Back to Journals » Journal of Experimental Pharmacology » Volume 18
Empagliflozin Improves Learning and Memory Deficits in Streptozotocin-Induced Hyperglycemic Male Wistar Rats
Authors Isawi IH ![]() , Al-Sawalha NA, Mahmoud F, Al-Khawaldeh I, Alzoubi KH
, Al-Sawalha NA, Mahmoud F, Al-Khawaldeh I, Alzoubi KH ![]()
Received 19 August 2025
Accepted for publication 29 January 2026
Published 11 February 2026 Volume 2026:18 561692
DOI https://doi.org/10.2147/JEP.S561692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Abdelwahab Omri
Israa H Isawi,1 Nour A Al-Sawalha,2 Fatema Mahmoud,2 Islam Al-Khawaldeh,1 Karem H Alzoubi3
1Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Clinical Pharmacy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan; 3Department of Pharmaceutical Sciences, College of Pharmacy, Qu Health, Qatar University, Doha, Qatar
Correspondence: Israa H Isawi, Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan, Tel +962791162595, Email [email protected]
Background: Sodium–glucose cotransporter 2 (SGLT2) inhibitors are a relatively new class of antidiabetic agents that lower blood glucose independently of insulin by inhibiting renal SGLT2 in the proximal tubule, thereby increasing urinary glucose and sodium excretion. Empagliflozin (Empa), an FDA-approved SGLT2 inhibitor, exhibits antioxidant, anti-inflammatory, and additional metabolic effects beyond glycemic control. Given the high expression of SGLT2 in the central nervous system and the established link between cognitive impairment, chronic hyperglycemia, oxidative stress, and inflammation, this study investigated Empa’s neuroprotective potential on learning and memory deficits in streptozotocin-induced hyperglycemic male Wistar rats.
Methods: Hyperglycemia was induced using streptozotocin (40 mg/kg/IP), followed by Empa treatment (10 mg/kg/day/PO). Cognitive performance was evaluated using the radial arm water maze, assessing learning and both short-term and long-term memory. Concurrently, hippocampal oxidative stress markers and key molecular mediators, including brain-derived neurotrophic factor (BDNF) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), were measured to elucidate possible underlying neuroprotective mechanisms.
Results: Hyperglycemic rats exhibited significant impairments in learning, short-term memory, and long-term memory compared to normoglycemic controls. Empa treatment significantly improved short-term memory, restoring performance to near-control levels. However, its impact on long-term memory was minimal. Unexpectedly, Empa induced only modest, non-statistically significant changes in hippocampal oxidative stress markers and BDNF and NF-κB levels.
Conclusion: The findings underscore the complexity of oxidative stress and inflammatory pathways involved in hyperglycemia-associated cognitive impairment. The beneficial effects of Empa on short-term memory may involve alternative mechanisms unrelated to oxidative stress modulation. Further studies involving extended durations, higher dosages, or larger sample sizes are warranted to better elucidate the neuroprotective mechanisms of Empa.
Keywords: SGLT2, cognitive impairment, hippocampus, animal model, BDNF
Introduction
Diabetes mellitus (DM) is a major global health concern, and its global prevalence continues to rise.1 In 2022, approximately 830 million individuals were diagnosed, with projections indicating that by 2050, over 1.31 billion people will be impacted by the disease. Chronic hyperglycemia is a hallmark of DM and, if not controlled, can lead to significant morbidity and mortality worldwide.2 These complications include neuropathy, retinopathy, nephropathy, cardiovascular disorders, lower-extremity amputation, and dermatological and gastrointestinal problems, among others.2–5 In terms of neuropathic complications, numerous studies have shown that chronic hyperglycemia adversely impacts cognitive function, hinders learning and memory, alters brain structure, and increases the risk of dementia.6,7 These modifications resembled signs akin to expedited cerebral aging. Researchers have proposed several mechanisms to explain this cognitive decline,8 with inflammation and oxidative stress being among the prevailing factors.9–11 In particular, oxidative stress plays a substantial role in disease onset and progression by inducing the development of insulin resistance, impairing glucose tolerance, and promoting β-cell dysfunction.12–14
Several therapeutic strategies that aim at controlling blood glucose levels, particularly those reducing hyperglycemia in individuals with type 2 diabetes mellitus (T2DM), have demonstrated a positive impact on cognitive abilities.15 For instance, lowering glycemic levels through glucagon-like peptide 1 (GLP-1) mimetics or GLP-1 receptor agonists has been linked to improved cognitive performance in DM and Alzheimer’s disease animal models.16 Similarly, dipeptidyl peptidase-4 (DPP-4) inhibitors have demonstrated a protective effect against cognitive deterioration in elderly individuals with DM.17,18 More recently, sodium–glucose cotransporter 2 (SGLT2) inhibitors—often referred to as gliflozins—have emerged as an effective treatment for T2DM.19,20 Gliflozins function by reducing the reabsorption of glucose and sodium in the renal tubules, increasing their urinary excretion, and ultimately reducing blood glucose levels in an insulin-independent manner.19,20 Beyond its ability to effectively manage hyperglycemia, the FDA-approved SGLT2 inhibitor empagliflozin (Empa) has been demonstrated to have significant cellular and metabolic benefits in both diabetic and non-diabetic populations.21–24 Empa has been shown to improve clinical outcomes and reduce mortality in diabetic patients with cardiovascular and chronic kidney diseases,25–28 while also decreasing inflammation and oxidative stress biomarkers in patients with heart failure.29 Moreover, a recent study also indicates that Empa attenuates hyperglycemia-induced testicular oxidative damage in diabetic rat models.30
Given the reported expression of SGLT2 in the central nervous system, researchers have explored potential neuroprotective roles of SGLT2 inhibitors.31–33 Recognizing that hyperglycemia-induced cognitive impairment has been linked to inflammation and oxidative stress as potential underlying causes,9–11 and considering the reported antioxidant and anti-inflammatory properties of Empa,34 examining its effect on cognitive function in hyperglycemia has emerged as a prominent area of research. For instance, Hierro-Bujalance et al showed that Empa mitigated the cognitive impairment induced by T2DM and AD in a mixed mouse model, reducing cortical thinning, neuronal loss, hemorrhage, microglial burden, and amyloid plaque formation.35 Furthermore, Yaribeygi et al also demonstrated that Empa enhanced cognitive performance in a Type 1 diabetes mellitus (T1DM) rat model by promoting an antioxidative defense system and reducing oxidative enzyme expression.36
Building on these findings, the current study investigates whether Empa can ameliorate cognitive impairment in streptozotocin (STZ)-induced hyperglycemia in a rat model, with emphasis on molecular regulators involved in hippocampus-dependent learning and memory (Figure 1). Because hyperglycemia-associated cognitive impairment is thought to arise, in part, from oxidative stress and inflammatory signaling that disrupt hippocampal synaptic function, we focused on two mechanistically relevant mediators: Brain-Derived Neurotrophic Factor (BDNF) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). BDNF is a key regulator of synaptic plasticity and synaptogenesis,37–40 and its expression is tightly regulated by neuronal activity, environmental enrichment, and metabolic states. However, chronic stressors or metabolic dysfunction, such as chronic hyperglycemia, are known to suppress its levels and subsequently impair hippocampal neurogenesis and memory consolidation. Meanwhile, NF-κB is a central transcriptional hub for stress-responsive inflammation that can influence BDNF-related pathways.41,42 In inflammatory settings like those seen in chronic hyperglycemia, NF-κB activation can paradoxically suppress BDNF by stimulating pro-inflammatory cytokine production, creating a feedback loop that intensifies synaptic dysfunction. This NF-κB–BDNF imbalance may therefore contribute to hippocampal dysfunction and cognitive impairment in hyperglycemia. Thus, chronic hyperglycemia may impair hippocampal memory through oxidative stress and NF-κB–linked inflammatory signaling that disrupts BDNF-dependent synaptic plasticity. To capture this broader pathophysiology, we also assessed the effects of Empa on hippocampal antioxidant markers. Given the established link among hyperglycemia-related cognitive impairment, oxidative stress, and inflammation, we hypothesized that Empa treatment may influence hippocampus-dependent learning and memory through the BDNF/NF-κB signaling axis. Accordingly, this study is primarily behavioral with targeted mechanistic profiling, relating RAWM outcomes to selected hippocampal oxidative stress and BDNF/NF-κB measures.
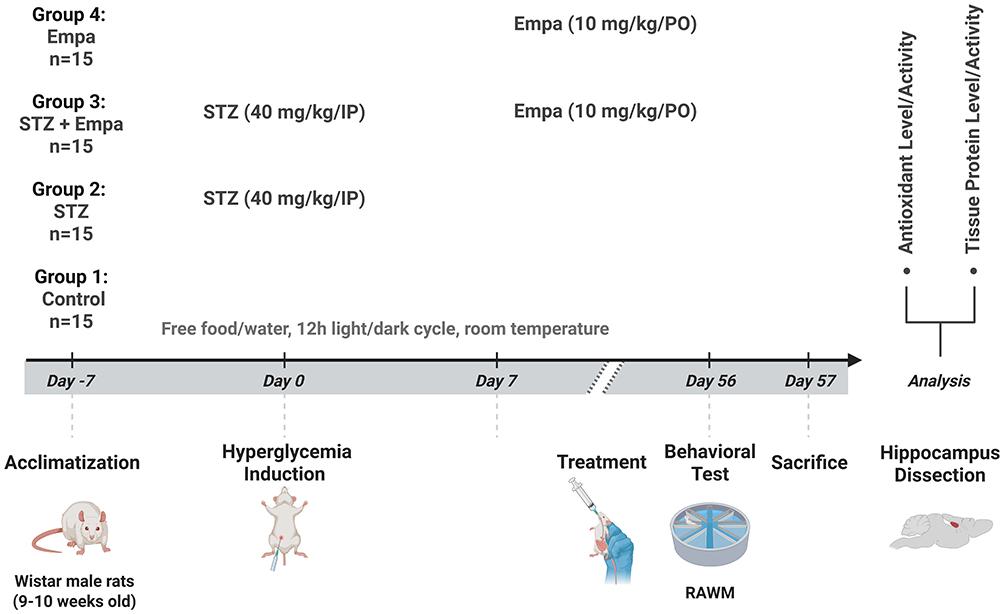 |
Figure 1 Graphical illustration of the experimental timeline. Abbreviations: STZ, streptozotocin; Empa, empagliflozin; RAWM, Radial arm water maze. |
Materials and Methods
Animals and Ethical Approval
Sixty Wistar male rats (9–10 weeks old) were acquired from Jordan University of Science and Technology, Irbid, Jordan, and housed in compliance with the recommendations of the Animal Care and Use Committee at the same institution (Approval No. 20210123). All procedures involving animals were conducted in accordance with institutional and national guidelines for the care and use of laboratory animals. The study was reviewed and approved by the Institutional Animal Care and Use Committee of Jordan University of Science and Technology, and all methods are reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org).
Rats were maintained under a twelve-hour light/dark cycle at room temperature, with unrestricted access to water and food. The animals were randomly allocated to one of the following groups: Group 1: control; Group 2: hyperglycemic rats (STZ); Group 3: hyperglycemic rats administered Empa (STZ + Empa); and Group 4: control rats administered Empa (Empa) (n=15 per group). The experimental timeline is illustrated in Figure 1.
Hyperglycemia Induction
Rats were fasted for 8 hrs and hyperglycemia was induced by a single intraperitoneal injection of a prepared solution of STZ (40 mg/kg) in sodium citrate buffer (pH 4.5). Following the STZ injection, the drinking water was augmented with sucrose (15 g/L) for 48 hours to reduce early mortality.43 Fasting plasma glucose was assessed one day post-STZ injection using a glucose analyzer (Accu-Chek Guide Blood Glucose Meter), and rats were deemed hyperglycemic if the fasting blood glucose level was above 150 mg/dL.43 Glucose levels were assessed weekly using a glucose analyzer. After 7 days, Empa treatment was initiated.44
Treatment
Empa was obtained from Sigma-Aldrich Corp. (MI, USA). The drug was freshly prepared daily by dissolving it in 5% hydroxymethyl cellulose (vehicle) and delivered via oral gavage at a dose of 10 mg/kg45,46 to the STZ + Empa and Empa groups once daily for a duration of 8 weeks following hyperglycemic induction.
Behavioral Assessments—Radial Arm Water Maze Test
Twenty-four hours following the conclusion of the 56-day period (8 weeks), short-term and long-term memory were evaluated using the radial arm water maze (RAWM) test.47,48 Briefly, the RAWM comprises a circular, black, water-filled tank containing six stainless-steel panels configured in a V-shape, creating a central area with six radial arms for animals to swim. The water temperature was maintained at 24 ± 1°C. Two distinct visual cues were strategically placed on the wall to serve as spatial orientation for the rats.
During acclimatization, animals completed six 1-min trials per day for two consecutive days before the RAWM. The goal was placed 2 cm above the water surface under normal lighting, with no arms installed in the maze and no visual cues present. Learning was then assessed in a 12-trial test, with a 5-min rest after the sixth trial. During testing, the goal was placed 2 cm below the water surface under dim lighting, with the arms installed and visual cues provided.
Learning was assessed by recording the number of errors (incorrect arm entries) per trial, which served as the primary behavioral variable. Each trial had a maximum duration of 60 seconds, and if the rat failed to locate the hidden platform, it was gently guided to it. During the acquisition phase, 12 trials were conducted in total. Short-term memory (STM) was examined for 30 minutes following the final acquisition session, whereas long-term memory (LTM) was analyzed at 5 hours and 24 hours post-training, with a singular test trial conducted at each interval. Behavioral testing and scoring were not performed under blinded conditions; this is acknowledged as a limitation.
Hippocampus Dissection and Measuring the Level/Activity of Antioxidants
After behavioral assessment, animals were euthanized by rapid decapitation without anesthesia to preserve brain lipid integrity and molecular biomarkers. The procedure was performed swiftly using a standard laboratory guillotine and is recognized as an acceptable method under the AVMA Guidelines for the Euthanasia of Animals (2020).49
After the behavioral assessment, all rats were sacrificed, and the hippocampus was dissected immediately and stored at −80°C until homogenization. Hippocampal tissue was manually homogenized with a plastic pestle in a solution of protease inhibitors (Sigma-Aldrich Corp., MI, USA) in phosphate buffer.
The activity of the antioxidative system in the brain was assessed by measuring the activity of superoxide dismutase (SOD) (Sigma-Aldrich Corp., USA, cat. # 19160), catalase (CAT) (Cayman Chemical, USA, cat. # 707002), and glutathione peroxidase (GPX) (Sigma-Aldrich Corp., USA, cat. # MBS2516156) enzymes, according to the instructions provided by each kit manufacturer. Antioxidant levels in the samples were quantified using a commercially available kit (Bio-RAD, Hercules, California, USA). Additionally, tissue lipid peroxidation was assessed by thiobarbituric acid reactive substances (TBARS) (Cayman Chemical, USA, cat. # 10009055).
Biochemical Assessments in Hippocampus Tissues
BDNF Protein Level
BDNF levels in the hippocampus were measured using a commercial Enzyme-Linked Immunosorbent Assay (ELISA) kit (R&D, UK, cat. # DBNT00), according to the manufacturer’s instructions.
NF-κB Activity
Hippocampal NF-κB activity was evaluated by quantifying NF-κB p65 levels using an ELISA kit (MyBioSource, USA, cat. # MBS453975), according to the manufacturer’s instructions. The supernatant containing the nuclear fraction was used to quantify NF-κB activity.
Statistical Analysis
Fifteen rats were initially allocated to each group; however, analyses were conducted on ten rats per group due to assay kit capacity. This reduced sample size may have limited statistical power to detect modest molecular differences; therefore, non-significant molecular findings should be interpreted with caution. Statistical analysis was performed using one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test to assess differences among groups. Results are expressed as mean ± standard error of the mean (SEM) for ten animals in each group. Mean differences between groups and their corresponding p-values were reported to demonstrate the magnitude and significance of observed effects. Statistical significance was defined as p-values less than 0.05. All analyses were conducted using GraphPad Prism version 8.0.2. Detailed pairwise group comparisons (Tukey’s test) are provided in Supplementary Table S1.
Results
The Effect of Empagliflozin on Learning Performance
Fasting blood glucose was measured one week after STZ injection, before the administration of Empa, and was 207.9 ± 22.23 mg/dL in the STZ group and 173.9 ± 17.75 mg/dL in the STZ + Empa group. After 8 weeks of Empa administration, fasting blood glucose levels were significantly decreased compared with STZ (301.3 ± 41.32 mg/dL in the STZ group versus 138.1 ± 14.29 mg/dL in the STZ + Empa group, P value = 0.0002).
Figure 2 presents cognitive performance data obtained from 12 successive trials of the RAWM test, shown by the number of errors committed. All groups exhibited progressive improvement over successive trials, signifying effective learning through repeated exposure. The STZ-induced hyperglycemic group had significantly elevated error rates, especially in trials 5–8, indicating compromised learning linked to hyperglycemia. Empa significantly attenuated cognitive impairment in hyperglycemic rats (STZ + Empa), nearing control levels by the end of the trials, thus underscoring Empa’s potential neuroprotective effects against hyperglycemic-induced cognitive decline. The Empa-only group demonstrated performance similar to the controls, suggesting that Empa alone does not negatively impact cognitive function in normoglycemic rats.
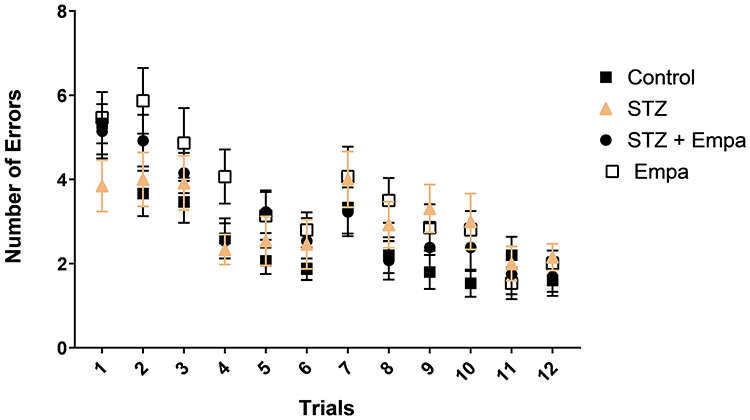 |
Figure 2 Effects of Empa on learning performance in STZ-induced hyperglycemic rats assessed by the Radial Arm Water Maze (RAWM). Statistics: one-way ANOVA performed for each trial with Tukey’s post hoc test as described in Statistical analysis; p < 0.05 considered significant. |
The Effect of Empagliflozin on the Short-Term and Long-Term Memory Performance
According to Figure 3, at the end of the 8th week, memory tests revealed significant differences between the experimental groups. In the STM tests, conducted 30 minutes after the last learning trial, and the LTM tests, conducted 5 and 24 hours after the last learning trial, the STZ group made significantly more errors compared to the control group. The results were as follows (Control vs STZ: STM: −2.453, p = 0.005; LTM at 5 hours: −2.608, p = 0.046; and at 24 hours: −2.718, p = 0.025). Empa treatment markedly reduced the number of errors in the STZ + Empa group relative to STZ alone; however, this improvement was only statistically significant in the STM test (STZ vs STZ + Empa: 2.222, p = 0.029). Similarly, the Empa-only group showed fewer errors than the STZ group in STM (STZ vs Empa: 2.313, p = 0.0115). However, no significant differences emerged for the 5-hour and 24-hour LTM tests in the Empa-treated hyperglycemic animals (STZ vs STZ + Empa: 0.354, p = 0.986 and 0.8462, p = 0.786, respectively).
 |
Figure 3 Effects of Empa on memory performance in the Radial arm water maze test (RAWM). (a) Short-term memory (STM) 30 minutes after the last learning trial, (b) long-term memory (LTM) at 5 hours post-trial, (c) long-term memory (LTM) test at 24 hours post-trial. The data are expressed as the number of errors made during these tests by the control, STZ, STZ + Empa, and Empa groups. Statistics: one-way ANOVA with Tukey’s post hoc test; *p < 0.05 considered significant. |
The Effect of Empagliflozin on the Level/Activity of Antioxidants
Figure 4 demonstrates activity of several antioxidants in the hippocampus across experimental groups. The data show that the levels of certain antioxidants, such as SOD and GPX, decreased following hyperglycemic induction with STZ compared to the control group (SOD: 11.16 ± 0.3891 vs 12.28 ± 0.6797; GPX: 0.1873 ± 0.01508 vs 0.2490 ± 0.007156); however, these differences were not statistically significant. While a marginally higher CAT activity in the STZ group compared to the control group was detected (CAT: 5.629 ± 0.5242 vs 4.806 ± 0.3734). Additionally, tissue lipid peroxidation, as assessed by measuring TBARS, was found to be lower in the STZ rats compared to controls (3.605 ± 0.5016 vs 6.225 ± 1.027), again without statistical significance.
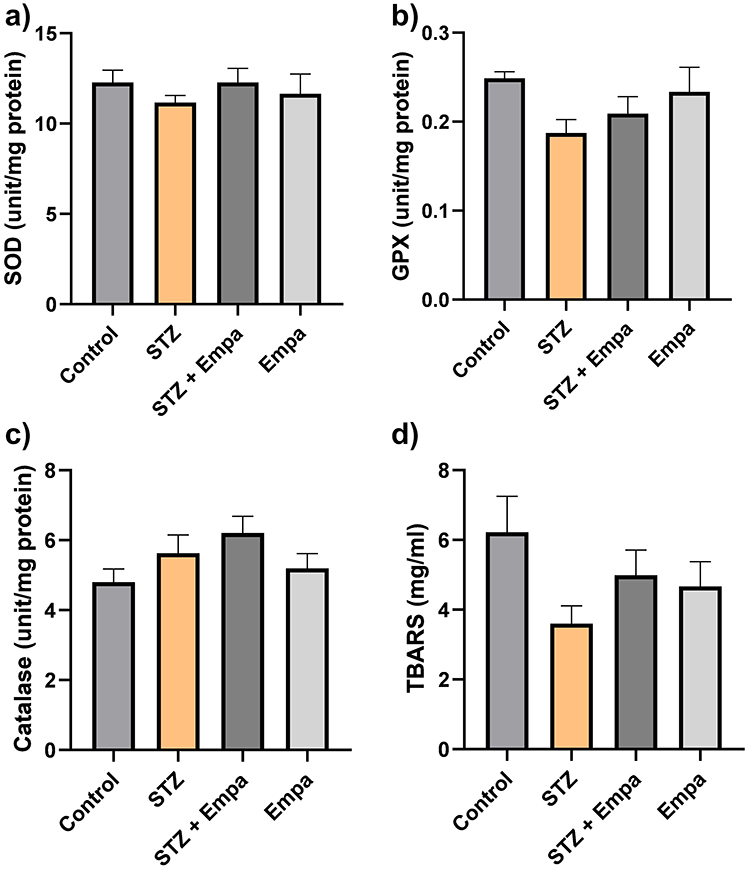 |
Figure 4 Effects of Empa on antioxidant enzyme activities in the hippocampus of hyperglycemic rats. Levels of the following antioxidant enzyme activity are represented (a) superoxide dismutase (SOD) in units/ mg of protein, (b) glutathione peroxidase (GPX) in unit/mL per mg of protein, (c) catalase (CAT) in units/ mg of protein, (d) thiobarbituric acid reactive substances (TBARS) in mg/mg of protein. Data are expressed as mean ± SEM for 10 rats per group. Statistics: one-way ANOVA with Tukey’s post hoc test; p < 0.05 considered significant. |
Treatment with Empa resulted in a small and non-statistically significant change in the activity of antioxidants among the STZ and control groups (Figure 4). For instance, treatment with Empa has increased the level of CAT in both the STZ and control groups (STZ vs STZ + Empa: 1.010, p = 0.801; Control vs Empa: −0.393, p = 0.927). It also increased the level of SOD, GPX, and TBARS in the STZ group (STZ vs STZ + Empa: −1.123, p = 0.755; −0.0219, p = 0.838; and −1.388, p = 0.576, respectively). However, the levels of these antioxidants decreased in the control group after treatment with Empa (Control vs Empa: 0.621, p = 0.942; 0.0153, p = 0.937; and 1.556, p = 0.480, respectively). Overall, none of these alterations reached statistical significance. Pairwise comparisons are summarized in Supplementary Table S1.
The Effect of Empagliflozin on Biochemical Factors Related to Hippocampus-Dependent Learning and Memory
Figure 5 demonstrates the mean levels of BDNF and NF-κB in the hippocampus across experimental groups. The data reveal that BDNF level was reduced in the STZ group compared to the control group (215.0 ± 16.94 vs 291.7 ± 21.64). Although this reduction was not statistically significant, a potential trend was noticed (76.73, p = 0.080). Treatment with Empa produced an increase in BDNF levels in the STZ group and a decrease in the control group; however, these alterations were not statistically significant (STZ vs STZ + Empa: −7.703, p = 0.994; Control vs Empa: 45.87, p = 0.434).
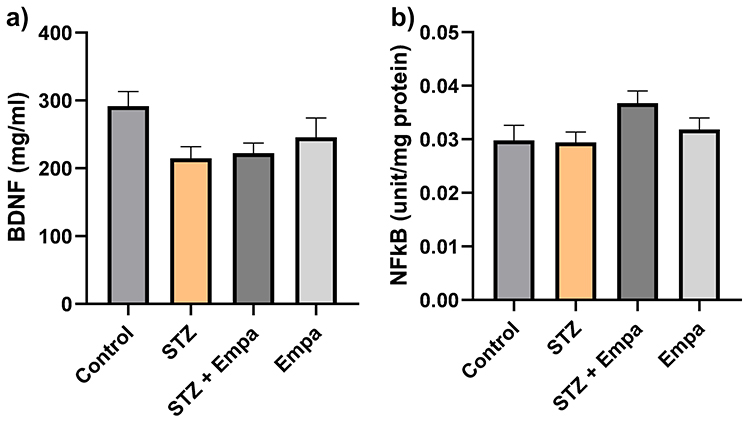 |
Figure 5 Effects of Empa on biochemical factors relevant to hippocampus-dependent learning and memory in hyperglycemic rats. Levels of the following biochemical factors are represented (a) brain-derived neurotrophic factor (BDNF) in pg/mg protein, (b) nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor in pg/mg protein. Data is expressed as mean ± SEM for 10 rats per group. Statistics: one-way ANOVA with Tukey’s post hoc test; p < 0.05 considered significant. |
The NF-κB levels were closely comparable between the control and STZ groups (0.02982 ± 0.002775 vs 0.02945 ± 0.001901). Treatment with Empa produced only minor, non-statistically significant differences in NF-κB levels compared to untreated groups (Control vs Empa: −0.002036, p = 0.927; STZ vs STZ + Empa: −0.007330, p = 0.110).
To facilitate interpretation across outcomes, a summary table of the main antioxidant and biochemical factors has been added (Table 1).
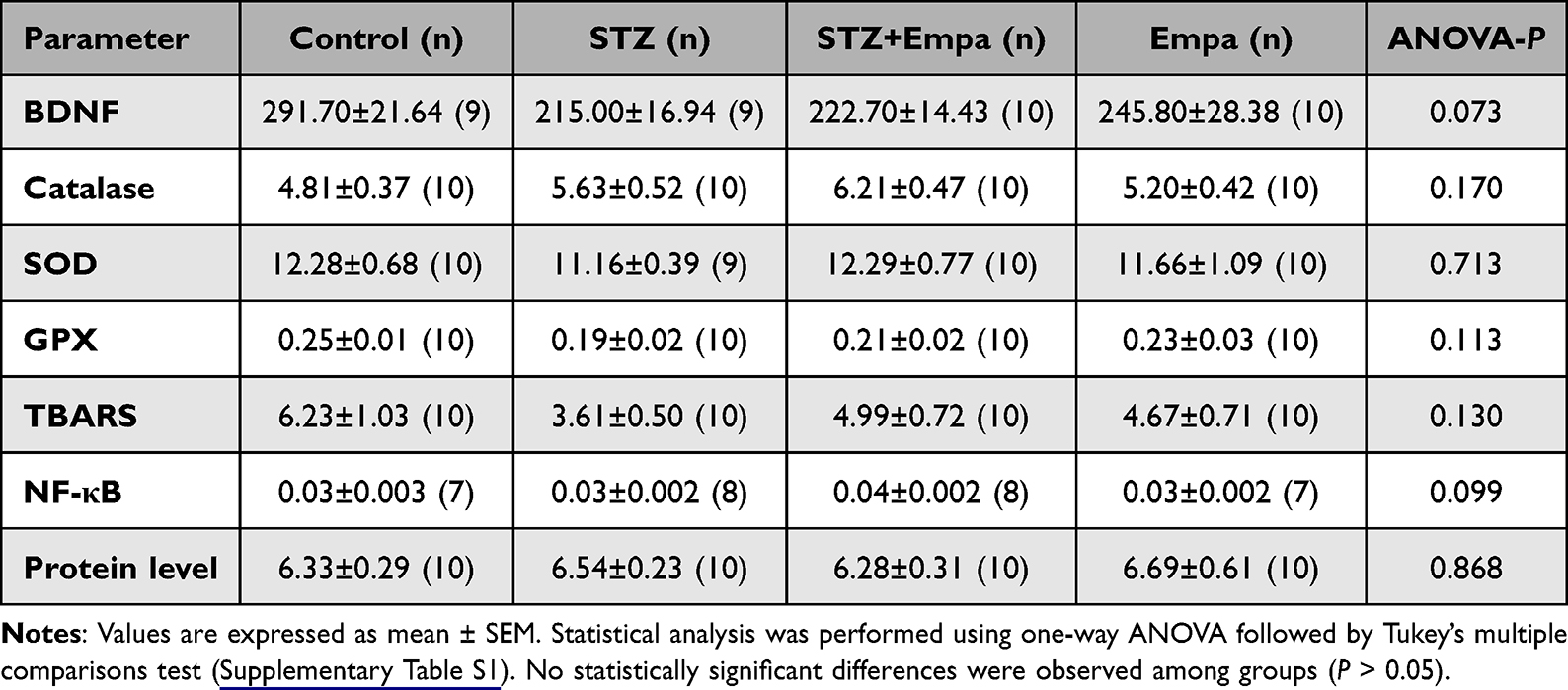 |
Table 1 Antioxidant Activity and Biomedical Factor Levels Across Different Experimental Groups |
Discussion
Uncontrolled hyperglycemia, as in DM, is known to negatively affect cognitive abilities, particularly learning and memory processes, which leads to a marked decrease in quality of life.2,8,50 Despite extensive research into the pathophysiology of hyperglycemic-induced cognitive decline, the multifactorial nature of this phenomenon has given rise to multiple hypotheses, with oxidative stress identified as a significant contributor.9–11 Within this context, SGLT2 inhibitors have increasingly been associated with cognitive benefits in experimental diabetes and mixed metabolic-neurodegenerative models, although the accompanying molecular signatures vary by model severity, dose, and sampling time.35,36
For example, Empa improved learning and memory and reduced neurovascular and neuropathological burden in a complex APP/PS1xdb/db (AD–T2D) mouse model after long-term treatment, accompanied by reduced microglial burden and improved cognitive performance.35 Other preclinical work indicates that SGLT2 inhibitors (including Empa and dapagliflozin) can ameliorate diabetes-associated cognitive impairment alongside changes in neurotrophin-related and neuroinflammatory gene-expression pathways (including BDNF-related signaling), supporting a multifactorial basis for neuroprotection.51
Importantly, mechanistic readouts are not uniformly observed across studies; in STZ-diabetic rats treated with Empa, improvements in cognition have been reported together with decreased oxidative stress drivers (eg, reduced Nox-4 expression) and enhanced antioxidant defenses, highlighting that the oxidative/inflammatory profile may depend on protocol and sampling strategy.36 Compared with other antidiabetic classes, GLP-1 receptor agonists have also been studied for cognitive outcomes; notably, a head-to-head preclinical study in obese mice evaluated both semaglutide and Empa and reported treatment-related improvements in cognition with hippocampal phosphoproteomic pathway changes, supporting the concept that different glucose-lowering agents may converge on brain-relevant signaling through partly distinct mechanisms.52
In this context, the present study investigated the effects of Empa on mitigating STZ-induced hyperglycemia-related cognitive impairment, relating RAWM outcomes to selected hippocampal oxidative stress markers and BDNF/NF-κB. Against this background, our distinctive finding is the dissociation between improved STM and only modest, non-significant endpoint changes in hippocampal oxidative stress markers and BDNF/NF-κB, suggesting that Empa’s STM benefit may reflect systemic metabolic normalization and/or alternative pathways not fully captured by the measured marker panel at a single terminal time point. Whereas several prior studies have linked cognitive improvement with robust antioxidant or neurotrophin/inflammatory shifts, our data suggest that Empa-related behavioral benefits can occur even when endpoint hippocampal oxidative markers and BDNF/NF-κB show only modest, non-significant changes. Moreover, few studies have specifically interrogated the BDNF/NF-κB axis in STZ-based hyperglycemia treated with Empa, underscoring the mechanistic relevance of our approach.
Consistent with this framework, we first interpret the behavioral effects and then consider mechanistic correlations. Our findings indicate that STZ-induced hyperglycemia impaired hippocampus-dependent cognition, as hyperglycemic rats made significantly more errors than controls in both STM and LTM evaluations. Empa treatment reduced error counts in STM assessments in hyperglycemic rats, suggesting an early or preferential benefit on immediate recall/acquisition; however, Empa did not significantly improve LTM at 5 or 24 hours. Because STM and LTM reflect partially distinct phases of memory processing (acquisition/early recall vs consolidation),53,54 this pattern may indicate that STM is more sensitive to the modest metabolic or neural adaptations induced by Empa, whereas measurable effects on consolidation may require higher doses, longer treatment duration, or more severe/chronic hyperglycemia. This STM–LTM dissociation may therefore help identify which phase of memory processing is most responsive to Empa under the present hyperglycemic conditions.
A further limitation is that locomotor activity and anxiety-like behavior were not independently assessed (eg, open-field55 or elevated plus maze).56 Because water-maze performance can be influenced by stress responsivity, anxiety, or locomotor changes, we cannot exclude non-cognitive contributions to performance differences.57 Future studies should include locomotor/anxiety assays alongside RAWM to better isolate memory-specific effects.
Another limitation of the present work is the experimental model. STZ-induced hyperglycemia is widely used and reliable;58 however, depending on dose and protocol, it primarily reflects β-cell injury and does not fully reproduce the chronic, progressive pathophysiology of T2DM, which is characterized by insulin resistance developing over time with later β-cell dysfunction.59 Therefore, although our model is appropriate for studying hyperglycemia-associated cognitive impairment, future studies should employ more clinically relevant T2DM paradigms, such as combining a high-fat diet (HFD) to induce insulin resistance with low-dose STZ to produce partial β-cell dysfunction (HFD/STZ).60,61 Using HFD/STZ models may better approximate the metabolic evolution of T2DM and may help clarify whether Empa’s cognitive effects differ under insulin-resistant conditions and over longer disease durations.
This model context is relevant when interpreting hippocampal redox outcomes, as disease severity and duration can strongly influence the detectability of oxidative stress markers. In contrast to the significant oxidative damage documented in various diabetic models,36,62 our study revealed only mild and non-significant changes in the levels of SOD, GPX, CAT, and TBARS in STZ-induced hyperglycemic rats when compared to control groups. This core panel was selected because it captures major enzymatic antioxidant defenses (SOD, CAT, GPX) together with lipid peroxidation (TBARS), which are widely used indices of hippocampal redox status in experimental hyperglycemia.63,64 In the STZ group, SOD and GPX levels decreased, and CAT levels slightly increased. In parallel, TBARS was also lower in the hyperglycemic group than in the controls, contrary to what some prior research in other models might predict.62 These patterns may indicate compensatory mechanisms, variability in disease severity, or the relatively mild hyperglycemic environment in our STZ model. Although higher-dose STZ protocols can exacerbate hyperglycemia, they are also associated with increased toxicity and mortality, negatively impacting animal welfare and survival;65 therefore, our STZ regimen may have produced a comparatively milder oxidative phenotype. However, we acknowledge that this marker set does not capture the full complexity of redox regulation (eg, glutathione cycling, non-enzymatic antioxidants, and overall antioxidant capacity), and therefore the oxidative status should be interpreted within the limits of this panel.66 After Empa administration, only minor, statistically non-significant alterations were observed in these antioxidant markers for both hyperglycemic (STZ + Empa) and control rats (Empa). Notably, other STZ-based studies have reported that Empa (20 mg/kg/PO, 5 weeks) has been associated with cognitive improvement together with clearer shifts in oxidative stress pathways (eg, reduced Nox-4 expression and improved antioxidant defenses), suggesting that differences in STZ protocol, treatment duration/dose, behavioral paradigm, and tissue sampling may account for variability in the detectability of oxidative markers across studies.36 Given that redox responses can be dynamic and time-dependent, a single terminal measurement may miss earlier transient changes. These findings suggest that either the dosing or duration of Empa was inadequate to produce measurable changes in oxidative stress status at the assessed endpoint, or that, under the tested conditions, direct antioxidant modulation is not the main mechanism through which Empa provides its benefits. However, to provide a more comprehensive assessment, future studies should assess other oxidative stress biomarkers, such as glutathione reductase or total antioxidant capacity, among others, and consider including glutathione-related indices (eg, GSH/GSSG) to better characterize glutathione cycling to draw definitive conclusions. Moreover, future work could explore whether combining Empa with complementary antioxidative strategies yields additive or synergistic effects that are not apparent with monotherapy.
With respect to neurotrophic signaling, the induction of hyperglycemia resulted in decreased BDNF levels relative to controls; however, this decrease did not achieve statistical significance. BDNF plays a critical role in hippocampal synaptic plasticity and cognitive function,37–39 and a non-significant upward trend in BDNF was observed in the STZ + Empa group. Linking behavioral and biochemical outcomes, the STM improvement in the STZ + Empa group may reflect functional benefits arising from systemic metabolic normalization together with subtle, sub-threshold hippocampal plasticity changes that are directionally consistent with increased BDNF signaling, even if bulk tissue BDNF levels did not differ significantly at the terminal time point.40,67 Thus, Empa-induced cognitive benefits may involve modest BDNF-related signaling changes that were not detectable with the current endpoint and biochemical sample size. Future studies should incorporate larger cohorts, time-course sampling, and complementary plasticity readouts (eg, CREB phosphorylation68 and synaptic protein markers),69 in addition to assessing BDNF expression and functional activity, to clarify whether BDNF-linked mechanisms contribute to the observed STM improvement.
Additionally, NF-κB, an essential modulator of inflammatory and stress responses,41,42 remained comparable across groups. Importantly, NF-κB “activity” is often driven by dynamic processes (eg, p65 phosphorylation, nuclear translocation, and DNA-binding) rather than changes in total p65 abundance measured in whole-tissue homogenates at a single endpoint.70 Therefore, unchanged NF-κB levels at the terminal time point may indicate that inflammatory signaling was modest in this model and/or that transient activation occurred earlier but was not captured by endpoint sampling. Accordingly, based on total NF-κB (p65) levels alone, Empa did not demonstrate robust anti-inflammatory effects under the present conditions, and inflammation-related interpretations have been moderated. Because cytokines (TNF-α, IL-1β, IL-6) were not measured, and total NF-κB (p65) does not fully represent pathway activation, inflammation-related conclusions should be interpreted cautiously.71,72 This does not exclude anti-inflammatory actions of Empa but suggests they were not detectable using total NF-κB (p65) at the current endpoint. Future studies should incorporate cytokine profiling and complementary NF-κB activation readouts (eg, phosphorylated p65, nuclear localization, and/or DNA-binding activity) to better characterize neuroinflammatory signaling and downstream mediators.
Beyond oxidative stress modulation, Empa may improve cognition through alternative pathways, which could explain the STM benefit despite modest changes in our oxidative/inflammatory readouts. Potential contributors include modulation of synaptic plasticity (eg, glutamatergic signaling and CREB/BDNF-related remodeling), improvements in mitochondrial function and brain energy metabolism (including enhanced metabolic flexibility and ketone utilization), and effects on neurotransmitter regulation and neurovascular coupling.73–75 Collectively, these possibilities suggest that the dissociation between improved STM and modest/non-significant endpoint biochemical changes may reflect mechanisms not fully captured by the selected marker panel at a single terminal time point.
A possible explanation for the modest and non-significant changes is that the selected Empa regimen (10 mg/kg/day for 8 weeks) may have been sufficient to improve glycemic control and STM but not optimal to elicit robust, measurable changes in terminal hippocampal redox/inflammatory readouts. Notably, 10 mg/kg/day is a commonly used “low-dose” regimen in rodent studies and has been employed for 8 weeks in several experimental models;35,76,77 however, other studies have used higher doses (eg, 30 mg/kg/day) and/or longer treatment windows to reveal clearer molecular effects.78 Accordingly, future studies should incorporate dose–response and time-course designs, intermediate sampling points, and larger biochemical sample sizes to better define molecular trajectories and mechanisms.
Hyperglycemia is associated with accelerated cognitive decline and increased dementia risk, making neurocognitive outcomes a clinically meaningful target alongside glycemic control. While our findings are preclinical, they support the translational hypothesis that SGLT2 inhibition may benefit cognition in diabetes, consistent with emerging human evidence; however, causality requires confirmation. Large observational analysis has reported lower incident dementia risk among SGLT2 inhibitor initiators compared with some other glucose-lowering therapies, although these designs cannot establish causality.79 Importantly, interventional data are beginning to appear: Empa has been reported to improve cognitive and physical impairment in frail older adults with diabetes in a clinical study, and additional trials are ongoing to evaluate cognitive endpoints with SGLT2 inhibitors.80 Taken together, these data highlight the need for adequately powered randomized clinical trials in patients with T2DM—using standardized cognitive outcomes, functional measures, and mechanistic biomarkers—to validate whether the STM-selective benefits observed here translate to clinically meaningful cognitive protection in humans and to define which patient populations may benefit most.81
Conclusion
In conclusion, STZ-induced hyperglycemia in rats led to a clear deterioration in both STM and LTM. Empa conferred a cognitive benefit only in STM, with only modest, non-statistically significant endpoint alterations of oxidative stress markers (SOD, GPX, CAT, TBARS) and key modulators (BDNF, NF-κB) in the hippocampus. This dissociation suggests that Empa’s STM benefit in this model may be mediated by mechanisms not captured by our terminal marker panel and/or by systemic metabolic improvement rather than robust hippocampal antioxidant or anti-inflammatory modulation at the endpoint. Further studies using larger cohorts, time-course sampling, and dose–response/longer-duration designs—potentially in more clinically relevant or severe hyperglycemic models—are warranted to better define Empa’s cognitive effects and underlying mechanisms.
Abbreviations
DM, diabetes mellitus; SGLT2, sodium–glucose cotransporter 2; STZ, streptozotocin; Empa, empagliflozin; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; BDNF, brain-derived neurotrophic factor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RAWM, radial arm water maze; SOD, superoxide dismutase; CAT, catalase; GPX, glutathione peroxidase; TBARS, thiobarbituric acid reactive substances; STM, short-term memory; LTM, long-term memory; HFD, high-fat diet.
Funding
This research was funded by the Deanship of Scientific Research at Jordan University of Science and Technology, grant number 20210123.
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Zhou B, Rayner AW, Gregg EW, et al. Worldwide trends in diabetes prevalence and treatment from 1990 to 2022: a pooled analysis of 1108 population-representative studies with 141 million participants. Lancet. 2024;404(10467):2077–15. doi:10.1016/S0140-6736(24)02317-1
2. Cloete L. Diabetes mellitus: an overview of the types, symptoms, complications and management. Nurs Stand. 2022;37(1):61–66. doi:10.7748/ns.2021.e11709
3. Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther. 2008;88(11):1254–1264. doi:10.2522/ptj.20080020
4. Păunică I, Giurgiu M, Dumitriu AS, et al. The bidirectional relationship between periodontal disease and diabetes mellitus—a review. Diagnostics. 2023;13(4):681. doi:10.3390/diagnostics13040681
5. Bhasin S, Enzlin P, Coviello A, Basson R. Sexual dysfunction in men and women with endocrine disorders. Lancet. 2007;369(9561):597–611. doi:10.1016/S0140-6736(07)60280-3
6. Milstein JL, Ferris HA. The brain as an insulin-sensitive metabolic organ. Mol Metab. 2021;52:101234. doi:10.1016/j.molmet.2021.101234
7. Michailidis M, Moraitou D, Tata DA, Kalinderi K, Papamitsou T, Papaliagkas V. Alzheimer’s disease as type 3 diabetes: common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int J Mol Sci. 2022;23(5):2687. doi:10.3390/ijms23052687
8. Zhang S, Zhang Y, Wen Z, et al. Cognitive dysfunction in diabetes: abnormal glucose metabolic regulation in the brain. Front Endocrinol. 2023;14:1192602. doi:10.3389/fendo.2023.1192602
9. Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev. 2014;2014:102158. doi:10.1155/2014/102158
10. Yaribeygi H, Panahi Y, Javadi B, Sahebkar A. The underlying role of oxidative stress in neurodegeneration: a mechanistic review. CNS Neurol Disord Drug Targets. 2018;17(3):207–215. doi:10.2174/1871527317666180425122557
11. Barone E, Di Domenico F, Perluigi M, Butterfield DA. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic Biol Med. 2021;176:16–33. doi:10.1016/j.freeradbiomed.2021.09.006
12. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50(5):567–575. doi:10.1016/j.freeradbiomed.2010.12.006
13. Pitocco D, Tesauro M, Alessandro R, Ghirlanda G, Cardillo C. Oxidative stress in diabetes: implications for vascular and other complications. Int J Mol Sci. 2013;14(11):21525–21550. doi:10.3390/ijms141121525
14. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev. 2020;2020:8609213. doi:10.1155/2020/8609213
15. Srikanth V, Sinclair AJ, Hill-Briggs F, Moran C, Biessels GJ. Type 2 diabetes and cognitive dysfunction—towards effective management of both comorbidities. Lancet Diabetes Endocrinol. 2020;8(6):535–545. doi:10.1016/S2213-8587(20)30118-2
16. Hölscher C. The incretin hormones glucagonlike peptide 1 and glucose‐dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014;10(suppl 1):S47–S54. doi:10.1016/j.jalz.2013.12.009
17. Rizzo MR, Barbieri M, Boccardi V, Angellotti E, Marfella R, Paolisso G. Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J Gerontol a Biol Sci Med Sci. 2014;69(9):1122–1131. doi:10.1093/gerona/glu032
18. Paolisso G, Monami M, Marfella R, Rizzo MR, Mannucci E. Dipeptidyl peptidase-4 inhibitors in the elderly: more benefits or risks? Adv Ther. 2012;29(3):218–233. doi:10.1007/s12325-012-0008-x
19. Whalen K, Miller S, Onge ES. The role of sodium-glucose co-transporter 2 inhibitors in the treatment of type 2 diabetes. Clin Ther. 2015;37(6):1150–1166. doi:10.1016/j.clinthera.2015.03.004
20. Fonseca-Correa JI, Correa-Rotter R. Sodium-glucose cotransporter 2 inhibitors mechanisms of action: a review. Front Med Lausanne. 2021;8:777861. doi:10.3389/fmed.2021.777861
21. Hou YC, Zheng CM, Yen TH, Lu KC. Molecular mechanisms of SGLT2 inhibitor on cardiorenal protection. Int J Mol Sci. 2020;21(21):7833. doi:10.3390/ijms21217833
22. Braunwald E. Gliflozins in the management of cardiovascular disease. N Engl J Med. 2022;386(21):2024–2034. doi:10.1056/NEJMra2115011
23. Lim GB. Empagliflozin improves outcomes in HFrEF regardless of diabetic status. Nat Rev Cardiol. 2020;17(11):681. doi:10.1038/s41569-020-00455-7
24. Bragagni A, Piani F, Borghi C. Surprises in cardiology: efficacy of gliflozines in heart failure even in the absence of diabetes. Eur Heart J Suppl. 2021;23(Suppl E):E40–E44. doi:10.1093/eurheartj/suab094
25. Wanner C, Lachin JM, Inzucchi SE, et al. Empagliflozin and clinical outcomes in patients with type 2 diabetes mellitus, established cardiovascular disease, and chronic kidney disease. Circulation. 2018;137(2):119–129. doi:10.1161/CIRCULATIONAHA.117.028268
26. Zou R, Shi W, Qiu J, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol. 2022;21(1):106. doi:10.1186/s12933-022-01532-6
27. Packer M, Anker SD, Butler J, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383(15):1413–1424. doi:10.1056/NEJMoa2022190
28. Herrington WG, Staplin N, Wanner C, et al. Empagliflozin in patients with chronic kidney disease. N Engl J Med. 2023;388(2):117–127. doi:10.1056/NEJMoa2204233
29. Kolijn D, Pabel S, Tian Y, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. 2021;117(2):495–507. doi:10.1093/cvr/cvaa123
30. Bdeir R, Al-Sawalha NA, Al-Fawares O, Hamadeneh L, Khawaldeh A. Effects of empagliflozin on gonadal functions of hyperglycemic male Wistar rats. PLoS One. 2024;19(6):e0305636. doi:10.1371/journal.pone.0305636
31. Wiciński M, Wódkiewicz E, Górski K, Walczak M, Malinowski B. Perspective of SGLT2 inhibition in treatment of conditions connected to neuronal loss: focus on Alzheimer’s disease and ischemia-related brain injury. Pharmaceuticals. 2020;13(11):379. doi:10.3390/ph13110379
32. Rizzo MR, Di Meo I, Polito R, et al. Cognitive impairment and type 2 diabetes mellitus: focus of SGLT2 inhibitors treatment. Pharmacol Res. 2022;176:106062. doi:10.1016/j.phrs.2022.106062
33. Hierro-Bujalance C, Garcia-Alloza M. Empagliflozin reduces brain pathology in Alzheimer’s disease and type 2 diabetes. Neural Regen Res. 2024;19(6):1189–1190. doi:10.4103/1673-5374.385865
34. Janić M, Cankar M, Šmid J, et al. Empagliflozin-metformin combination has antioxidative and anti-inflammatory properties that correlate with vascular protection in adults with type 1 diabetes. J Diabetes Res. 2022;2022:6796470. doi:10.1155/2022/6796470
35. Hierro-Bujalance C, Infante-Garcia C, Del Marco A, et al. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimers Res Ther. 2020;12(1):40. doi:10.1186/s13195-020-00607-4
36. Yaribeygi H, Hemmati MA, Nasimi F, Pakdel R, Jamialahmadi T, Sahebkar A. Empagliflozin alleviates diabetes-induced cognitive impairments by lowering nicotinamide adenine dinucleotide phosphate oxidase-4 expression and potentiating the antioxidant defense system in brain tissue of diabetic rats. Behav Brain Res. 2024;460:114830. doi:10.1016/j.bbr.2023.114830
37. Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1. doi:10.3389/neuro.02.001.2010
38. Azman KF, Zakaria R. Recent advances on the role of brain-derived neurotrophic factor (BDNF) in neurodegenerative diseases. Int J Mol Sci. 2022;23(12):6827. doi:10.3390/ijms23126827
39. Rozanska O, Uruska A, Zozulinska-Ziolkiewicz D. Brain-derived neurotrophic factor and diabetes. Int J Mol Sci. 2020;21(3):841. doi:10.3390/ijms21030841
40. Miranda M, Morici JF, Zanoni MB, Bekinschtein P. Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci. 2019;13:363. doi:10.3389/fncel.2019.00363
41. Kairisalo M, Korhonen L, Sepp M, et al. NF‐κB‐dependent regulation of brain‐derived neurotrophic factor in hippocampal neurons by X‐linked inhibitor of apoptosis protein. Eur J Neurosci. 2009;30(6):958–966. doi:10.1111/j.1460-9568.2009.06898.x
42. Xu T, Liu J, Li XR, et al. The mTOR/NF-κB pathway mediates neuroinflammation and synaptic plasticity in diabetic encephalopathy. Mol Neurobiol. 2021;58(8):3848–3862. doi:10.1007/s12035-021-02390-1
43. Kitada M, Ogura Y, Koya D. Rodent models of diabetic nephropathy: their utility and limitations. Int J Nephrol Renovasc Dis. 2016;9:279–290. doi:10.2147/IJNRD.S103784
44. Mostafavinia A, Amini A, Ghorishi SK, Pouriran R, Bayat M. The effects of dosage and the routes of administrations of streptozotocin and alloxan on induction rate of type1 diabetes mellitus and mortality rate in rats. Lab Anim Res. 2016;32(3):160. doi:10.5625/lar.2016.32.3.160
45. Jojima T, Tomotsune T, Iijima T, Akimoto K, Suzuki K, Aso Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol Metab Syndr. 2016;8(1):45. doi:10.1186/s13098-016-0169-x
46. Verma S, Rawat S, Ho KL, et al. Empagliflozin increases cardiac energy production in diabetes. JACC Basic Transl Sci. 2018;3(5):575–587. doi:10.1016/j.jacbts.2018.07.006
47. Burešová O, Bureš J, Oitzl MS, Zahálka A. Radial maze in the water tank: an aversively motivated spatial working memory task. Physiol Behav. 1985;34(6):1003–1005. doi:10.1016/0031-9384(85)90028-9
48. Diamond DM, Park CR, Heman KL, Rose GM. Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus. 1999;9(5):542–552. doi:10.1002/(SICI)1098-1063(1999)9:5<542::AID-HIPO8>3.0.CO;2-N
49. Leary SL. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition. American Veterinary Medical Association; 2020.
50. Ong KL, Stafford LK, McLaughlin SA, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2023;402(10397):203–234. doi:10.1016/S0140-6736(23)01301-6
51. Piątkowska-Chmiel I, Herbet M, Gawrońska-Grzywacz M, Pawłowski K, Ostrowska-Leśko M, Dudka J. Molecular and neural roles of sodium-glucose cotransporter 2 inhibitors in alleviating neurocognitive impairment in diabetic mice. Psychopharmacology. 2023;240(4):983–1000. doi:10.1007/s00213-023-06341-7
52. Chen X, Chen S, Li Z, et al. Effect of semaglutide and empagliflozin on cognitive function and hippocampal phosphoproteomic in obese mice. Front Pharmacol. 2023;14:975830. doi:10.3389/fphar.2023.975830
53. Joo HR, Frank LM. The hippocampal sharp wave–ripple in memory retrieval for immediate use and consolidation. Nat Rev Neurosci. 2018;19(12):744–757. doi:10.1038/s41583-018-0077-1
54. Cotton K, Ricker TJ. Examining the relationship between working memory consolidation and long-term consolidation. Psychon Bull Rev. 2022;29(5):1625–1648. doi:10.3758/s13423-022-02084-2
55. Kraeuter AK, Guest PC, Sarnyai Z. The open field test for measuring locomotor activity and anxiety-like behavior. In: Guest PC editor, Pre-Clinical Models. Methods Mol Biol. Vol. 1916. Humana Press; 2019:99–103. doi:10.1007/978-1-4939-8994-2_9
56. Danduga RCSR, Kola PK. Elevated plus maze for assessment of anxiety and memory in rodents. In: Ray SK editor, Neuroprotection. Methods Mol Biol. Vol. 2761. Humana Press; 2024:93–96. doi:10.1007/978-1-0716-3662-6_8
57. Othman MZ, Hassan Z, Has ATC. Morris water maze: a versatile and pertinent tool for assessing spatial learning and memory. Exp Anim. 2022;71(3):264–280. doi:10.1538/expanim.21-0120
58. Ghasemi A, Jeddi S. Streptozotocin as a tool for induction of rat models of diabetes: a practical guide. EXCLI J. 2023;22:274–294. doi:10.17179/excli2022-5720
59. Singh R, Gholipourmalekabadi M, Shafikhani SH. Animal models for type 1 and type 2 diabetes: advantages and limitations. Front Endocrinol. 2024;15:1359685. doi:10.3389/fendo.2024.1359685
60. Brito AKDS, Mendes AVDS, Timah Acha B, et al. Experimental models of type 2 diabetes mellitus induced by combining hyperlipidemic diet (HFD) and streptozotocin administration in rats: an integrative review. Biomedicines. 2025;13(5):1158. doi:10.3390/biomedicines13051158
61. Attrill EH, Scharapow O, Perera S, et al. Controlled induction of type 2 diabetes in mice using high fat diet and osmotic-mini pump infused streptozotocin. Sci Rep. 2025;15(1):8812. doi:10.1038/s41598-025-89162-2
62. Khan T, Khan S, Akhtar M, Ali J, Najmi AK. Empagliflozin nanoparticles attenuates type2 diabetes induced cognitive impairment via oxidative stress and inflammatory pathway in high fructose diet induced hyperglycemic mice. Neurochem Int. 2021;150:105158. doi:10.1016/j.neuint.2021.105158
63. Giribabu N, Srinivasarao N, Swapna Rekha S, Muniandy S, Salleh N. Centella asiatica attenuates diabetes induced hippocampal changes in experimental diabetic rats. Evid Based Complement Alternat Med. 2014;2014(1):592062. doi:10.1155/2014/592062
64. Omer AB, Afzal O, Altamimi ASA, et al. Neuroprotective effect of barbaloin on streptozotocin-induced cognitive dysfunction in rats via inhibiting cholinergic and neuroinflammatory cytokines pathway─TNF-α/IL-1β/IL-6/NF-κB. ACS Omega. 2023;8(8):8110–8118. doi:10.1021/acsomega.2c08277
65. Wang-Fischer Y, Garyantes T. Improving the reliability and utility of streptozotocin-induced rat diabetic model. J Diabetes Res. 2018;2018:8054073. doi:10.1155/2018/8054073
66. Kotha RR, Tareq FS, Yildiz E, Luthria DL. Oxidative stress and antioxidants—a critical review on in vitro antioxidant assays. Antioxidants. 2022;11(12):2388. doi:10.3390/antiox11122388
67. Davarpanah M, Shokri-mashhadi N, Ziaei R, Saneei P. A systematic review and meta-analysis of association between brain-derived neurotrophic factor and type 2 diabetes and glycemic profile. Sci Rep. 2021;11(1):13773. doi:10.1038/s41598-021-93271-z
68. Wang IF, Wang Y, Yang YH, Huang GJ, Tsai KJ, Shen CKJ. Activation of a hippocampal CREB-pCREB-miRNA-MEF2 axis modulates individual variation of spatial learning and memory capability. Cell Rep. 2021;36(5):109477. doi:10.1016/j.celrep.2021.109477
69. Zhu X, Han S, Geng Y, Ren W, Quan F. Brain-derived neurotrophic factor-TrkB pathway on synaptic plasticity in ischemic stroke rats. Int Heart J. 2024;65(6):1095–1106. doi:10.1536/ihj.24-312
70. Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9
71. Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflamm Res. 2018;11:407–419. doi:10.2147/JIR.S140188
72. Mao H, Zhao X, Sun SC. NF-κB in inflammation and cancer. Cell Mol Immunol. 2025;22(8):811–839. doi:10.1038/s41423-025-01310-w
73. Hayden MR, Grant DG, Aroor AR, Demarco VG. Empagliflozin ameliorates type 2 diabetes-induced ultrastructural remodeling of the neurovascular unit and neuroglia in the female db/db mouse. Brain Sci. 2019;9(3):57. doi:10.3390/brainsci9030057
74. Mei J, Li Y, Niu L, et al. SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous system. Transl Neurodegener. 2024;13(1):41. doi:10.1186/s40035-024-00431-y
75. Kostrzewska P, Kuca P, Witek P, Małyszko J, Madetko Alster N, Alster P. SGLT-2 inhibitors in the prevention and progression of neurodegenerative diseases: a narrative review. Neurol Ther. 2025;14(6):2295–2312. doi:10.1007/s40120-025-00832-9
76. Xue M, Li T, Wang Y, et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin Sci. 2019;133(15):1705–1720. doi:10.1042/CS20190585
77. Ozbek EN, Yesilyurt Dirican ZE, Makal M, Arioglu Inan E, Yetik-Anacak G. Empagliflozin protects against oxidative stress in the diabetic brain by inducing H2S formation. Pharmaceuticals. 2025;18(9):1259. doi:10.3390/ph18091259
78. Shao Q, Meng L, Lee S, et al. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019;18(1):165. doi:10.1186/s12933-019-0964-4
79. Shin A, Koo BK, Lee JY, Kang EH. Risk of dementia after initiation of sodium-glucose cotransporter-2 inhibitors versus dipeptidyl peptidase-4 inhibitors in adults aged 40-69 years with type 2 diabetes: population based cohort study. BMJ. 2024;386e079475. doi:10.1136/bmj-2024-079475
80. Mone P, Lombardi A, Gambardella J, et al. Empagliflozin improves cognitive impairment in frail older adults with type 2 diabetes and heart failure with preserved ejection fraction. Diabetes Care. 2022;45(5):1247–1251. doi:10.2337/dc21-2434
81. Seminer A, Mulihano A, O’Brien C, et al. Cardioprotective glucose-lowering agents and dementia risk: a systematic review and meta-analysis. JAMA Neurol. 2025;82(5):450–460. doi:10.1001/jamaneurol.2025.0360
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Dexmedetomidine Alleviates Hippocampal Tissue Damage in Rapid Eye Movement Sleep-Deprived Rats by Activating BDNF/TrkB Signaling Pathway
Zheng B, Li Y, Liu C
Nature and Science of Sleep 2025, 17:2423-2435
Published Date: 1 October 2025