y, Jia Y, Wang D, et al. Programmed cell death 4 as an endogenous suppressor of BDNF translation is involved in stress-induced depression. Mol Psychiatry. 2021;26(6):2316–2333. doi:10.1038/s41380-020-0692-x
105. Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol. 2007;18(5–6):391–418. doi:10.1097/FBP.0b013e3282ee2aa8
106. Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33(5):699–771. doi:10.1016/j.neubiorev.2009.01.004
107. Kang HJ, Voleti B, Hajszan T, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18(9):1413–1417. doi:10.1038/nm.2886
108. Marchetti L, Lauria M, Caberlotto L, et al. Gene expression signature of antidepressant treatment response/non-response in Flinders Sensitive Line rats subjected to maternal separation. Eur Neuropsychopharmacol. 2020;31:69–85. doi:10.1016/j.euroneuro.2019.11.004
109. Marshe VS, Maciukiewicz M, Hauschild AC, et al. Genome-wide analysis suggests the importance of vascular processes and neuroinflammation in late-life antidepressant response. Transl Psychiatry. 2021;11(1):127. doi:10.1038/s41398-021-01248-3
110. Moschny N, Zindler T, Jahn K, et al. Novel candidate genes for ECT response prediction-A pilot study analyzing the DNA methylome of depressed patients receiving electroconvulsive therapy. Clin Epigenetics. 2020;12(1):114. doi:10.1186/s13148-020-00891-9
111. Brus O, Cao Y, Gustafsson E, et al. Self-assessed remission rates after electroconvulsive therapy of depressive disorders. Eur Psychiatry. 2017;45:154–160. doi:10.1016/j.eurpsy.2017.06.015
112. Wu W, Howard D, Sibille E, French L. Differential and spatial expression meta-analysis of genes identified in genome-wide association studies of depression. Transl Psychiatry. 2021;11(1):8. doi:10.1038/s41398-020-01127-3
113. Hu TM, Chung HS, Ping LY, et al. Differential expression of multiple disease-related protein groups induced by valproic acid in human SH-SY5Y neuroblastoma cells. Brain Sci. 2020;10(8). doi:10.3390/brainsci10080545
114. You X, Zhang Y, Long Q, et al. Does single gene expression omnibus data mining analysis apply for only tumors and not mental illness? A preliminary study on bipolar disorder based on bioinformatics methodology. Medicine. 2020;99(35):e21989. doi:10.1097/MD.0000000000021989
115. Liu W, Zhang L, Zheng D, Zhang Y. Umbilical cord blood-based gene signatures related to prenatal major depressive disorder. Medicine. 2019;98(28):e16373. doi:10.1097/MD.0000000000016373
116. Filipovic D, Novak B, Xiao J, Yan Y, Yeoh K, Turck CW. Chronic fluoxetine treatment of socially isolated rats modulates prefrontal cortex proteome. Neuroscience. 2022;501:52–71. doi:10.1016/j.neuroscience.2022.08.011
117. Belaish S, Israel-Elgali I, Shapira G, et al. Genome wide analysis implicates upregulation of proteasome pathway in major depressive disorder. Transl Psychiatry. 2021;11(1):409. doi:10.1038/s41398-021-01529-x
118. Minelli A, Magri C, Barbon A, et al. Proteasome system dysregulation and treatment resistance mechanisms in major depressive disorder. Transl Psychiatry. 2015;5(12):e687. doi:10.1038/tp.2015.180
119. Tomida S, Mamiya T, Sakamaki H, et al. Usp46 is a quantitative trait gene regulating mouse immobile behavior in the tail suspension and forced swimming tests. Nat Genet. 2009;41(6):688–695. doi:10.1038/ng.344
120. Yoon S, Parnell E, Kasherman M, et al. Usp9X controls ankyrin-repeat domain protein homeostasis during dendritic spine development. Neuron. 2020;105(3):506–521 e507. doi:10.1016/j.neuron.2019.11.003
121. Imai S, Mamiya T, Tsukada A, et al. Ubiquitin-specific peptidase 46 (Usp46) regulates mouse immobile behavior in the tail suspension test through the GABAergic system. PLoS One. 2012;7(6):e39084. doi:10.1371/journal.pone.0039084
122. Fukuo Y, Kishi T, Kushima I, et al. Possible association between ubiquitin-specific peptidase 46 gene and major depressive disorders in the Japanese population. J Affect Disord. 2011;133(1–2):150–157. doi:10.1016/j.jad.2011.04.020
123. Boo YJ, Park CI, Kim HW, Kim SJ, Kang JI. Possible association of the ubiquitin-specific peptidase 46 gene (USP46) with affective temperamental traits in healthy Korean volunteers. Psychiatry Investig. 2019;16(1):87–92. doi:10.30773/pi.2018.10.02
124. Karam EG, Itani L, Fayyad J, et al. Temperament and suicide: a national study. J Affect Disord. 2015;184:123–128. doi:10.1016/j.jad.2015.05.047
125. Mullins N, Forstner AJ, O’Connell KS, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53(6):817–829. doi:10.1038/s41588-021-00857-4
126. Jenkins PM, Kim N, Jones SL, et al. Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A. 2015;112(4):957–964. doi:10.1073/pnas.1416544112
127. Smith KR, Kopeikina KJ, Fawcett-Patel JM, et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron. 2014;84(2):399–415. doi:10.1016/j.neuron.2014.10.010
128. Tseng WC, Jenkins PM, Tanaka M, Mooney R, Bennett V. Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABAA receptors. Proc Natl Acad Sci U S A. 2015;112(4):1214–1219. doi:10.1073/pnas.1417989112
129. Hughes T, Sonderby IE, Polushina T, et al. Elevated expression of a minor isoform of ANK3 is a risk factor for bipolar disorder. Transl Psychiatry. 2018;8(1):210. doi:10.1038/s41398-018-0175-x
130. Konopaske GT, Lange N, Coyle JT, Benes FM. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71(12):1323–1331. doi:10.1001/jamapsychiatry.2014.1582
131. Cubillos-Ruiz A, Guo T, Sokolovska A, et al. Engineering living therapeutics with synthetic biology. Nat Rev Drug Discov. 2021;20(12):941–960. doi:10.1038/s41573-021-00285-3
132. Becker K, Klarner H, Nowicka M, Siebert H. Designing miRNA-based synthetic cell classifier circuits using answer set programming. Front Bioeng Biotechnol. 2018;6:70. doi:10.3389/fbioe.2018.00070
133. Matsuyama H, Suzuki HI. Systems and synthetic microRNA Biology: from biogenesis to disease pathogenesis. Int J Mol Sci. 2019;21(1). doi:10.3390/ijms21010132
134. Lin MW, Tseng YW, Shen CC, et al. Synthetic switch-based baculovirus for transgene expression control and selective killing of hepatocellular carcinoma cells. Nucleic Acids Res. 2018;46(15):e93. doi:10.1093/nar/gky447
135. Liu Y, Han Y, Zhang H, et al. Synthetic miRNA-mowers targeting miR-183-96-182 cluster or miR-210 inhibit growth and migration and induce apoptosis in bladder cancer cells. PLoS One. 2012;7(12):e52280. doi:10.1371/journal.pone.0052280
136. Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021;296:100647. doi:10.1016/j.jbc.2021.100647
137. Takahashi D, Moriyama J, Nakamura T, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019;76(5):797–810 e710. doi:10.1016/j.molcel.2019.09.009
138. Ramadas B, Kumar Pain P, Manna D. LYTACs: an emerging tool for the degradation of non-cytosolic proteins. ChemMedChem. 2021;16(19):2951–2953. doi:10.1002/cmdc.202100393
139. Post RM. Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev. 2007;31(6):858–873. doi:10.1016/j.neubiorev.2007.04.003
140. Schmidt HD, Duman RS. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. 2010;35(12):2378–2391. doi:10.1038/npp.2010.114
141. Yu H, Wan L, Tang Z, et al. TRIM27 regulates the expression of PDCD4 by the ubiquitin‑proteasome pathway in ovarian and endometrial cancer cells. Oncol Rep. 2022;48(1). doi:10.3892/or.2022.8331
142. Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314(5798):467–471. doi:10.1126/science.1130276
143. Matsuhashi S, Manirujjaman M, Hamajima H, Ozaki I. Control mechanisms of the tumor suppressor PDCD4: expression and functions. Int J Mol Sci. 2019;20(9):2304. doi:10.3390/ijms20092304
144. Casarotto PC, Girych M, Fred SM, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021;184(5):1299–1313 e1219. doi:10.1016/j.cell.2021.01.034
145. Guo YY, Lu Y, Zheng Y, et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) promotes hippocampus-dependent memory via its deubiquitinating effect on TrkB. J Neurosci. 2017;37(25):5978–5995. doi:10.1523/JNEUROSCI.3148-16.2017
146. Martin-Rodriguez C, Song M, Anta B, et al. TrkB deubiquitylation by USP8 regulates receptor levels and BDNF-dependent neuronal differentiation. J Cell Sci. 2020;133(24). doi:10.1242/jcs.247841
147. Erbay LG, Karlidag R, Oruc M, Cigremis Y, Celbis O. Association of BDNF / TrkB and NGF / TrkA levels in postmortem brain with major depression and suicide. Psychiatr Danub. 2021;33(4):491–498. doi:10.24869/psyd.2021.491
148. Pandya C, Kutiyanawalla A, Turecki G, Pillai A. Glucocorticoid regulates TrkB protein levels via c-Cbl dependent ubiquitination: a decrease in c-Cbl mRNA in the prefrontal cortex of suicide subjects. Psychoneuroendocrinology. 2014;45:108–118. doi:10.1016/j.psyneuen.2014.03.020
149. Troubat R, Barone P, Leman S, et al. Neuroinflammation and depression: a review. Eur J Neurosci. 2021;53(1):151–171. doi:10.1111/ejn.14720
150. Jiang X, Zhou J, Wang Y, et al. PROTACs suppression of GSK-3beta, a crucial kinase in neurodegenerative diseases. Eur J Med Chem. 2021;210:112949. doi:10.1016/j.ejmech.2020.112949
151. Sirerol-Piquer M, Gomez-Ramos P, Hernandez F, et al. GSK3beta overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus. 2011;21(8):910–922. doi:10.1002/hipo.20805
152. Albert U, De Cori D, Blengino G, Bogetto F, Maina G. Trattamento con litio e potenziali effetti collaterali a lungo termine: una revisione sistematica della letteratura [Lithium treatment and potential long-term side effects: a systematic review of the literature]. Riv Psichiatr. 2014;49(1):12–21. Italian. doi:10.1708/1407.15620
153. Wang C, Zhang Y, Wu Y, Xing D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur J Med Chem. 2021;225:113749. doi:10.1016/j.ejmech.2021.113749
154. Henning NJ, Boike L, Spradlin JN, et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat Chem Biol. 2022;18(4):412–421. doi:10.1038/s41589-022-00971-2
155. Xi JY, Zhang RY, Chen K, et al. Advances and perspectives of proteolysis targeting chimeras (PROTACs) in drug discovery. Bioorg Chem. 2022;125:105848. doi:10.1016/j.bioorg.2022.105848
156. Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012;8(7):639–645. doi:10.1038/nchembio.995
157. Phillips ML, Swartz HA. A critical appraisal of neuroimaging studies of bipolar disorder: toward a new conceptualization of underlying neural circuitry and a road map for future research. Am J Psychiatry. 2014;171(8):829–843. doi:10.1176/appi.ajp.2014.13081008
158. Pizzagalli DA, Roberts AC. Prefrontal cortex and depression. Neuropsychopharmacology. 2022;47(1):225–246. doi:10.1038/s41386-021-01101-7
159. Liu X, Ciulli A. DUB be good to me. Nat Chem Biol. 2022;18(4):358–359. doi:10.1038/s41589-022-00978-9
160. Ciuculete DM, Voisin S, Kular L, et al. meQTL and ncRNA functional analyses of 102 GWAS-SNPs associated with depression implicate HACE1 and SHANK2 genes. Clin Epigenetics. 2020;12(1):99. doi:10.1186/s13148-020-00884-8
161. Li X, Luo Z, Gu C, et al. Common variants on 6q16.2, 12q24.31 and 16p13.3 are associated with major depressive disorder. Neuropsychopharmacology. 2018;43(10):2146–2153. doi:10.1038/s41386-018-0078-9
162. Ryan MM, Lockstone HE, Huffaker SJ, Wayland MT, Webster MJ, Bahn S. Gene expression analysis of bipolar disorder reveals downregulation of the ubiquitin cycle and alterations in synaptic genes. Mol Psychiatry. 2006;11(10):965–978. doi:10.1038/sj.mp.4001875
163. Sayad A, Taheri M, Azari I, Oskoei VK, Ghafouri-Fard S. PIAS genes as disease markers in bipolar disorder. J Cell Biochem. 2019;120(8):12937–12942. doi:10.1002/jcb.28564
164. Teyssier JR, Rey R, Ragot S, Chauvet-Gelinier JC, Bonin B. Correlative gene expression pattern linking RNF123 to cellular stress-senescence genes in patients with depressive disorder: implication of DRD1 in the cerebral cortex. J Affect Disord. 2013;151(2):432–438. doi:10.1016/j.jad.2013.04.010
165. Tripathi A, Scaini G, Barichello T, Quevedo J, Pillai A. Mitophagy in depression: pathophysiology and treatment targets. Mitochondrion. 2021;61:1–10. doi:10.1016/j.mito.2021.08.016
166. FloresSantibáñez F, Medel B, Bernales JI, Osorio F. Understanding the Role of the Unfolded Protein Response Sensor IRE1 in the Biology of Antigen Presenting Cells. Cells. 2019;8(12):1563. doi: 10.3390/cells8121563.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.