")
Back to Journals » OncoTargets and Therapy » Volume 13
Emerging Roles and Therapeutic Interventions of Aerobic Glycolysis in Glioma
Authors Han W , Shi J, Cao J, Dong B, Guan W
Received 7 May 2020
Accepted for publication 26 June 2020
Published 16 July 2020 Volume 2020:13 Pages 6937—6955
DOI https://doi.org/10.2147/OTT.S260376
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Wei Han, Jia Shi, Jiachao Cao, Bo Dong, Wei Guan
Department of Neurosurgery, The Third Affiliated Hospital of Soochow University, Changzhou, People’s Republic of China
Correspondence: Wei Guan
Department of Neurosurgery, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, People’s Republic of China
Email [email protected]
Abstract: Glioma is the most common type of intracranial malignant tumor, with a great recurrence rate due to its infiltrative growth, treatment resistance, intra- and intertumoral genetic heterogeneity. Recently, accumulating studies have illustrated that activated aerobic glycolysis participated in various cellular and clinical activities of glioma, thus influencing the efficacy of radiotherapy and chemotherapy. However, the glycolytic process is too complicated and ambiguous to serve as a novel therapy for glioma. In this review, we generalized the implication of key enzymes, glucose transporters (GLUTs), signalings and transcription factors in the glycolytic process of glioma. In addition, we summarized therapeutic interventions via the above aspects and discussed promising clinical applications for glioma.
Keywords: aerobic glycolysis, glioma, biological roles, therapeutic interventions, clinical application
Introduction
Glioma, deriving from neuroepithelial cells, is the most prevalent primary tumor in the central nervous system (CNS), with a proportion of 80% of intracranial malignancies.1 According to World Health Organization (WHO) classification and cellular morphology, gliomas of WHO Ⅰ-Ⅳ could be categorized into several classes including astrocytoma, oligodendroglia, ependymoma, etc.2 Notably, gliomas are characterized by their rapid proliferation, infiltrative growth, treatment resistance, intra- and intertumoral genetic heterogeneity.3 Despite the fact that most glioma patients could receive maximal safe surgical resection with adjuvant chemotherapy and radiotherapy, the recurrence rate of them is still high and the prognosis is poor, which is still less than 15 months.4,5
Glycolysis refers to a biological process that glucose or glycogen is decomposed into lactic acid accompanied by moderate production of ATP without ample oxygen.6 Despite the presence of abundant oxygen, cancer cells tend to produce energy via glycolysis even in a higher pace, which was put forward by Otto Warburg, namely the Warburg effect.7 Based on previous studies, key enzymes (HKs, PFK-1, and PKs), glucose transporters (GLUTs) and transcript factors (HIF-1α, c-myc, and p53) have been recognized as main regulators in the glycolytic activities.8 In addition, PI3K/Akt, mTOR, and AMPK signalings were also strongly relevant to glycolysis in multiple solid tumors.9,11 More importantly, the glycolytic process was tightly correlated with various cellular activities, evoking promising therapeutic targets for various tumors.12,13 For instance, lncRNA maternally expressed gene 3 (MEG3) suppressed proliferation and invasion via regulation of glycolysis in colorectal cancer.14 Similarly, the curcumin analogue WZ35 inhibited glycolysis and facilitated the generation of reactive oxygen species (ROS), promoting JNK-dependent apoptosis of gastric cancer cells.15 Xi et al16 also reported that human equilibrative nucleoside transporter 1 (hENT1) was involved in modulating chemotherapy sensitivity of pancreatic cancer cells by inhibiting glycolysis. Recently, gathering investigations have intensively focused on the roles and therapeutic interventions of the glycolytic process in glioma. In this review, we have summarized the roles of key glycolytic enzymes, GLUTs, main signaling pathways, and transcription factors detected in glycolysis of glioma, which may offer possibilities for novel therapies.
Implication of Key Enzymes and GLUTs in Aerobic Glycolysis
Hexokinases (HKs)
HKs catalyze the first step of glycolytic procedure by phosphorylating glucose in the mitochondrial outer membrane of brain and tumor cells, ultimately generating glucose-6-phosphate (G-6-P).17,18 Further gene detection has revealed that HKs exist as five HK isoforms including HKI-IV and HK domain-containing protein 1 (HKDC1), with separate locations of different chromosomes.19 Interestingly, HKII, identified as a “housekeeping enzyme”, is highly expressed in all mammalian tissues, while the other HKs were characterized with distinct tissue-specificity and differential expression.20 Additionally, HKII has been verified to facilitate glycolysis via multiple central metabolic pathways.21 It was also acknowledged that malignant transformation of neural stem cells was paralleled by overexpression of HKII.22 Recently, accumulating trials demonstrated that aberrant expression of HKII triggered multiple mechanisms to regulate the progression of multiple solid tumors, especially in glioma.23,25
Noteworthily, HKII knockdown transformed the glycolytic process to oxidative phosphorylation (OXPHOS), accompanied by the production of ROS in glioma.26 Conversely, a higher glycolytic index along with activated procedures of lipid and protein synthesis was induced by HKII overexpression.27 Nie et al28 also reported that the elevated HKII contributed to an increase in glucose uptake and lactate production in glioma cells with IDH1R132H mutation. Further in vitro experiments illustrated that HKII was significantly upregulated in gliomas and related to proliferation, invasion, apoptosis, and angiogenesis.29 The clonogenic power and cell-cycle progression of glioma cells were also mediated by misregulation of HKII.27,30 Regarding autophagic death, HKII was confirmed its relevance with glioma cells treated by RSL3, a novel compound of small molecules targeting glutathione peroxidase 4 (GPX4).31
Subsequent functional investigation has been carried out for roles of HKII in glioma, which may emerge as a promising therapeutic target for glioma treatment. For example, X box-binding protein 1 (XBP1) knockdown promoted decreases of cellular viability, tumor formation capacity, and the production of ATP/lactate by inhibition of HKII expression.32 Concurrently, some signalings were correlated with biological activities of HKII. The silence of PERK signaling, usually activated upon the lack of oxygen and glucose, decreased tumor formation capacity via mitochondria translocation of HKII.33 Nodal signaling was also involved in boosting xenograft development through upregulation of HKII.34 Moreover, miRNAs could serve as critical regulators of glycolysis in glioma due to their roles in regulating gene expression. Overexpression of miR-1297 markedly inhibited HKII targeting karyopherin α2 (KPNA2), thus promoting cellular proliferation and glycolysis in glioma.35 Similar activities could also be detected in glioma with elevated expression of miR-378e via targeting ribophorin-II (RPN2).36 In addition, it was confirmed that HKII was regulated by miR-218/Bmi1 pathway to exert its tumorigenic effects on glioma.25 Zhao et al37 also reported that miR-143 laid suppressive effects on glycolysis, tumor formation capacity, and promoted differentiation by directly targeting HKII. Notably, 2-deoxy-D-glucose (2-DG), known as a suppressor for glycolysis, was identified to have synergistic effects in the inhibition of HKII by miR-143. Collectively, it has been verified that HKII exerted a pivotal role in the glycolytic pathway and could be a reliable target for glioma.
Phosphofructokinase-1 (PFK-1)
It has been widely recognized that PFK-1 catalyzes the second rate-limiting procedure of glycolysis, transforming fructose-6-phosphate (F-6-P) to fructose-1, 6-biphosphate (F-1, 6-BP) mediated by Mg2+ and ATP.38 Interestingly, PFK-1 could be allosterically activated by ADP, AMP and F-2, 6-BP, and inhibited by citric acid and ATP.39 PFK-1 is mainly composed of three different isoforms, including PFKM (muscle), PFKP (platelet), and PFKL (liver).40 Noteworthily, other isoforms including PKM1, PKM2, PKMR, and PFKFB1-4 have been explored in the glycolytic process.41 Gathering evidence has revealed that PFK-1 mediates the metabolic process by regulating the oxidation of glucose.42 More importantly, the glycolytic flux of tumor cells could be modulated by PFK-1, usually activated in a significant number of tumor types, thus affecting cellular growth, invasiveness, and survival.43,44
Clinically, it was reported that the elevated expression of PFKFB4 was found in glioma patients and also associated with a shorter survival.45 Similar increased activity of PFK-1 was also detected in the glioma cell lines.46 In the glycolytic process of glioma, PFKFB3 mRNA and protein expression could be motivated by TGF-β1, thus upregulating glycolytic flux.47 Further in vitro experiments showed that the inhibition of PFK-1 consistently decreased glioma cell viability and migration.48 More significantly, glioma cells with PFK-1 knockdown induced more caspase-dependent cell death.48 Regarding angiogenesis, the biological effects of lacking PFK-1 was reflected by the decreased number and length vascular tubules.49
Accumulating studies have illustrated that PFK-1 could serve as a surprising target for glioma therapy. For example, citrate, a natural organic acid, targeted PFK-1 to exert anti-tumor effects via inhibiting glycolysis.49 Due to the relevance of glycolysis with mitochondria, voltage-dependent anion channel 2 (VDAC2) coupled PFKP to influence the glycolytic process.50 Moreover, the Smad, p38/MAPK, and PI3K/Akt signalings also participated in the activities of PFKFB3 in glioma.47 Similarly, multiple lines of evidence suggested that gene transcription regulation was involved in the biological activities of PFK-1. LncRNA urothelial cancer associated 1 (UCA1) downregulated the expression of miR-182, which was directly bound to PFKFB2, modulating C-X-C motif ligand 14 (CXCL14) secretion, glycolysis, and invasion of glioma cells.51 Li et al35 also found that KPNA2, a key regulator of glycolysis in glioma, would be targeted by miR-1297 decreasing the functions of PFK-1. In summary, all the above results have suggested that targeting the PFK-1 might be a promising therapeutic target for glioma.
Pyruvate Kinases (PKs)
PKs, the third and last key rate-limiting enzymes of glycolysis, irreversibly catalyze the production of pyruvic acid and ATP from phosphoenolpyruvate (PEP) and ADP.52 Four different subtypes of PKs, including PKL, PKR, PKM1, and PKM2, have been detected in the glycolytic process.53 In addition, these isoforms with definite or indefinite biological activity are distributed in diverse tissues and organs.54 It has been verified that PKs play vital roles in the glycolytic process via controlling the metabolic flux and ATP production.55 Apart from their biological effects in glycolysis, PKs regulate major cell signalings by phosphorylating key proteins to adjust gene expression in the nucleus.56 Recently, PKM1 and PKM2, two main subtypes of PKs in the brain tissue, have become hotpots in glioma treatment for its correlation with radiation sensitivity.57
PKM2 was involved in adapting glioma cells to a different microenvironment through regulating nutrients and growth signaling pathways allosterically.58 More clinical databases have revealed that the translocation of PKM2, induced by epidermal growth factor receptor (EGFR), transactivated β-catenin signaling in promoting brain tumor development.59 It was also qualified that the expression of PKM2 was related to malignancy grades and prognosis of patients with glioma.60 Mechanically, the low expression of PKM2 was consistent with glycolysis dysfunction, accompanied by the decreased concentration of intracellular ATP and pyruvate.31 In vitro, PKM2 also participated in regulating cellular proliferation, migration, and invasion.61 Moreover, the cell-cycle progression could be activated by PKM2 along with elevated contents of cyclin D1 and c-myc.60 More importantly, the upregulation of PKM2 contributed to an increased apoptosis rate in glioma cells via stabilizing Bcl2.62,63 Besides, the activation of autophagic responses was found to be relevant with PKM2.64
Surprisingly, gathering functional regulators for PKM1 and PKM2 have emerged, providing a new insight into the investigation of therapeutic interventions for glioma treatment. HSP90α1’s ATPase could activate PKM2 and subsequently facilitate combination with Bcl2 in ROS adaptation of glioma cells.63 G9a knockdown induced accumulation of Akt and HIF-1α expression, which drive PKM2-Yes-associated protein 1 (YAP1) cross talk in glioma cells.64 Yang et al60,65 also revealed that PKM2, activated by EGFR, exerted its pro-tumor effects via being directly bound with histone H3 or the distinct regulation of NF-κB signaling pathways. Moreover, β-catenin signaling was also correlated with biological activities of PKM2 in glioma.59 Besides, lncRNAs or miRNAs were found to be relevant with biological activities of PKs. MiRNA let-7a modulated c-myc/hnRNPA1 axis to promote glucose metabolism and cell growth by PKM2.66 Surprisingly, miR-1297 silencing facilitated the activities of PKM in glioma via downregulation of KPNA2.35 It was also detected by Liu et al61 that linc00689 competitively interacted with miR-338-3p to boost PKM2 expression, therefore influencing the malignant phenotypes of glioma cells. Overall, PKs have been verified to participate in the glycolytic process, forecasting the potential application for glioma treatment.
Glucose Transporters (GLUTs)
GLUTs, a sort of membrane protein controlling glycolytic flux, mainly take part in transmembrane transport of glucose.67,68 GLUTs share a similar structure with 12 transmembrane domains, with amino and carboxy terminal domains on the cytoplasmic side.69 According to sequence similarity and substrate specificity, GLUTs family of 14 members could be classified into three categories.70 Additionally, different affinities for glucose of GLUTs in different organs revealed their specificity tissue distribution.71 For instance, GLUT1 and GLUT3 were distributed in the brain which demands more glucose than other organs.72,73 GLUT3 and GLUT10 were predominantly detected in the muscle tissues.74,75 Moreover, GLUT4 is mainly observed in muscle and fat cells due to its role in glucose transportation.76 Surprisingly, it has been reported that upregulation of GLUTs was correlated with poor survival, therapy resistance in tumor patients, accompanied by promotion effects on cellular growth and invasion in vitro.67
In glioma, overexpression of GLUT1 and GLUT3, related to higher pathological grades, have been detected in glioma patients.77 It was also reported that GLUT3 was mainly in the plasma membrane, whereas GLUT1 could be detected in both plasma membrane and cytoplasm.78 Further studies have disclosed that GLUT1 regulated glycolysis through extracellular deoxyglucose uptake and glucose transport,79,80 while GLUT3, predominantly in neurons, might play a pivotal role in neuronal glucose influx.78,81 Interestingly, GLUT1 and GLUT3 were paralleled with the expression of key enzymes of glycolysis including HKII, PKM2, pyranose dehydrogenase (PDH), and lactate dehydrogenase A (LDH-A).29,82 While in vitro, GLUT1 exerted its pro-tumor effects in proliferation, invasion, and tumorigenesis.83,84 Dai et al85 also indicated that the proliferation and cell-cycle of glioma cells were also mediated by GLUT3. When it regards angiogenesis, targeting GLUT1 or GLUT3 could unlock the potential for clinical application.86,87 A study by Kuang et al86 also verified that GLUT3 overexpression promoted metabolic reprogramming associated with antiangiogenic therapy resistance. Surprisingly, the inhibition of GLUT1 could suppress tumor initiating cells, thus facilitating the capacity of temozolomide (TMZ) against glioma.88,89
Further clinical investigations have uncovered that KPNA2 overexpression significantly upregulated the expression of GLUT-1 residing on the plasma membrane and 2-DG uptake.79 A study by Chen et al80 also revealed that cAMP-response element binding protein 1 (CREB1) affected the expression of GLUT1 and was involved in the metabolic process of glioma. In addition, some signalings participated in the activities of GLUTs in glioma. The activation of GLUT1 expression and Warburg effect could be mediated by PI3K/Akt signaling pathway.83 The Nodal signaling has also been verified to regulate the energy metabolism in glioma cells.34 Similar biological effects were also observed in IDH1R132H mutant or Ras knockdown glioma cells via the HIF-1α/GLUT1 axis.28,90,91 Besides, miRNA-451, targeting recombinant human calcium binding protein 39 (CAB39) to regulate GLUT1 inhibited the glucose metabolism, the proliferation, and invasion of glioma cells.84 Similarly, miR-495 increased GLUT1 to promote glucose uptake and lactate production in glioma.92 Regarding the functional activities of GLUT3, the same regulating role of HIF-1α was explored, which may be reliable predictors of the biological behavior of glioma.93 MiR-106a knockdown also inhibited glioma cell glucose uptake and proliferation by targeting GLUT3.85 Taken together, GLUT1 could be a therapeutic target and predictor for glioma treatment, while the biological roles of GLUT3 and other glucose transporters are still ambiguous, which needs further exploration (Figure 1 and Table 1).
 |
Table 1 Roles of RNAs Modulating Glycolytic Key Regulators in Glioma |
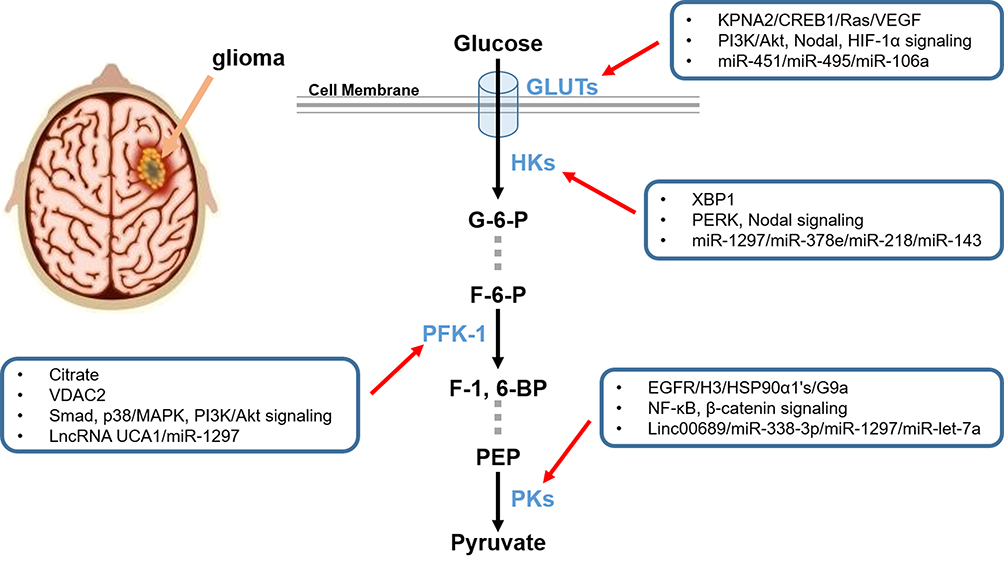 |
Figure 1 Schematic diagram of key enzymes and GLUTs in aerobic glycolysis. HKs, PFK-1, and PKs are the three key rate-limiting enzymes for glycolysis, catalyzing the three irreversible steps in the glycolytic process. GLUTs are a large cluster of membrane proteins that facilitate the transport of glucose through the cellular plasma membrane. All these promising therapeutic targets mentioned above and their regulators are synthetically exhibited. |
Roles of Main Signaling Pathways in Aerobic Glycolysis
PI3K/Akt Signaling
The dysregulation of PI3K/Akt signaling has been found in most of the human cancers, which plays a vital role in cellular proliferation, invasion, migration, apoptosis, and angiogenesis.94 The PI3Ks are a family of kinase enzymes that could phosphorylate phosphatidylinositol 4, 5-bisphosphate (PIP2) to generate phosphatidylinositol 3, 4, 5-triphosphate (PIP3), thus promoting phosphorylation of Akt and activating several downstream targets.95 The PI3Ks could be divided into three subtypes in accordance with structural features and substrate specificities.96 Class IA PI3K and class IB PI3K are heterodimers, both comprising of a regulatory subunit and a catalytic subunit. While class II PI3Ks consisted of three isoforms (PI3KC2α, PI3KC2β, and PI3KC2γ), which could be stimulated by cytokine receptors, receptor tyrosine kinases (RTKs), and integrins. Besides, class III PI3Ks mainly containa catalytic VPS34 subunit.97 Akt, also named protein kinase B, is a serine/threonine protein kinase that could be activated by PI3Ks inducing downstream effector proteins including mTOR, Bcl‐2 family, glycogen synthase kinase 3, E2F, forkhead transcription factor (FKHR), and S6 protein kinase.98,99 Currently, metabolic pathways, especially aerobic glycolysis, could be modulated by PI3K/Akt signaling, which may provide evidence for clinical interventions in glioma.100,101
Gathering evidence has confirmed the relevance of glycolysis with PI3K/Akt signaling in glioma. It has been found that the activities of glycolysis were modulated by PI3K/Akt signaling in regulating glucose uptake, lactic acid production, and nicotinamide adenine dinucleotide (NAD) level.102 In addition, the ATP level was also involved in the biological functions of PI3K/Akt signaling.84 As one of the three key enzymes, PFKFB3 could be elevated significantly through activation of PI3K/Akt signaling, thus resulting in an increase in F-2, 6-BP concentration, glucose uptake, glycolytic flux, and lactate production.47 Similarly, the inhibition of HKII mitochondria translocation, mediated by partial block of Akt, inhibited glioma cell growth under low glucose stress.33 Qian et al103 also found that the autophosphorylated phosphoglycerate kinase 1 (PGK1) was dephosphorylated and inhibited in the suppression of glycolysis via PI3K/Akt signaling. Besides, the pyruvate dehydrogenase kinase 1 (PDK1) on Thr346 would be phosphorylated by active Akt accumulates in the mitochondria.104 Interestingly, several cellular activities have been found to be relevant with glycolysis modulated by PI3K/Akt signaling. The suppression of PI3K/Akt signaling significantly inhibited glycolysis to participate in regulating the proliferation, invasion, and migration of glioma cells.84 The involvement of epithelial-mesenchymal transition (EMT) was also in PI3K/Akt signaling and glycolysis.105 Moreover, the cell-cycle progression and autophagic responses could be detected in PI3K/Akt signaling and glycolysis.64,106 More importantly, the repression of PI3K/Akt signaling promoted the accumulation of ROS and subsequent induction of apoptosis in syk-positive or syk-negative gliomas.107 Furthermore, the ERK pathway, an apoptosis-related signaling, could also be activated by PI3K/Akt signaling.102
Further investigation was performed for clinical application of PI3K/Akt signaling in modulation of glycolysis of glioma. Petunidin-3-Oglucoside (Pt3glc), an anthocyanin in the red grape and derived beverages, caused significant changes in glioma cell morphology, leading to more pronounced apoptotic cell death via PI3K/Akt signaling.102 A study by Guo et al84 revealed that miR-451 repressed PI3K/Akt signaling targeting CAB39 via downregulation of GLUT1 in glioma. In addition, some biological molecules were also found in the functional effects of PI3K/Akt signaling in glioma. Phosphatase and tensin homolog (PTEN) suppressed glycolysis targeting PGK1 or glycogen synthase kinase-3β (GSK3β) via PI3K/Akt signaling.103,108 It was also reported that WW domain-binding protein 2 (WBP2) exerted oncogenic effects by modulating PI3K/Akt signaling and targeting glycolytic enzyme enolase 1 (ENO1) in glioma.109 Rodríguez-Garcia et al47 still found that the expression of PFKFB3, upregulated by TGF-β1, was involved in cell proliferation via its role in carbohydrate metabolism. Besides, PX-866 (an effective specific inhibitor of PI3K) and YZ129 (a class of inhibitors of the calcineurin-NFAT pathway) regulated glycolysis and other cellular activities via PI3K/Akt signaling.106,110 Intriguingly, there were still a certain number of regulators targeting Akt to affect the glycolytic process. Tricyclodecan-9-yl-xanthogenate (D609) influenced cell proliferation and invasion targeting CXCR4 via interfering with AKT and EGFR.111 The pMU and pMC treatments laid an inhibitory effect on Akt, ROS induction, and an increase of cytosolic cytochrome c accumulation.26 In addition, Tp53 induced glycolysis and apoptosis regulator (TIGAR) promoted Akt activation and bound to Akt, thus promoting glycolysis of glioma cells.105 Upon G9a inhibition, PKM2-YAP1 activated autophagic activities via the Akt-HIF-1α axis in glioma.64 The activation of Akt was involved in the platelet-derived growth factor (PDGF)/PDGF receptor (PDGFR) axis controlling of glioma glycolysis.112 Moreover, signalings or miRNAs were also related to modulation of Akt in glycolysis of glioma. PERK silencing repressed HKⅡ mitochondria translocation through p-AKT knockdown, thus inhibiting glioma cell growth.33 MiR-7 suppressed colony formation of glioma cells in vitro and decreased the p-Akt expression level via insulin-like growth factor 1 receptor (IGF-1R).113 Therefore, PI3K/Akt signaling has shown its significance in modulating the glycolysis of glioma, which may be a promising therapy for glioma treatment.
3.2 mTOR Signaling
The mechanistic target of rapamycin (mTOR) signaling, usually activated in tumors, plays a significant role in tumor metabolism modulating gene transcription and protein synthesis to influence cellular activities.114,115 The mTOR, expert in integrating environmental stimulations into cellular responses, is an evolutionary conserved serine/threonine kinase.116 Additionally, mTOR could be classified into two complexes, including mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).117 The conserved components of mTORC1 include mTOR, regulatory-associated protein of mTOR (Raptor), proline-rich AKT substrate 40 kDa (PRAS40), DEP-domain-containing mTOR-interacting protein (Deptor), and mammalian lethal with Sec13 protein 8 (mLST8) whereas mTORC2 consists of mTOR, rapamycin-insensitive companion of mTOR (Rictor), stress-activated protein kinase-interacting protein (SIN1) and mLST8.118,119 The biological activities mediated by the two complexes are different due to their unique binding proteins; mTORC1 mainly influences protein, lipid, nucleotide, and glucose metabolism, autophagy, energy metabolism, lysosome biogenesis, cell survival, and cytoskeletal organization, whereas mTORC2 is involved in cell cytoskeletal remodeling, cell migration, glucose metabolism, ion transport, and cell survival.120
The activation of mTOR signaling in clinical samples were highly related to shorter survival time in glioma patients.121 Mechanically, activated glycolysis along with high glucose consumption, extracellular lactate flow, and upregulation of glycolysis-associated genes, have been detected in mTOR signaling.122 In addition, the expression of glycolytic enzymes, including HKII and PKFP, were modulated by mTOR signaling as consequences of metabolic adaptation.123 As one of the two main forms of mTOR, it has been verified that mTORC1 was paralleled with the process of glycolysis.124 Besides, mTORC2 targeted the c-myc/GFAT1 axis to regulate the interaction between glycolysis and glutaminolysis in glioma.125 mTORC2 signaling also positively promoted H3K56Ac to regulate cellular activities via phosphorylating AGC kinases.126 Clinically, chemoresistance of 1,3Bis (2-chloroethyl)-1-nitrosourea (BCNU) and cisplatin to glioma might be regulated by mTOR signaling.122 Further in vitro experiments have shown that mTOR knockdown suppressed the motility, migration, and invasive behavior of glioma cells.124 The proliferation and clone formation of glioma cells were also involved in mTOR signaling.127 More importantly, the mTOR signaling played a pivotal role in the cell-cycle arrest and induction of apoptosis.106
Surprisingly, preliminary clinical studies illustrated that rapamycin in combination with doxycycline might exert promising anti-tumor effects on glioma via mTOR signaling.123 It was also reported that metformin decreased ATP production by glycolytic process via inhibition of mTOR signaling. Interestingly, metformin in combination with temozolomide or irradiation would induce a synergistic anti-tumor response.128 Furthermore, biological molecules were found to be relevant with mTOR signaling in glycolysis of glioma. Carboxypeptidase E (CPE) activated mTORC1 signaling in glioma cells and subsequently phosphorylated ribosomal protein S6 (RPS6), thus promoting glycolysis.124 Monocarboxylic acid transporter 1 (MCT1) and YZ129 were also found to exert regulating effects on mTOR signaling axis, thus inhibiting glycolysis.106,127 Zhang et al122 disclosed that knockdown of Clock 1 (Clk1) elevated HIF-1α expression of glioma cells, which was mediated by mTOR signaling. In summary, the multiple roles of mTOR signaling have been studied in regulating glycolysis of glioma, which could be utilized for glioma treatment.
AMPK Signaling
The AMP-activated protein kinase (AMPK), a cellular metabolic sensor, participates in maintaining energy homeostasis under external stresses in multiple human cancers.129 The AMPK, expressed extensively in all eukaryotic cells, comprises of a catalytic subunit (α) and two regulatory subunits (β and γ).130,132 According to different inputs, outputs, functions, and sub-cellular localizations, a total of 12 potential AMPK complexes were assembled by various isoforms of each subunit.133 Moreover, the α subunit, phosphorylated at Thr-172, translocates a phosphate group from ATP to the Ser/Thr sites, known as a biomarker for AMPK signaling.132 The γ subunit serves as an allosteric activator, while the β subunit provides a structural bridge between α and γ subunits.134,136 Therefore, upon binding to γ-subunit, AMPK would be elevated via the allosteric activation of AMP, which could also be precisely modulated by upstream kinases.137,138 More importantly, phosphorylating of AMPK signaling depends on adenosine triphosphate ATP/AMP ratio to a great extent.139,140
Interestingly, AMPK signaling expressed a dual role to protect normal brain cells from energy stress while repressing the tumor cells.141 It has been reported that activities of glycolysis including glucose uptake, ATP levels, and lactate production were regulated by AMPK signaling in glioma cells.84 Zhang et al122 also found that the extracellular acidification rate (ECAR), an indirect indicator of lactate production and enhanced glycolytic metabolism, was significantly increased in the activation of AMPK signaling. In addition, key enzymes of the glycolytic process, such as HKII, PFK, PDK1, LDH-A, and GLUT1, depended on the vitality of AMPK signaling.122,142 Further in vitro experiments revealed that AMPK signaling was tightly related to cellular proliferation, migration, and invasion of glioma.84 Sesen et al128 also delivered a bulletin that cellular activities including cell-cycle arrest, autophagy, apoptosis, and cell death were mediated by AMPK signaling. As for tumor survival and angiogenesis, AMPK α2, one of the AMPK alpha isoforms, exerted pro-tumor effects via modulating vascular endothelial growth factor (VEGF) production on glioma patients.143 Besides, the chemoresistance of glioma cells was verified to be correlated with AMPK signaling.122 Notably, the cytotoxicity induced by EGFR inhibition could also be eliminated by AMPK signaling in glioma.144
More regulators concerning AMPK signaling have been further studied. It was verified that amentoflavone increased miR-124-3p by inhibiting DNA cytosine-5-methyltransferase 1 (DNMT1) through Sp1.145 Besides, methylene blue activated AMPK signaling and subsequently inhibited downstream targets, thus reversing the Warburg effect.146 Sesen et al128 also revealed that metformin decreased oxygen consumption and ATP production, and upregulated lactate and glycolytic ATP production through AMPK signaling. The cell cycle arrest at G0/G1 phase could also be blocked by metformin, thus promoting mitochondria-dependent apoptosis through activation of AMPK signaling.147 Moreover, 7 beta-hydroxycholesterol (7b-HC) provoked metabolic response to change lipid draft composition via AMPK activation in glioma. Interestingly, 7b-HC also influenced the affinity of PKs to their substrates.142 Surprisingly, several chemical molecules were connected with regulation of glycolysis in glioma. Clk1, encoding an enzyme concerning ubiquinone biosynthesis, elevated HIF-1α expression via AMPK signaling in glioma.122 Additionally, a mitochondrial adenylate kinase 4 (AK4) regulated ATP levels and AMPK signaling, which was connected with overall survival of glioma patients.148 Research by Guo et al149 pointed out that the AMPK agonist AICAR suppressed glioma cell proliferation via inhibiting the synthesis of cholesterol and fatty acid. Similarly, miR-451 regulated metabolism of glucose via AMPK signaling to decrease GLUT1 expression in glioma.84 JCV, a human neurotropic virus, targeted T-antigen to regulate the expression level of ROS and ATP production via AMPK signaling.150 Above all, the AMPK signaling could also be a promising target for glioma treatment for its distinctive roles in regulation of glycolysis (Figure 2).
 |
Figure 2 Interaction of major signaling pathways in aerobic glycolysis. PI3K/Akt, mTOR, and AMPK signalings have been detected to modulate the glycolytic process, thus controlling multiple cellular activities. Related intracellular and extracellular regulators were also exhibited. |
Interplay of Transcription Factors in Aerobic Glycolysis
HIF-1α
Hypoxia, the most common biological feature of most solid tumors, plays a prominent role in tumor metabolisms, especially in glycolysis.151 Hypoxia-inducible factor 1 (HIF-1), known as one of the major regulators for hypoxia, is a heterodimeric transcription factor composed of an inducible HIF-1α subunit and a constitutive HIF-1β subunit.152 Importantly, activity of HIF-1 is mainly determined by HIF-1α subunit, which is an oxygen-sensitive subunit of 120 kDa diffusely expressed in all tissues.153 Recently, more and more studies have been carried out focusing on the roles of HIF-1α in glycolysis, while glioma is no exception.154,156
HIF-1α was found to direct glucose away from mitochondria, resulting in regulatory T cells (Tregs) utilizing fatty acids for mitochondrial metabolism under hypoxia.157 It has also been recognized that HIF-1α transformed glycolysis to pentose phosphate pathway.158 Mechanically, HIF-1α induced glucose uptake and lactate accumulation, lowered intracellular ATP concentrations, and elevated mitochondrial membrane potential, which was paralleled with the expression level GLUTs, HKⅡ, PKM2, PDK1, and LDH-A.29 Interestingly, HIF-1α overexpression facilitated the VEGF pathway of vasculogenesis.29 Subsequent in vitro experiments have illustrated that a decreased cell proliferative activity and an elevated apoptosis rate were found in the activity of HIF-1α in glioma.34,62 Moreover, growth inhibition was also associated with tumor vessels reduction, enhanced levels of ROS, and autophagic responses.64,158,159 HIF-1α signaling was also involved in regulating the metabolism, tumor-specific immunity, and tumor growth in glioma.160 Regarding the effects of chemotherapy and radiotherapy, HIF-1α signaling participated in regulating effectiveness of irradiation and temozolomide of glioma via the glycolytic process.158,161
Surprisingly, more and more regulators have been detected in the functional activities of HIF-1α in glioma. The hypoxic condition could contribute to the upregulation of HIF-1α via elevating the expression of TIGAR, while prolyl hydroxylation of HIF-1α would happen under normoxic conditions.29,158 Conversely, glioma cells with IDH mutant produced more 2-hydroxyglutarate to facilitate HIF-1α degradation.162 Signalings were also key components in the activities of HIF-1α. The Wnt/β-catenin and AHR signaling could be modulated by HIF-1α regulating the tumor growth.29,160 Nodal signaling also regulated energy metabolism in glioma by elevating HIF-1α.34 Ahmad et al64 still found that G9a inhibition activated autophagic responses targeting the Akt-HIF-1α axis in glioma. Further, the von Hippel-Lindau tumor suppressor protein (pVHL) suppressed tumor development by binding to HIF-1α, subsequently suppressing HIF-1, VEGF, and Bcl-2 partially.159 Besides, miR-448 could inhibit HIF-1α signaling and then negatively control the glycolysis process in glioma.161 Hence, HIF-1α might be a promising biomarker and target for the glycolytic process of glioma.
c-Myc
Myc family proteins, comprising of c-myc, l-myc, and n-myc, are nuclear transcription factors that regulate the metabolic process, cellular proliferation, and cell-cycle progression.163 Due to their high conservation in genome, ribonucleic acid, and protein sequences, they share a similar general presentation.164 Interestingly, c-myc is expressed in many kinds of adult tissues while n-myc is found mostly in early developmental stages of neuronal tissues. L-myc is prominently located in embryonic brain, lung, and kidney, which needs further investigation.165 Noteworthily, c-myc exhibits many important functions modulating signaling transduction of cancerous cells, involving cell growth, differentiation, genome stability, survival, and angiogenesis.166,168
In metabolic imaging, glycolytic activity had a positive correlation with c-myc expression in xenograft models of glioma.169 c-myc was also reported to be involved in deoxyglucose uptake, lactate production, level of OXPHOS, and activities of the key glycolytic enzymes including HKⅡ, PFK1, PKM2, and GLUT1.79 In addition, the increased expression of amino-acid transporter 2 (ASCT2), ENO1, LDH-A, LDH-B, PDK1, and MCT1 were detected in c-myc-overexpressed glioma cells.170 The c-myc signaling is also involved in modulating the interaction between glycolysis and glutaminolysis of glioma.125 Further in vitro investigation has demonstrated that cellular proliferation, migration, and cell cycle arrest at different checkpoints participated in the activities of c-myc.171 More importantly, the expression of c-myc was relevant to brain tumorigenesis in vivo.60
In the glycolytic process, PKM2 promoted glucose metabolism of glioma targeting miRNA let-7a, promoting c-myc/hnRNPA1 axis.66 Moreover, PKM2 could induce histone H3 modifications, upregulating cyclin D1 and c-myc via EGF.60 Clinically, diclofenac and ibuprofen reduced the expression of c-myc and LDH-A via signal transducers and activators of transcription 3 (STAT-3) phosphorylation.171 Several signalings were also involved in the modulation of the expression level of c-myc. c-myc relatively elevated expression of fructose-6-phosphate aminotransferase 1 (GFAT1) targeting mTORC2 signaling.125 It was also observed that Wnt/β-catenin signaling could be activated by c-myc.29 Besides, KPNA2 elevated the expression of c-myc by translocating E2F1, thus influencing the glycolytic process.79 Kim et al170 also found that the K+ channel tetramerization domain-containing 2 (KCTD2) affected glycolysis via c-myc degradation by ubiquitination. Overall, given the crucial roles of c-myc in the progression of glioma, therapeutic interventions elevating expression of c-myc might be a reliable therapy for glioma.
p53
The p53 gene family, named as an important tumor suppressor, is composed of p53, p63, and p73, which avoid damage from widely mutated genomes under outer stress.172,173 As one of main transcription factors, p53 exerts suppressive effects on tumor cells via modulating gene transcription, facilitating cell-cycle arrest, apoptosis, and even death.174 More importantly, it is observed that p53 represses the glycolytic process and boosts mitochondrial respiration, which also participates in the regulation of several metabolic processes.175 However, p53 mutation is an event with high frequency in tumorigenesis, leading to key roles of its downstream target TIGAR in glycolysis of malignancy.175 TIGAR, highly expressed in cancers, is involved in regulating glycolysis and tumor growth redox balance mediated by pentose phosphate pathway (PPP).176,177 Therefore, p53 and TIGAR would be regulators of glycolysis, which needs to be summarized and further explored.
TIGAR overexpression has been acknowledged in gliomas, which may reduce glycolysis and induce the PPP pathway, protecting glioma cells away from oxidative damage.158,178 Moreover, generation of F-2, 6-P, lactate, and ROS levels were reduced with a concomitant elevation in glutathione (GSH) levels in TIGAR-expressed cells.178 Further in vitro experiments revealed that the cell growth and capacity of forming colonies was inhibited in TIGAR-knockdown glioma cell lines.179 As the main regulator of cell cycle, p53 could induce cell cycle arrest, which in turn facilitated Bax-dependent apoptosis.180,181 Clinically, TIGAR sensitized glioma cells to hypoxia, irradiation, and TMZ.158,182
Accumulating evidence demonstrated that the expression of TIGAR was significantly decreased in glioma patients with IDH1 mutations, accompanied by elevated expression of H3K9me3 in the initial transcription of TIGAR.182 TIGAR, upregulated by HIF-1α, could also modulate metabolic processes resulting from spontaneous homeostasis and outer stimulations.158 Interestingly, it has been detected that the expression level of TIGAR was apparently upregulated under exposure to irradiation.179 IFNγ was found to increase the expression level of retinoic acid inducible gene (RIG-I), further elevating p53 and TIGAR.183 Other interventions of manumycin or TGF-β elevated hCG-β expression of glioma cells, which abrogated expression of TIGAR.184 In addition, repressing transketolase isoenzyme transketolase-like 1 (TKTL1) could diminish activities of TIGAR.178 The functional activities of DCA were also connected with DCA-induced Foxo3 and p53 expression, contributing to high expression of Bad, Noxa, and Puma.180 Notably, Bartesaghi et al185 also reported that p53 mutations were observed alterations in ETC subunit composition and activity in glioma-initiating neural stem cells. Therefore, the suppressive roles of p53 and its downstream target TIGAR have been confirmed preliminarily, which still needs more investigation (Figure 3 and Table 2).
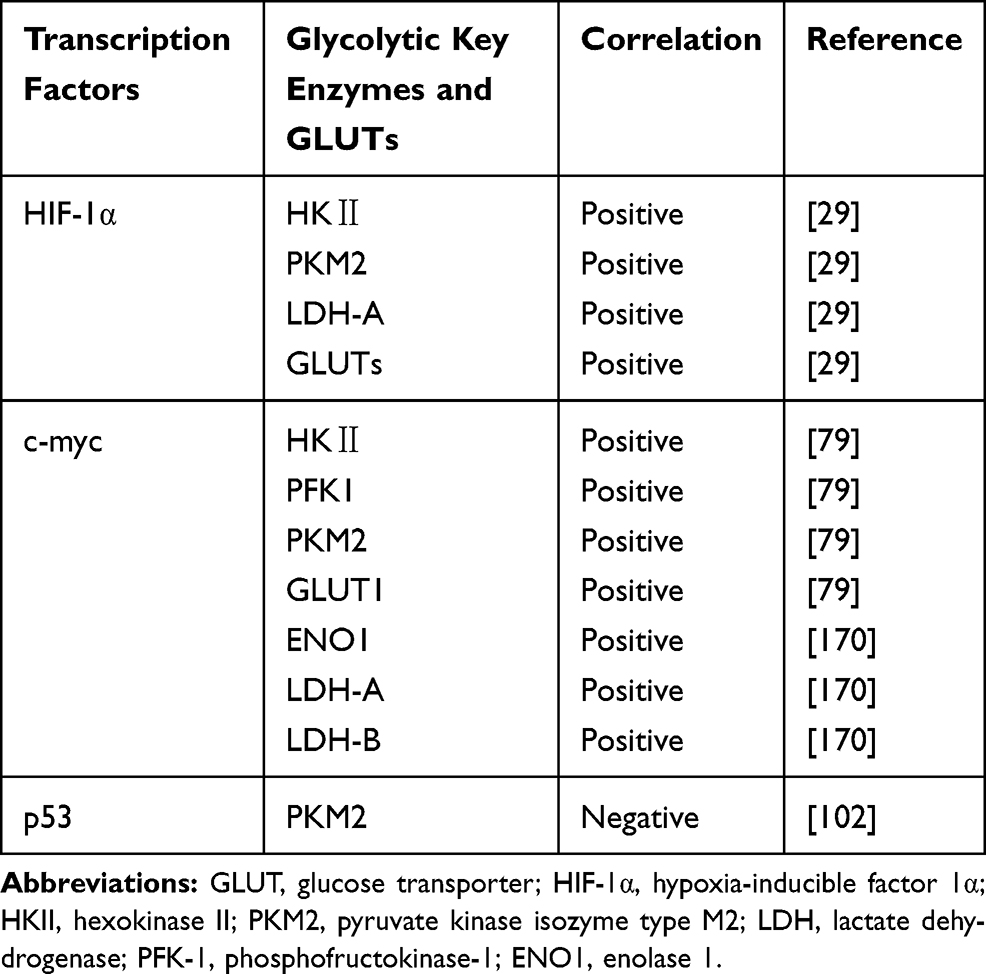 |
Table 2 Roles of Transcription Factors Modulating Glycolytic Key Regulators in Glioma |
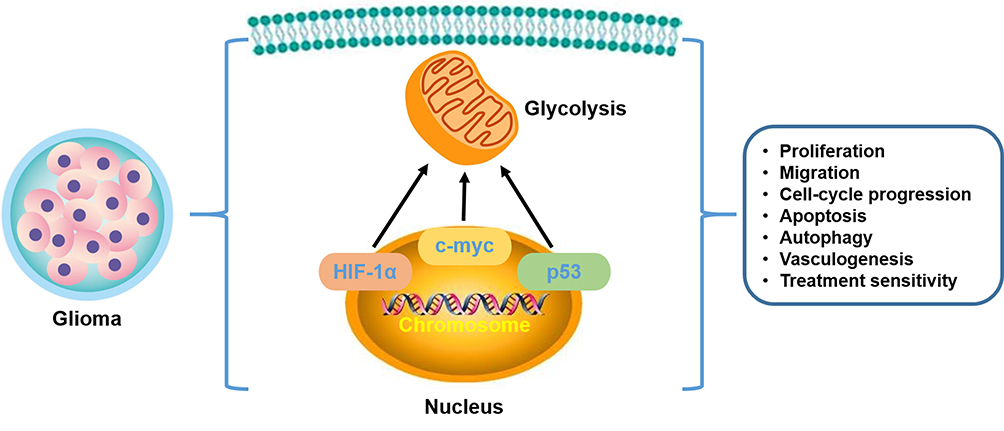 |
Figure 3 Transcription factors in aerobic glycolysis of glioma. HIF-1α, c-myc, and p53, rooted in the nucleus, play a vital role in affecting the glycolysis from the transcription level. Additionally, cellular activities are also generalized from the current investigations. |
Discussion
Over the past 50 years, disturbances in the crosstalk between glycolysis and the progression of glioma have received much attention and investigation. Accumulating evidence has illustrated that it would be promising to apply the glycolytic process for diagnosis and treatment of glioma.186 With increased number of lncRNAs, miRNAs, biological molecules and chemical drugs found in modulating glycolysis of glioma, it is of great significance to summarize the interactions between glycolysis and glioma for clinical application.
However, there are still several deficiencies and limitations in the clinical application of glycolysis for glioma treatment. Firstly, lncRNAs and miRNAs play pivotal roles in regulating glycolysis for glioma treatment, of which the clinical application still poses an obstacle in glioma. Surprisingly, lncRNAs or miRNAs themselves could serve as therapeutic molecules for treatment individualization.187,188 Apart from this, nucleic acid analogs also facilitated the application of these RNAs. Ray et al189 have found that peptide nucleic acid (PNA) specifically recognized DNA or RNA and invaded into duplex homopurine sequences of DNA, thus modulating gene expression level. Interestingly, the lncRNA-miRNA-mRNA network may also offer promising interventions for gene therapy for glioma.190 In addition, some small molecule drugs targetan offending RNA molecule or utilize anti-sense oligos to specifically interfere with RNA transcription.191
Secondly, owing to the fact that glycolysis was observed in both tumor and normal cells, targeting key enzymes and GLUTs of the glycolytic process may exert uncertain side-effects on normal tissues. For example, diclofenac and ibuprofen, as suppressive drugs in glioma treatment, may induce a series of symptoms in the upper gastrointestinal tract.192,193 Oteiza et al194,195 and Fukuda et al also reported that citrate could contribute to hypocalcemia and neurotoxicity of CNS and the peripheral nervous system (PNS). Hence, aiming at the systemic toxicity, the emergence of selective targeted delivery may provide a promising choice to settle the awkward situation.196 Moreover, it is a challenge to confirm treatment prescription according to clinical responses and toxic effects.197
Thirdly, though targeting glycolysis has been testified scientific and reasonable for glioma treatment, it is still far from acheiving clinical success before entering clinical arena. Notably, metformin was verified to possess both an inhibitory effect of glioma cells and a protective effect of neurons.128,198 Conversely, rapamycin and doxycycline were reported to exert suppressive effects via inhibition of glycolysis through mTOR signaling.123 McMahon et al199 found the neurotoxicity of rapamycin may cause abnormal activation of mTOR signaling and subsequent epilepsy in human or animal models.Therefore, it is imperative to carry out more clinical trials to detect the biological function of chemical drugs, providing evidence for further application of these drugs.
Fourthly, gathering experiments have explored the potential of combining targeted glycolysis with other treatment strategies. A study by Meng et al200,201 unfolded that decreased glycolysis could gain increased anti-cancer effects of immunotherapy via induction of immune sensitive surface ligands. Functionally, the activated glycolytic process is positively correlated with immune evasion,202,203 activities of tumor microenvironment (TME),204 and cytokine storm,205 contributing to an anti-PD-1/PD-L1 immunotherapy response.201,204 Besides, glycolysis also boosts activated immune cell proliferation, including T cells and monocytes, leading to immune tolerance and resistance.206,207 Surprisingly, the combination of glycolysis suppression with radiotherapy or chemotherapy led to the elevated sensitivity and anti-tumor effects.208,210 In addition, Fiorillo et al.211 also probed the possible application of targeting glycolysis for endocrine therapy and photothermal therapy.,212 Thus, targeting glycolysis might sensitize glioma cells to other treatment strategies, which could predict another application of the glycolytic process for glioma.
In conclusion, distinctly activated glycolysis is an inherent feature of metabolic reprogramming in glioma, which could be regulated by key enzymes (HKs, PFK-1, and PKs), GLUTs, signalings (PI3K/Akt, mTOR, and AMPK), and transcription factors (HIF-1α, c-myc, and p53). More importantly, targeted therapy, gene therapy, and immunotherapy might act as novel therapeutic approaches for application of glycolysis in glioma treatment, which still needs more clinical experiments.
Abbreviations
HK, hexokinase; PFK-1, phosphofructokinase-1; PK, pyruvate kinase; GLUT, glucose transporter; mTOR, mammalian target of rapamycin; AMPK, adenosine 5ʹ-monophosphate (AMP)-activated protein kinase; HIF-1α, hypoxia inducible factor-1α; CNS, central nervous system; WHO, World Health Organization; ATP, adenosine triphosphate; MEG3, maternally expressed gene 3; ROS, reactive oxygen species; hENT1, human equilibrative nucleoside transporter 1; JNK, c-Jun N-terminal kinase; hENT1, human equilibrated nucleoside transporter 1; G-6-P, glucose-6-phosphate; HKDC1, HK domain-containing protein 1; OXPHOS, oxidative phosphorylation; IDH, isocitrate dehydrogenase; GPX4, glutathione peroxidase 4; XBP1, X box-binding protein 1; PERK, phosphorylated extracellular regulated protein kinase; KPNA2, karyopherin α2; RPN2, ribophorin-II; 2-DG, 2-deoxy-D-glucose; F-6-P, fructose-6-phosphate; F-1, 6-BP, fructose-1, 6-biphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; PFKM, phosphofructokinase-muscle; PFKP, phosphofructokinase-platelet; PFKL, phosphofructokinase-liver; TGF-β1, transforming growth factor-β1; VDAC2, voltage-dependent anion channel 2; UCA1, urothelial cancer associated 1; CXCL14, C-X-C motif ligand 14; PEP, phosphoenolpyruvate; EGFR, epidermal growth factor receptor; HSP, heat shock protein; YAP1, Yes-associated protein 1; PDH, pyranose dehydrogenase; LDH, lactate dehydrogenase; TMZ, temozolomide; CREB1, cAMP-response element binding protein 1; CAB39, recombinant human calcium binding protein 39; PIP2, phosphatidylinositol 4, 5-bisphosphate; PIP3, phosphatidylinositol 3, 4, 5-triphosphate; RTK, receptor tyrosine kinase; FKHR, forkhead transcription factor; NAD, nicotinamide adenine dinucleotide; PGK1, phosphoglycerate kinase 1; PDK1, pyruvate dehydrogenase kinase 1; EMT, epithelial-mesenchymal transition; ERK, extracellular regulated protein kinase; Pt3glc, Petunidin-3-Oglucoside; PTEN, phosphatase and tensin homolog; GSK3β, glycogen synthase kinase-3β; WBP2, WW domain-binding protein 2; ENO1, enolase 1; NFAT, nuclear factor of activated T cells; D609, tricyclodecan-9-yl-xanthogenate; CXCR4, C-X-C motif receptor 4; TIGAR, Tp53 induced glycolysis and apoptosis regulator; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; IGF-1R, insulin-like growth factor 1 receptor; mTOR, mechanistic target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; Raptor, regulatory-associated protein of mTOR; PRAS40, proline-rich AKT substrate 40 kDa; Deptor, DEP-domain-containing mTOR-interacting protein; mLST8, mammalian lethal with Sec13 protein 8; Rictor, rapamycin-insensitive companion of mTOR; SIN1, stress-activated protein kinase-interacting protein; BCNU, 1,3Bis (2-chloroethyl)-1-nitrosourea; CPE, carboxypeptidase E; RPS6, ribosomal protein S6; MCT1, monocarboxylic acid transporter 1; Clk1, clock 1; AMPK, AMP-activated protein kinase; CBM, carbohydrate-binding module; ECAR, extracellular acidification rate; VEGF, vascular endothelial growth factor; DNMT1, DNA (cytosine-5-)-methyltransferase 1; 7b-HC, 7 beta-hydroxycholesterol; AK4, adenylate kinase 4; Tregs, regulatory T cells; PDX, patient-derived xenograft; ASCT2, amino-acid transporter 2; STAT-3, signal transducers and activators of transcription 3; GFAT1, fructose-6-phosphate aminotransferase 1; KCTD2, K+ channel tetramerization domain-containing 2; PPP, pentose phosphate pathway; GSH, glutathione; RIG-I, retinoic acid inducible gene; TKTL1, transketolase-like 1; DCA, Dichloroacetate; ETC, electron transport chain; PNS, peripheral nervous system; PNA, peptide nucleic acid; TME, tumor microenvironment.
Disclosure
The authors declare that they have no competing interests.
References
1. Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019;15(7):405–417. doi:10.1038/s41582-019-0220-2
2. Basu B, Ghosh MK. Extracellular vesicles in glioma: from diagnosis to therapy. BioEssays. 2019;41(7):e1800245. doi:10.1002/bies.201800245
3. Jung E, Alfonso J, Osswald M, Monyer H, Wick W, Winkler F. Emerging intersections between neuroscience and glioma biology. Nat Neurosci. 2019;22(12):1951–1960. doi:10.1038/s41593-019-0540-y
4. Cheng J, Meng J, Zhu L, Peng Y. Exosomal noncoding RNAs in Glioma: biological functions and potential clinical applications. Mol Cancer. 2020;19(1):66. doi:10.1186/s12943-020-01189-3
5. Chen X, Mao YG, Yu ZQ, Wu J, Chen G. Potential rules of anesthetic gases on glioma. Med Gas Res. 2020;10(1):50–53. doi:10.4103/2045-9912.279984
6. Huang M, Xiong H, Luo D, Xu B, Liu H. CSN5 upregulates glycolysis to promote hepatocellular carcinoma metastasis via stabilizing the HK2 protein. Exp Cell Res. 2020;388(2):111876. doi:10.1016/j.yexcr.2020.111876
7. Xia M, Feng S, Chen Z, Wen G, Zu X, Zhong J. Non-coding RNAs: key regulators of aerobic glycolysis in breast cancer. Life Sci. 2020;250:117579. doi:10.1016/j.lfs.2020.117579
8. Marin-Hernandez A, Gallardo-Perez JC, Rodriguez-Enriquez S, Encalada R, Moreno-Sanchez R, Saavedra E. Modeling cancer glycolysis. Biochim Biophys Acta. 2011;1807(6):755–767. doi:10.1016/j.bbabio.2010.11.006
9. Yu X, Gao X, Mao X, et al. Knockdown of FOXO6 Inhibits Glycolysis and Reduces Cell Resistance to Paclitaxel in HCC Cells via PI3K/Akt Signaling Pathway. Onco Targets Ther. 2020;13:1545–1556. doi:10.2147/OTT.S233031
10. Shi D, Zhao D, Niu P, Zhu Y, Zhou J, Chen H. Glycolysis inhibition via mTOR suppression is a key step in cardamonin-induced autophagy in SKOV3 cells. BMC Complement Altern Med. 2018;18(1):317. doi:10.1186/s12906-018-2380-9
11. Fujiwara H, Tateishi K, Misumi K, et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci Rep. 2019;9(1):18859. doi:10.1038/s41598-019-55211-w
12. Chang CC, Zhang C, Zhang Q, et al. Upregulation of lactate dehydrogenase a by 14-3-3zeta leads to increased glycolysis critical for breast cancer initiation and progression. Oncotarget. 2016;7(23):35270–35283. doi:10.18632/oncotarget.9136
13. Wikoff WR, Grapov D, Fahrmann JF, et al. Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prev Res. 2015;8(5):410–418. doi:10.1158/1940-6207.CAPR-14-0329
14. Zuo S, Wu L, Wang Y, Yuan X. Long Non-coding RNA MEG3 Activated by Vitamin D Suppresses Glycolysis in Colorectal Cancer via Promoting c-Myc Degradation. Front Oncol. 2020;10:274. doi:10.3389/fonc.2020.00274
15. Chen T, Zhao L, Chen S, et al. The curcumin analogue WZ35 affects glycolysis inhibition of gastric cancer cells through ROS-YAP-JNK pathway. Food Chem Toxicol. 2020;137:111131. doi:10.1016/j.fct.2020.111131
16. Xi Y, Yuan P, Li T, Zhang M, Liu MF, Li B. hENT1 reverses chemoresistance by regulating glycolysis in pancreatic cancer. Cancer Lett. 2020;479:112–122. doi:10.1016/j.canlet.2020.03.015
17. Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–964. doi:10.1080/15548627.2015.1042195
18. Miccoli L, Oudard S, Sureau F, Poirson F, Dutrillaux B, Poupon MF. Intracellular pH governs the subcellular distribution of hexokinase in a glioma cell line. Biochem J. 1996;313(Pt 3):957–962. doi:10.1042/bj3130957
19. Irwin DM, Tan H. Molecular evolution of the vertebrate hexokinase gene family: identification of a conserved fifth vertebrate hexokinase gene. Comp Biochem Physiol Part D Genomics Proteomics. 2008;3(1):96–107. doi:10.1016/j.cbd.2007.11.002
20. Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging roles of aerobic glycolysis in breast cancer. Clin Transl Oncol. 2019:1–16.
21. Garcia SN, Guedes RC, Marques MM. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr Med Chem. 2019;26(41):7285–7322. doi:10.2174/0929867326666181213092652
22. Saga I, Shibao S, Okubo J, et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014;16(8):1048–1056. doi:10.1093/neuonc/nou096
23. Cao L, Wang M, Dong Y, et al. Circular RNA circRNF20 promotes breast cancer tumorigenesis and Warburg effect through miR-487a/HIF-1alpha/HK2. Cell Death Dis. 2020;11(2):145. doi:10.1038/s41419-020-2336-0
24. Dai W, Meng X, Mo S, et al. FOXE1 represses cell proliferation and Warburg effect by inhibiting HK2 in colorectal cancer. Cell Commun Signal. 2020;18(1):7. doi:10.1186/s12964-019-0502-8
25. Liu H, Liu N, Cheng Y, et al. Hexokinase 2 (HK2), the tumor promoter in glioma, is downregulated by miR-218/Bmi1 pathway. PLoS One. 2017;12(12):e0189353. doi:10.1371/journal.pone.0189353
26. Ponnala S, Chetty C, Veeravalli KK, Dinh DH, Klopfenstein JD, Rao JS. Metabolic remodeling precedes mitochondrial outer membrane permeabilization in human glioma xenograft cells. Int J Oncol. 2012;40(2):509–518. doi:10.3892/ijo.2011.1255
27. Valentin-Guillama G, Lopez S, Kucheryavykh YV, et al. HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers. 2018;10(9):9. doi:10.3390/cancers10090301
28. Nie Q, Guo P, Guo L, et al. Overexpression of isocitrate dehydrogenase-1R(1)(3)(2)H enhances the proliferation of A172 glioma cells via aerobic glycolysis. Mol Med Rep. 2015;11(5):3715–3721. doi:10.3892/mmr.2015.3187
29. Vallee A, Guillevin R, Vallee JN. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/beta-catenin pathway in gliomas. Rev Neurosci. 2018;29(1):71–91. doi:10.1515/revneuro-2017-0032
30. El Sayed SM, Abou El-Magd RM, Shishido Y, et al. D-amino acid oxidase gene therapy sensitizes glioma cells to the antiglycolytic effect of 3-bromopyruvate. Cancer Gene Ther. 2012;19(1):1–18. doi:10.1038/cgt.2011.59
31. Wang X, Lu S, He C, et al. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem Biophys Res Commun. 2019;518(3):590–597. doi:10.1016/j.bbrc.2019.08.096
32. Liu Y, Hou X, Liu M, et al. XBP1 silencing decreases glioma cell viability and glycolysis possibly by inhibiting HK2 expression. J Neurooncol. 2016;126(3):455–462. doi:10.1007/s11060-015-2003-y
33. Hou X, Liu Y, Liu H, et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci Rep. 2015;5:9065. doi:10.1038/srep09065
34. Lai JH, Jan HJ, Liu LW, et al. Nodal regulates energy metabolism in glioma cells by inducing expression of hypoxia-inducible factor 1alpha. Neuro Oncol. 2013;15(10):1330–1341. doi:10.1093/neuonc/not086
35. Li H, Yuan H. MiR-1297 negatively regulates metabolic reprogramming in glioblastoma via repressing KPNA2. Hum Cell. 2020:2:1.
36. Ding C, Wu Z, You H, et al. CircNFIX promotes progression of glioma through regulating miR-378e/RPN2 axis. J Exp Clin Cancer Res. 2019;38(1):506. doi:10.1186/s13046-019-1483-6
37. Zhao S, Liu H, Liu Y, et al. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013;333(2):253–260. doi:10.1016/j.canlet.2013.01.039
38. Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381(Pt 3):561–579. doi:10.1042/BJ20040752
39. Schmitz JP, Groenendaal W, Wessels B, et al. Combined in vivo and in silico investigations of activation of glycolysis in contracting skeletal muscle. Am J Physiol Cell Physiol. 2013;304(2):C180–193. doi:10.1152/ajpcell.00101.2012
40. Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol. 2011;76:211–216. doi:10.1101/sqb.2011.76.010868
41. Mulukutla BC, Yongky A, Daoutidis P, Hu WS. Bistability in glycolysis pathway as a physiological switch in energy metabolism. PLoS One. 2014;9(6):e98756. doi:10.1371/journal.pone.0098756
42. Lang L, Chemmalakuzhy R, Shay C, Teng Y. PFKP Signaling at a Glance: an Emerging Mediator of Cancer Cell Metabolism. Adv Exp Med Biol. 2019;1134:243–258.
43. Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86(3):174–179. doi:10.1016/j.yexmp.2009.01.003
44. Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr. 2007;39(3):223–229. doi:10.1007/s10863-007-9080-3
45. Goidts V, Bageritz J, Puccio L, et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012;31(27):3235–3243. doi:10.1038/onc.2011.490
46. Dominguez JE, Graham JF, Cummins CJ, et al. Enzymes of glucose metabolism in cultured human gliomas: neoplasia is accompanied by altered hexokinase, phosphofructokinase, and glucose-6-phosphate dehydrogenase levels. Metab Brain Dis. 1987;2(1):17–30. doi:10.1007/BF00999506
47. Rodriguez-Garcia A, Samso P, Fontova P, et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017;284(20):3437–3454. doi:10.1111/febs.14201
48. El Sayed SM, El-Magd RM, Shishido Y, et al. 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J Bioenerg Biomembr. 2012;44(1):61–79. doi:10.1007/s10863-012-9409-4
49. El Sayed SM, El-Magd RM, Shishido Y, et al. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J Bioenerg Biomembr. 2012;44(5):513–523. doi:10.1007/s10863-012-9455-y
50. Zhou K, Yao YL, He ZC, et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018;9(10):988. doi:10.1038/s41419-018-1015-x
51. He Z, You C, Zhao D. Long non-coding RNA UCA1/miR-182/PFKFB2 axis modulates glioblastoma-associated stromal cells-mediated glycolysis and invasion of glioma cells. Biochem Biophys Res Commun. 2018;500(3):569–576. doi:10.1016/j.bbrc.2018.04.091
52. Xu D, Liang J, Lin J, Yu C. PKM2: A Potential Regulator of Rheumatoid Arthritis via Glycolytic and Non-Glycolytic Pathways. Front Immunol. 2019;10:2919. doi:10.3389/fimmu.2019.02919
53. Gupta V, Bamezai RN. Human pyruvate kinase M2: a multifunctional protein. Protein Sci. 2010;19(11):2031–2044. doi:10.1002/pro.505
54. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, function and expression and regulation. Cell Biosci. 2019;9(1):52. doi:10.1186/s13578-019-0317-8
55. Schormann N, Hayden KL, Lee P, Banerjee S, Chattopadhyay D. An overview of structure, function, and regulation of pyruvate kinases. Protein Sci. 2019;28(10):1771–1784.
56. Alquraishi M, Puckett DL, Alani DS, et al. Pyruvate kinase M2: A simple molecule with complex functions. Free Radic Biol Med. 2019;143:176–192. doi:10.1016/j.freeradbiomed.2019.08.007
57. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108(38):16062–16067. doi:10.1073/pnas.1106704108
58. Liu VM, Vander Heiden MG. The Role of Pyruvate Kinase M2 in Cancer Metabolism. Brain Pathol. 2015;25(6):781–783. doi:10.1111/bpa.12311
59. Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480(7375):118–122. doi:10.1038/nature10598
60. Yang W, Xia Y, Hawke D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150(4):685–696. doi:10.1016/j.cell.2012.07.018
61. Liu X, Zhu Q, Guo Y, Xiao Z, Hu L, Xu Q. LncRNA LINC00689 promotes the growth, metastasis and glycolysis of glioma cells by targeting miR-338-3p/PKM2 axis. Biomed Pharmacother. 2019;117:109069. doi:10.1016/j.biopha.2019.109069
62. Xu G, Bai X, Wang M, Xie W, Li R, Li C. [Characterization of glycolytic phenotype of SHG44 human glioma cells under hypoxic conditions and its association with cell proliferation and apoptosis]. Nan Fang Yi Ke Da Xue Xue Bao. 2013;33(3):406–411. Chinese.
63. Liang J, Cao R, Wang X, et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017;27(3):329–351. doi:10.1038/cr.2016.159
64. Ahmad F, Dixit D, Joshi SD, Sen E. G9a inhibition induced PKM2 regulates autophagic responses. Int J Biochem Cell Biol. 2016;78:87–95. doi:10.1016/j.biocel.2016.07.009
65. Yang W, Xia Y, Cao Y, et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell. 2012;48(5):771–784. doi:10.1016/j.molcel.2012.09.028
66. Luan W, Wang Y, Chen X, et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015;6(15):13006–13018. doi:10.18632/oncotarget.3514
67. Zhao M, Zhang Z. Glucose Transporter Regulation in Cancer: A Profile and the Loops. Crit Rev Eukaryot Gene Expr. 2016;26(3):223–238. doi:10.1615/CritRevEukaryotGeneExpr.2016016531
68. Alam F, Islam MA, Khalil MI, Gan SH. Metabolic Control of Type 2 Diabetes by Targeting the GLUT4 Glucose Transporter: intervention Approaches. Curr Pharm Des. 2016;22(20):3034–3049. doi:10.2174/1381612822666160307145801
69. Pujol-Gimenez J, Barrenetxe J, Gonzalez-Muniesa P, Lostao MP. The facilitative glucose transporter GLUT12: what do we know and what would we like to know? J Physiol Biochem. 2013;69(2):325–333. doi:10.1007/s13105-012-0213-8
70. Ancey PB, Contat C, Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018;285(16):2926–2943. doi:10.1111/febs.14577
71. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121–138. doi:10.1016/j.mam.2012.07.001
72. Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res. 1997;49(5):617–626.
73. Yeh WL, Lin CJ, Fu WM. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol. 2008;73(1):170–177. doi:10.1124/mol.107.038851
74. Bilan PJ, Mitsumoto Y, Maher F, Simpson IA, Klip A. Detection of the GLUT3 facilitative glucose transporter in rat L6 muscle cells: regulation by cellular differentiation, insulin and insulin-like growth factor-I. Biochem Biophys Res Commun. 1992;186(2):1129–1137. doi:10.1016/0006-291X(92)90864-H
75. McVie-Wylie AJ, Lamson DR, Chen YT. Molecular cloning of a novel member of the GLUT family of transporters, SLC2a10 (GLUT10), localized on chromosome 20q13.1: a candidate gene for NIDDM susceptibility. Genomics. 2001;72(1):113–117. doi:10.1006/geno.2000.6457
76. Sayem ASM, Arya A, Karimian H, Krishnasamy N, Ashok Hasamnis A, Hossain CF. Action of Phytochemicals on Insulin Signaling Pathways Accelerating Glucose Transporter (GLUT4) Protein Translocation. Molecules. 2018;23(2):2. doi:10.3390/molecules23020258
77. Boado RJ, Black KL, Pardridge WM. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Brain Res Mol Brain Res. 1994;27(1):51–57. doi:10.1016/0169-328X(94)90183-X
78. Maher F, Davies-Hill TM, Lysko PG, Henneberry RC, Simpson IA. Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron-specific expression of GLUT3. Mol Cell Neurosci. 1991;2(4):351–360. doi:10.1016/1044-7431(91)90066-W
79. Li J, Liu Q, Liu Z, et al. KPNA2 promotes metabolic reprogramming in glioblastomas by regulation of c-myc. J Exp Clin Cancer Res. 2018;37(1):194. doi:10.1186/s13046-018-0861-9
80. Chen J, Zhang C, Mi Y, Chen F, Du D. CREB1 regulates glucose transport of glioma cell line U87 by targeting GLUT1. Mol Cell Biochem. 2017;436(1–2):79–86. doi:10.1007/s11010-017-3080-3
81. Libby CJ, Zhang S, Benavides GA, et al. Identification of Compounds That Decrease Glioblastoma Growth and Glucose Uptake in Vitro. ACS Chem Biol. 2018;13(8):2048–2057. doi:10.1021/acschembio.8b00251
82. Charnley N, Airley R, Du Plessis D, et al. No relationship between 18F-fluorodeoxyglucose positron emission tomography and expression of Glut-1 and −3 and hexokinase I and II in high-grade glioma. Oncol Rep. 2008;20(3):537–542.
83. Lee JH, Liu R, Li J, et al. EGFR-Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol Cell. 2018;70(2):197–210.
84. Guo H, Nan Y, Zhen Y, et al. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour Biol. 2016;37(10):13751–13761. doi:10.1007/s13277-016-5219-3
85. Dai DW, Lu Q, Wang LX, et al. Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2A3 in GBM. BMC Cancer. 2013;13:478. doi:10.1186/1471-2407-13-478
86. Kuang R, Jahangiri A, Mascharak S, et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight. 2017;2(2):e88815. doi:10.1172/jci.insight.88815
87. Gorin F, Harley W, Schnier J, Lyeth B, Jue T. Perinecrotic glioma proliferation and metabolic profile within an intracerebral tumor xenograft. Acta Neuropathol. 2004;107(3):235–244. doi:10.1007/s00401-003-0803-1
88. Shibuya K, Okada M, Suzuki S, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015;6(2):651–661. doi:10.18632/oncotarget.2892
89. Azzalin A, Nato G, Parmigiani E, Garello F, Buffo A, Magrassi L. Inhibitors of GLUT/SLC2A Enhance the Action of BCNU and Temozolomide against High-Grade Gliomas. Neoplasia. 2017;19(4):364–373. doi:10.1016/j.neo.2017.02.009
90. Lehman JA, Hauck PM, Gendron JM, et al. Serdemetan antagonizes the Mdm2-HIF1alpha axis leading to decreased levels of glycolytic enzymes. PLoS One. 2013;8(9):e74741. doi:10.1371/journal.pone.0074741
91. Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005;65(3):999–1006.
92. Nie S, Li K, Huang Y, Hu Q, Gao X, Jie S. miR-495 mediates metabolic shift in glioma cells via targeting Glut1. J Craniofac Surg. 2015;26(2):e155–158. doi:10.1097/SCS.0000000000001385
93. Liu Y, Li YM, Tian RF, et al. The expression and significance of HIF-1alpha and GLUT-3 in glioma. Brain Res. 2009;1304:149–154. doi:10.1016/j.brainres.2009.09.083
94. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: biological and therapeutic significance. Semin Cancer Biol. 2019;59:147–160. doi:10.1016/j.semcancer.2019.05.012
95. Rahmani F, Ferns GA, Talebian S, Nourbakhsh M, Avan A, Shahidsales S. Role of regulatory miRNAs of the PI3K/AKT signaling pathway in the pathogenesis of breast cancer. Gene. 2020;737:144459. doi:10.1016/j.gene.2020.144459
96. Xu Z, Han X, Ou D, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–587. doi:10.1007/s00253-019-10257-8
97. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. doi:10.1038/nrd2926
98. Rahmani F, Ziaeemehr A, Shahidsales S, et al. Role of regulatory miRNAs of the PI3K/AKT/mTOR signaling in the pathogenesis of hepatocellular carcinoma. J Cell Physiol. 2020;235(5):4146–4152. doi:10.1002/jcp.29333
99. Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–395. doi:10.1016/S0898-6568(01)00271-6
100. Chamcheu JC, Roy T, Uddin MB, et al. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells. 2019;8(8):8. doi:10.3390/cells8080803
101. Mirza-Aghazadeh-Attari M, Ekrami EM, Aghdas SAM, et al. Targeting PI3K/Akt/mTOR signaling pathway by polyphenols: implication for cancer therapy. Life Sci. 2020;255:117481. doi:10.1016/j.lfs.2020.117481
102. Wang G, Fu XL, Wang JJ, Guan R, Sun Y, Tony To -S-S. Tony To SS: inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt-MAPK pathway. J Cell Physiol. 2019;234(5):5888–5903. doi:10.1002/jcp.26474
103. Qian X, Li X, Shi Z, et al. PTEN Suppresses Glycolysis by Dephosphorylating and Inhibiting Autophosphorylated PGK1. Mol Cell. 2019;76(3):516–527. doi:10.1016/j.molcel.2019.08.006
104. Chae YC, Vaira V, Caino MC, et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell. 2016;30(2):257–272. doi:10.1016/j.ccell.2016.07.004
105. Tang Z, He Z. TIGAR promotes growth, survival and metastasis through oxidation resistance and AKT activation in glioblastoma. Oncol Lett. 2019;18(3):2509–2517. doi:10.3892/ol.2019.10574
106. Liu Z, Li H, He L, et al. Discovery of Small-Molecule Inhibitors of the HSP90-Calcineurin-NFAT Pathway against Glioblastoma. Cell Chem Biol. 2019;26(3):352–365. doi:10.1016/j.chembiol.2018.11.009
107. Sun S, Xue D, Chen Z, et al. R406 elicits anti-Warburg effect via Syk-dependent and -independent mechanisms to trigger apoptosis in glioma stem cells. Cell Death Dis. 2019;10(5):358. doi:10.1038/s41419-019-1587-0
108. Beckner ME, Gobbel GT, Abounader R, et al. Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab Invest. 2005;85(12):1457–1470. doi:10.1038/labinvest.3700355
109. Chen S, Zhang Y, Wang H, et al. WW domain-binding protein 2 acts as an oncogene by modulating the activity of the glycolytic enzyme ENO1 in glioma. Cell Death Dis. 2018;9(3):347. doi:10.1038/s41419-018-0376-5
110. Koul D, Shen R, Kim YW, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol. 2010;12(6):559–569. doi:10.1093/neuonc/nop058
111. Mercurio L, Cecchetti S, Ricci A, et al. Phosphatidylcholine-specific phospholipase C inhibition down- regulates CXCR4 expression and interferes with proliferation, invasion and glycolysis in glioma cells. PLoS One. 2017;12(4):e0176108. doi:10.1371/journal.pone.0176108
112. Ran C, Liu H, Hitoshi Y, Israel MA. Israel MA: proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res. 2013;73(6):1831–1843. doi:10.1158/0008-5472.CAN-12-2460
113. Wang B, Sun F, Dong N, et al. MicroRNA-7 directly targets insulin-like growth factor 1 receptor to inhibit cellular growth and glucose metabolism in gliomas. Diagn Pathol. 2014;9(1):211. doi:10.1186/s13000-014-0211-y
114. Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10(1):31. doi:10.1186/s13578-020-00396-1
115. Mirabilii S, Ricciardi MR, Tafuri A. mTOR Regulation of Metabolism in Hematologic Malignancies. Cells. 2020;9(2):2. doi:10.3390/cells9020404
116. Ferrin G, Guerrero M, Amado V, Rodriguez-Peralvarez M, De la Mata M. De la Mata M: activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int J Mol Sci. 2020;21(4):4. doi:10.3390/ijms21041266
117. Hodges SL, Lugo JN. Therapeutic role of targeting mTOR signaling and neuroinflammation in epilepsy. Epilepsy Res. 2020;161:106282. doi:10.1016/j.eplepsyres.2020.106282
118. Woo Y, Lee HJ, Jung YM, Jung YJ. mTOR-Mediated Antioxidant Activation in Solid Tumor Radioresistance. J Oncol. 2019;2019:5956867. doi:10.1155/2019/5956867
119. Magaway C, Kim E, Jacinto E. Targeting mTOR and Metabolism in Cancer: lessons and Innovations. Cells. 2019;8(12):12. doi:10.3390/cells8121584
120. Zhang T, Guo J, Gu J, Chen K, Li H, Wang J. Protective Role of mTOR in Liver Ischemia/Reperfusion Injury: involvement of Inflammation and Autophagy. Oxid Med Cell Longev. 2019;2019:7861290. doi:10.1155/2019/7861290
121. Masui K, Tanaka K, Akhavan D, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–739. doi:10.1016/j.cmet.2013.09.013
122. Zhang L, Yang H, Zhang W, et al. Clk1-regulated aerobic glycolysis is involved in glioma chemoresistance. J Neurochem. 2017;142(4):574–588. doi:10.1111/jnc.14096
123. Petovari G, Hujber Z, Krencz I, et al. Targeting cellular metabolism using rapamycin and/or doxycycline enhances anti-tumour effects in human glioma cells. Cancer Cell Int. 2018;18(1):211. doi:10.1186/s12935-018-0710-0
124. Ilina EI, Armento A, Sanchez LG, et al. Effects of soluble CPE on glioma cell migration are associated with mTOR activation and enhanced glucose flux. Oncotarget. 2017;8(40):67567–67591. doi:10.18632/oncotarget.18747
125. Liu B, Huang ZB, Chen X, See YX, Chen ZK, Yao HK. Mammalian Target of Rapamycin 2 (MTOR2) and C-MYC Modulate Glucosamine-6-Phosphate Synthesis in Glioblastoma (GBM) Cells Through Glutamine: fructose-6-Phosphate Aminotransferase 1 (GFAT1). Cell Mol Neurobiol. 2019;39(3):415–434. doi:10.1007/s10571-019-00659-7
126. Vadla R, Haldar D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation. Cell Cycle. 2018;17(1):110–123. doi:10.1080/15384101.2017.1404207
127. Zhou HG, Zhang JD, Zhang YF. [The effect of downregulation of MCT1 on the proliferation of glioma cells]. Zhonghua Zhong Liu Za Zhi. 2019;41(3):208–213. Chinese. doi:10.3760/cma.j.issn.0253-3766.2019.03.010
128. Sesen J, Dahan P, Scotland SJ, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS One. 2015;10(4):e0123721. doi:10.1371/journal.pone.0123721
129. Yung MM, Ngan HY, Chan DW. Targeting AMPK signaling in combating ovarian cancers: opportunities and challenges. Acta Biochim Biophys Sin (Shanghai). 2016;48(4):301–317. doi:10.1093/abbs/gmv128
130. Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20(6):939–952. doi:10.1016/j.cmet.2014.09.013
131. Hardie DG, Ross FA, Hawley SA. Hawley SA: AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi:10.1038/nrm3311
132. Joshi T, Singh AK, Haratipour P, et al. Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications. J Cell Physiol. 2019;234(10):17212–17231. doi:10.1002/jcp.28528
133. Curry DW, Stutz B, Andrews ZB, Elsworth JD. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J Parkinson’s Dis. 2018;8(2):161–181. doi:10.3233/JPD-171296
134. Liu J, Li X, Lu Q, et al. AMPK: a balancer of the renin-angiotensin system. Biosci Rep. 2019;39(9):9. doi:10.1042/BSR20181994
135. Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472(7342):230–233. doi:10.1038/nature09932
136. Gu C, Li T, Jiang S, et al. AMP-activated protein kinase sparks the fire of cardioprotection against myocardial ischemia and cardiac ageing. Ageing Res Rev. 2018;47:168–175. doi:10.1016/j.arr.2018.08.002
137. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403(1):139–148. doi:10.1042/BJ20061520
138. Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346(Pt 3):659–669. doi:10.1042/bj3460659
139. Camici M, Allegrini S, Tozzi MG. Interplay between adenylate metabolizing enzymes and AMP-activated protein kinase. FEBS J. 2018;285(18):3337–3352. doi:10.1111/febs.14508
140. Tain YL, Hsu CN. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. Int J Mol Sci. 2018;19:6. doi:10.3390/ijms19061744
141. Mukherjee P, Mulrooney TJ, Marsh J, Blair D, Chiles TC, Seyfried TN. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol Cancer. 2008;7(1):37. doi:10.1186/1476-4598-7-37
142. de Weille J, Fabre C, Gaven C, Bakalara N. Similar pyruvate kinase modifications in glioblastoma cells by 7beta-hydroxycholesterol and glutamine withdrawal. Biochem Pharmacol. 2013;86(1):161–167. doi:10.1016/j.bcp.2013.03.012
143. Neurath KM, Keough MP, Mikkelsen T, Claffey KP. AMP-dependent protein kinase alpha 2 isoform promotes hypoxia-induced VEGF expression in human glioblastoma. Glia. 2006;53(7):733–743. doi:10.1002/glia.20326
144. Hartel I, Ronellenfitsch M, Wanka C, Wolking S, Steinbach JP, Rieger J. Activation of AMP-activated kinase modulates sensitivity of glioma cells against epidermal growth factor receptor inhibition. Int J Oncol. 2016;49(1):173–180. doi:10.3892/ijo.2016.3498
145. Zhaohui W, Yingli N, Hongli L, et al. Amentoflavone induces apoptosis and suppresses glycolysis in glioma cells by targeting miR-124-3p. Neurosci Lett. 2018;686:1–9. doi:10.1016/j.neulet.2018.08.032
146. Yang SH, Li W, Sumien N, Forster M, Simpkins JW, Liu R. Alternative mitochondrial electron transfer for the treatment of neurodegenerative diseases and cancers: methylene blue connects the dots. Prog Neurobiol. 2017;157:273–291. doi:10.1016/j.pneurobio.2015.10.005
147. Isakovic A, Harhaji L, Stevanovic D, et al. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci. 2007;64(10):1290–1302. doi:10.1007/s00018-007-7080-4
148. Lanning NJ, Looyenga BD, Kauffman AL, et al. A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep. 2014;7(3):907–917. doi:10.1016/j.celrep.2014.03.065
149. Guo D, Hildebrandt IJ, Prins RM, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009;106(31):12932–12937. doi:10.1073/pnas.0906606106
150. Noch E, Sariyer IK, Gordon J, Khalili K. JC virus T-antigen regulates glucose metabolic pathways in brain tumor cells. PLoS One. 2012;7(4):e35054. doi:10.1371/journal.pone.0035054
151. Peng X, Gao H, Xu R, Wang H, Mei J, Liu C. The interplay between HIF-1alpha and noncoding RNAs in cancer. J Exp Clin Cancer Res. 2020;39(1):27. doi:10.1186/s13046-020-1535-y
152. Zhao Z, Mu H, Li Y, Liu Y, Zou J, Zhu Y. Clinicopathological and prognostic value of hypoxia-inducible factor-1alpha in breast cancer: a meta-analysis including 5177 patients. Clin Transl Oncol. 2020. doi:10.1007/s12094-020-02332-8
153. Tang W, Zhao G. Small molecules targeting HIF-1alpha pathway for cancer therapy in recent years. Bioorg Med Chem. 2020;28(2):115235. doi:10.1016/j.bmc.2019.115235
154. Sharma M, Boytard L, Hadi T, et al. Enhanced glycolysis and HIF-1alpha activation in adipose tissue macrophages sustains local and systemic interleukin-1beta production in obesity. Sci Rep. 2020;10(1):5555. doi:10.1038/s41598-020-62272-9
155. Lu Y, Wang L, Ding W, et al. Ammonia mediates mitochondrial uncoupling and promotes glycolysis via HIF-1 activation in human breast cancer MDA-MB-231cells. Biochem Biophys Res Commun. 2019;519(1):153–159. doi:10.1016/j.bbrc.2019.08.152
156. Zhu W, Li Y, Zhao D, et al. Dihydroartemisinin suppresses glycolysis of LNCaP cells by inhibiting PI3K/AKT pathway and downregulating HIF-1alpha expression. Life Sci. 2019;233:116730. doi:10.1016/j.lfs.2019.116730
157. Miska J, Lee-Chang C, Rashidi A, et al. HIF-1alpha Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019;27(1):226–237. doi:10.1016/j.celrep.2019.03.029
158. Maurer GD, Heller S, Wanka C, Rieger J, Steinbach JP. Knockdown of the TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) Sensitizes Glioma Cells to Hypoxia, Irradiation and Temozolomide. Int J Mol Sci. 2019;20(5):5. doi:10.3390/ijms20051061
159. Sun X, Liu M, Wei Y, et al. Overexpression of von Hippel-Lindau tumor suppressor protein and antisense HIF-1alpha eradicates gliomas. Cancer Gene Ther. 2006;13(4):428–435. doi:10.1038/sj.cgt.7700907
160. Gabriely G, Wheeler MA, Takenaka MC, Quintana FJ. Role of AHR and HIF-1alpha in Glioblastoma Metabolism. Trends Endocrinol Metab. 2017;28(6):428–436. doi:10.1016/j.tem.2017.02.009
161. Lan F, Qin Q, Yu H, Yue X. Effect of glycolysis inhibition by miR-448 on glioma radiosensitivity. J Neurosurg. 2019;1–9.
162. Chesnelong C, Chaumeil MM, Blough MD, et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014;16(5):686–695. doi:10.1093/neuonc/not243
163. Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int J Mol Sci. 2019;20(9):9. doi:10.3390/ijms20092340
164. Chanvorachote P, Sriratanasak N, Nonpanya N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020;40(2):609–618. doi:10.21873/anticanres.13990
165. Bragelmann J, Bohm S, Guthrie MR, Mollaoglu G, Oliver TG, Sos ML. Family matters: how MYC family oncogenes impact small cell lung cancer. Cell Cycle. 2017;16(16):1489–1498. doi:10.1080/15384101.2017.1339849
166. Baudino TA, McKay C, Pendeville-Samain H, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16(19):2530–2543. doi:10.1101/gad.1024602
167. Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, Felsher DW. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc Natl Acad Sci U S A. 2003;100(17):9974–9979. doi:10.1073/pnas.1732638100
168. Sipos F, Firneisz G, Muzes G. Therapeutic aspects of c-MYC signaling in inflammatory and cancerous colonic diseases. World j Gastroenterol. 2016;22(35):7938–7950. doi:10.3748/wjg.v22.i35.7938
169. Mair R, Wright AJ, Ros S, et al. Metabolic Imaging Detects Low Levels of Glycolytic Activity That Vary with Levels of c-Myc Expression in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2018;78(18):5408–5418. doi:10.1158/0008-5472.CAN-18-0759
170. Kim EJ, Kim SH, Jin X, Jin X, Kim H. KCTD2, an adaptor of Cullin3 E3 ubiquitin ligase, suppresses gliomagenesis by destabilizing c-Myc. Cell Death Differ. 2017;24(4):649–659. doi:10.1038/cdd.2016.151
171. Leidgens V, Seliger C, Jachnik B, et al. Ibuprofen and Diclofenac Restrict Migration and Proliferation of Human Glioma Cells by Distinct Molecular Mechanisms. PLoS One. 2015;10(10):e0140613. doi:10.1371/journal.pone.0140613
172. Schmitt CA, Fridman JS, Yang M, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–346. doi:10.1016/S0092-8674(02)00734-1
173. Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303(5660):1010–1014. doi:10.1126/science.1092734
174. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356(2Pt A):197–203. doi:10.1016/j.canlet.2013.12.025
175. Hashimoto N, Nagano H, Tanaka T. The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases. Endocr J. 2019;66(6):485–496. doi:10.1507/endocrj.EJ18-0565
176. Geng J, Yuan X, Wei M, Wu J, Qin ZH. The diverse role of TIGAR in cellular homeostasis and cancer. Free Radic Res. 2018;52(11–12):1240–1249. doi:10.1080/10715762.2018.1489133
177. Itahana Y, Itahana K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int J Mol Sci. 2018;19:3.
178. Wanka C, Steinbach JP, Rieger J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem. 2012;287(40):33436–33446. doi:10.1074/jbc.M112.384578
179. Pena-Rico MA, Calvo-Vidal MN, Villalonga-Planells R, et al. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol. 2011;101(1):132–139. doi:10.1016/j.radonc.2011.07.002
180. Morfouace M, Lalier L, Bahut M, et al. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J Biol Chem. 2012;287(40):33664–33674. doi:10.1074/jbc.M111.320028
181. Kambach DM, Halim AS, Cauer AG, et al. Disabled cell density sensing leads to dysregulated cholesterol synthesis in glioblastoma. Oncotarget. 2017;8(9):14860–14875. doi:10.18632/oncotarget.14740
182. Yin N, Xie T, Zhang H, Chen J, Yu J, Liu F. IDH1-R132H mutation radiosensitizes U87MG glioma cells via epigenetic downregulation of TIGAR. Oncol Lett. 2020;19(2):1322–1330. doi:10.3892/ol.2019.11148
183. Ghildiyal R, Sen E. CK2 induced RIG-I drives metabolic adaptations in IFNgamma-treated glioma cells. Cytokine. 2017;89:219–228. doi:10.1016/j.cyto.2015.10.009
184. Ahmad F, Ghosh S, Sinha S, Joshi SD, Mehta VS, Sen E. TGF-beta-induced hCG-beta regulates redox homeostasis in glioma cells. Mol Cell Biochem. 2015;399(1–2):105–112. doi:10.1007/s11010-014-2237-6
185. Bartesaghi S, Graziano V, Galavotti S, et al. A D, Karlsson A, Martins LM et al: inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci U S A. 2015;112(4):1059–1064. doi:10.1073/pnas.1413165112
186. Shao Y, Chen HT, Ma QR, Zhang YW, He YQ, Liu J. Long non-coding RNA PVT1 regulates glioma proliferation, invasion, and aerobic glycolysis via miR-140-5p. Eur Rev Med Pharmacol Sci. 2020;24(1):274–283. doi:10.26355/eurrev_202001_19922
187. Bon C, Luffarelli R, Russo R, et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019;47(20):10728–10743. doi:10.1093/nar/gkz798
188. Grunt M, Failla AV, Stevic I, Hillebrand T, Schwarzenbach H. A novel assay for exosomal and cell-free miRNA isolation and quantification. RNA Biol. 2020;17(4):425–440. doi:10.1080/15476286.2020.1721204
189. Ray A, Norden B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB j. 2000;14(9):1041–1060. doi:10.1096/fasebj.14.9.1041
190. Dong Y, Xiao Y, Shi Q, Jiang C. Dysregulated lncRNA-miRNA-mRNA Network Reveals Patient Survival-Associated Modules and RNA Binding Proteins in Invasive Breast Carcinoma. Front Genet. 2019;10:1284. doi:10.3389/fgene.2019.01284
191. Velagapudi SP, Cameron MD, Haga CL, et al. Design of a small molecule against an oncogenic noncoding RNA. Proc Natl Acad Sci U S A. 2016;113(21):5898–5903. doi:10.1073/pnas.1523975113
192. Alekseeva LI. [Comparative evaluation of the safety and efficacy of etoricoxib and diclofenac on the upper gastrointestinal tract in patients with osteoarthrosis and rheumatoid arthritis (the multinational etoricoxib and diclofenac arthritis long-term (MEDAL) study program)]. Ter Arkh. 2010;82(8):57–62. Russian.
193. Hawkey CJ. Naproxen sodium did not lead to substantially more upper gastrointestinal tract bleeding than ibuprofen during short term use as an analgesic. Gut. 1998;43(3):315. doi:10.1136/gut.43.3.315
194. Oteiza PI, Keen CL, Han B, Golub MS. Aluminum accumulation and neurotoxicity in Swiss-Webster mice after long-term dietary exposure to aluminum and citrate. Metabolism. 1993;42(10):1296–1300. doi:10.1016/0026-0495(93)90128-B
195. Fukuda T, Nakashima Y, Harada M, et al. Effect of whole blood clotting time in rats with ionized hypocalcemia induced by rapid intravenous citrate infusion. J Toxicol Sci. 2006;31(3):229–234. doi:10.2131/jts.31.229
196. Yan C, Wang J, Yang Y, Ma W, Chen X. Molecular biomarker-guided anti-angiogenic targeted therapy for malignant glioma. J Cell Mol Med. 2019;23(8):4876–4882. doi:10.1111/jcmm.14417
197. Wang L, Qin W, Huo YJ, et al. Advances in targeted therapy for malignant lymphoma. Signal Transduction Targeted Ther. 2020;5(1):15.
198. Chen X, Wu W, Gong B. et al. Metformin attenuates cadmium-induced neuronal apoptosis in vitro via blocking ROS-dependent PP5/AMPK-JNK signaling pathway. Neuropharmacology;2020. 108065. doi:10.1016/j.neuropharm.2020.108065
199. McMahon J, Huang X, Yang J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. 2012;32(45):15704–15714. doi:10.1523/JNEUROSCI.2392-12.2012
200. Meng G, Li B, Chen A, et al. Yu D and Wei J: targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br J Cancer. 2020;122(1):111–120. doi:10.1038/s41416-019-0639-7
201. Ganapathy-Kanniappan S. Taming Tumor Glycolysis and Potential Implications for Immunotherapy. Front Oncol. 2017;7:36. doi:10.3389/fonc.2017.00036
202. Lim SO, Li CW, Xia W, et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016;76(5):1284–1296. doi:10.1158/0008-5472.CAN-15-2478
203. Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: emerging concepts and therapeutic opportunities. Biochimica Et Biophysica Acta Rev Cancer. 2017;1868(1):212–220. doi:10.1016/j.bbcan.2017.04.002
204. Jiang Z, Liu Z, Li M, Chen C, Wang X. Chen C and Wang X: increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine. 2019;42:431–442. doi:10.1016/j.ebiom.2019.03.068
205. Reiter RJ, Sharma R, Ma Q, Dominquez-Rodriguez A, Marik PE, Abreu-Gonzalez P. Melatonin Inhibits COVID-19-induced Cytokine Storm by Reversing Aerobic Glycolysis in Immune Cells: A Mechanistic Analysis. Med Drug Discovery. 2020;6:100044. doi:10.1016/j.medidd.2020.100044
206. Cascone T, McKenzie JA, Mbofung RM, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018;27(5):977–987. doi:10.1016/j.cmet.2018.02.024
207. Sukumar M, Gattinoni L. The short and sweet of T-cell therapy: restraining glycolysis enhances the formation of immunological memory and antitumor immune responses. Oncoimmunology. 2014;3(1):e27573. doi:10.4161/onci.27573
208. Dholakia AS, Chaudhry M, Leal JP, et al. Baseline metabolic tumor volume and total lesion glycolysis are associated with survival outcomes in patients with locally advanced pancreatic cancer receiving stereotactic body radiation therapy. Int J Radiat Oncol Biol Phys. 2014;89(3):539–546. doi:10.1016/j.ijrobp.2014.02.031
209. Xiao L, Hu ZY, Dong X, et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33(37):4568–4578. doi:10.1038/onc.2014.32
210. Beneteau M, Zunino B, Jacquin MA, et al. Carles M and Ricci JE: combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc Natl Acad Sci U S A. 2012;109(49):20071–20076. doi:10.1073/pnas.1206360109
211. Fiorillo M, Sanchez-Alvarez R, Sotgia F, Lisanti MP. Sotgia F and Lisanti MP: the ER-alpha mutation Y537S confers Tamoxifen-resistance via enhanced mitochondrial metabolism, glycolysis and Rho-GDI/PTEN signaling: implicating TIGAR in somatic resistance to endocrine therapy. Aging (Albany NY). 2018;10(12):4000–4023. doi:10.18632/aging.101690
212. Golding JP, Kemp-Symonds JG, Dobson JM. Kemp-Symonds JG and Dobson JM: glycolysis inhibition improves photodynamic therapy response rates for equine sarcoids. Vet Comp Oncol. 2017;15(4):1543–1552. doi:10.1111/vco.12299
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.