Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Emerging pharmaceutical therapies for COPD
Authors Lakshmi SP ![]() , Reddy AT
, Reddy AT ![]() , Reddy RC
, Reddy RC
Received 3 September 2016
Accepted for publication 16 January 2017
Published 21 July 2017 Volume 2017:12 Pages 2141—2156
DOI https://doi.org/10.2147/COPD.S121416
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Sowmya P Lakshmi,1,2 Aravind T Reddy,1,2 Raju C Reddy1,2
1Department of Medicine, Division of Pulmonary, Allergy and Critical Care Medicine, University of Pittsburgh School of Medicine, 2Veterans Affairs Pittsburgh Healthcare System, Pittsburgh, PA, USA
Abstract: COPD, for which cigarette smoking is the major risk factor, remains a worldwide burden. Current therapies provide only limited short-term benefit and fail to halt progression. A variety of potential therapeutic targets are currently being investigated, including COPD-related proinflammatory mediators and signaling pathways. Other investigational compounds target specific aspects or complications of COPD such as mucus hypersecretion and pulmonary hypertension. Although many candidate therapies have shown no significant effects, other emerging therapies have improved lung function, pulmonary hypertension, glucocorticoid sensitivity, and/or the frequency of exacerbations. Among these are compounds that inhibit the CXCR2 receptor, mitogen-activated protein kinase/Src kinase, myristoylated alanine-rich C kinase substrate, selectins, and the endothelin receptor. Activation of certain transcription factors may also be relevant, as a large retrospective cohort study of COPD patients with diabetes found that the peroxisome proliferator-activated receptor γ (PPARγ) agonists rosiglitazone and pioglitazone were associated with reduced COPD exacerbation rate. Notably, several therapies have shown efficacy only in identifiable subgroups of COPD patients, suggesting that subgroup identification may become more important in future treatment strategies. This review summarizes the status of emerging therapeutic pharmaceuticals for COPD and highlights those that appear most promising.
Keywords: pulmonary, PPAR, phosphodiesterase, emphysema, cigarette, mucus
A Letter to the Editor has been received and published for this article.
Introduction
COPD affects ~200 million people worldwide1 and is the third leading cause of death in the US, claiming ~150,000 lives in 2013.2 Approximately 6.3% of US adults aged ≥18 years have COPD,3 and its combined indirect and direct costs in 2010 surpassed $52 billion. More effective treatments for COPD are needed to address this serious health problem. This review summarizes the status of potential therapies currently or recently in clinical trials and highlights those that appear most promising.
COPD pathology
COPD, a complex and heterogeneous chronic progressive inflammatory disease of the distal airways characterized by persistent airflow limitations, typically results from inhaling noxious gases and particles, especially cigarette smoke.4–6 The resulting inflammatory response involves increased numbers of neutrophils, macrophages, B cells, and CD4+ and CD8+ T lymphocytes in the airways.7 Histopathological changes include edema, loss of alveoli, and tissue remodeling involving smooth muscle hypertrophy and fibrosis, which are associated with bronchoconstriction and airway mucus hypersecretion exacerbated by reduced clearance.5–8 The accompanying systemic inflammation and increased reactive oxygen species (ROS) in patients may contribute to such manifestations as cardiovascular complications, loss of skeletal muscle, osteoporosis, and psychosocial effects.
COPD exacerbations
COPD exacerbations can significantly accelerate lung function decline and overall health. In patients with severe COPD, acute exacerbations requiring hospitalization account for up to 50% of COPD health care costs. Exacerbation risk appears to be associated with serum eosinophilia: in patients with clinical COPD and serum eosinophils above 0.34×109 cells/L, risk of severe and moderate exacerbations were increased compared to those in patients with fewer eosinophils.9 Causes of COPD exacerbations appear multifactorial and elusive but may include bacterial or viral infections, exposure to environmental pollutants, and unidentified factors.5 Exacerbations are accompanied by a rapid rise of proinflammatory cytokines and chemokines that trigger and sustain rapid influx of neutrophils and their products such as neutrophil elastase into the airways.10,11 Reduction in the frequency of COPD exacerbation is a primary or secondary end point of several current clinical trials of emerging therapies.12,13
Treatment of stable COPD
The American Thoracic Society and other leading pulmonary disease organizations recommend that COPD patients cease smoking and use one or more inhaled bronchodilators.14 The wide choice of inhaled bronchodilators includes short-acting beta2-agonists (SABA) and long-acting beta2-agonists (LABA), short-acting muscarinic antagonists (SAMA) and long-acting muscarinic antagonists (LAMA), and combinations of β-agonists and antimuscarinic agents. Inhaled corticosteroids (ICS) and combinations of corticosteroids with other agents may also be used. Oral medications, including methylxanthines and a phosphodiesterase 4 (PDE4) inhibitor, may also be used as adjunctive agents or, in the case of systemic corticosteroids, for acute exacerbations.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) recently categorized COPD patients into four groups, in which the familiar spirometry-based grades (GOLD I–IV) are supplemented by patients’ symptom burden (assessed by the modified British Medical Research Council questionnaire or COPD Assessment Test) and exacerbation history: spirometry and exacerbation history are jointly used to determine the risk of adverse health events.5 Groups A and C have relatively mild symptoms, while Groups A and B have relatively low risk. GOLD recommends that Groups A and B receive SABA, LABA, SAMA, or LAMA bronchodilators. It recommends that Groups C and D also receive ICS or possibly the PDE4 inhibitor roflumilast. Despite symptomatic relief and reduced rates of exacerbations with current medications, long-term studies of treated COPD patients suggest that medications do not halt the decline of lung function4 and resulting mortality, although there is limited evidence that fluticasone and/or salmeterol may slow the rate of decline.15,16 Several groups are pursuing identification of biomarkers for COPD subgroups based on either the etiological agent/event triggering the exacerbation or the physiological targets that may indicate response to a given therapy.6,17 For example, COPD patients with higher levels of exhaled nitric oxide (FeNO) had a greater probability of the enhanced eosinophilic infiltration18 that is associated with a greater response to corticosteroids.19–21 These studies carry significant implications both for current treatment and for the development of novel therapies that may be targeted to specific groups of patients.
Inflammation in COPD
Smoke, from cigarettes, biomass fuel, or air pollution, is a common inducer of inflammation in COPD. The inhaled irritants provoke significant oxidative stress and activate inflammatory cells.22,23 Excess neutrophils and macrophages, along with T cells, B cells, and dendritic cells, infiltrate the peripheral airways,24–26 concurrent with the narrowing or obliteration of small bronchioles.27 A number of inflammatory markers are significantly elevated in the serum of patients with COPD, notably including CD40 ligand, epidermal growth factor (EGF), brain-derived neurotrophic factor (BDNF), a variety of acute-phase proteins, and neutrophil-associated proteins.28 These molecules may represent targets for future COPD therapies.
Glucocorticoid (GC) resistance
COPD is relatively resistant to modulation by corticosteroids, as even high doses of GCs do not delay or inhibit COPD progression. Histological analysis of lung tissue removed during lung volume reduction surgery of COPD patients indicated that GC treatment significantly reduced the number of airways with lymphoid follicles but had no long-term effect on luminal occlusion or airway wall thickening.29
Corticosteroids suppress inflammation both by activating transcription of anti-inflammatory and suppressing transcription of proinflammatory genes.30 Binding of corticosteroids to the classic glucocorticoid receptor (GRα) leads to activation, which allows release from its cytoplasmic anchor. The receptor is then acetylated, allowing binding to response elements in the promoter elements of target genes, and recruitment of coactivators such as CREB-binding protein, with consequent transcriptional activation. GC-mediated gene repression requires histone deacetylase 2 (HDAC2) at two distinct steps.31 First, this acetyl group must be removed by HDAC2 to allow binding to proinflammatory transcription factors such as nuclear factor-κB (NF-κB). Second, activated GR recruits HDAC2, which then deacetylates histones and thus inactivates transcription. Alveolar macrophages (AMs), lungs, and bronchi of COPD patients express lower levels of HDAC2 than those in healthy controls,32 however, and theophylline treatment, which enhances HDAC activity in AMs, also restores AM GC sensitivity.33 These effects may underlie the reported ability of theophylline treatment (200–400 mg/day, depending on weight), added to the LABA formoterol and the GC budesonide in a 50-patient (study completion) single-blinded, placebo-controlled study, to improve 6-minute walk distance and forced expiratory volume in 1 second (FEV1).34 However, a double-blind, placebo-controlled study in 46 per-protocol patients found no effect of low-dose (100 mg bid) theophylline added to fluticasone plus salmeterol on HDAC activity, inflammatory biomarkers, or frequency of exacerbation.35
Observed COPD-associated reductions in HDAC2 are believed to result from oxidative and nitrosative stress,32 and levels of the antioxidant transcription factor NF-E2–related factor 2 (Nrf2) are similarly reduced in AMs of COPD patients.36 Treatment with the Nrf2 activator sulforaphane, similar to theophylline treatment, increases HDAC2 activity and restores GC sensitivity.37
A number of other mechanisms of GC resistance have also been suggested.30 Among these may be increased expression of the translationally inactive GRβ, although this remains controversial30 and GR expression has been found to be reduced in lungs of healthy smokers and COPD patients.38 Another possibility is posttranslational modifications of GRα by phosphorylation or nitrosylation.39 Such phosphorylation may be due to p38 mitogen-activated protein kinase (MAPK) activation, and p38 MAPK inhibitors have been shown to reduce the GC resistance seen in some patients with severe asthma.40
Restoration of GC sensitivity is an attractive strategy for the development of novel pharmaceutical therapies. As previously noted, the ability of theophylline treatment to accomplish this is controversial.34,35 However, roflumilast has been shown ex vivo to improve GC sensitivity of neutrophils from COPD patients,41 and subgroup analysis of two clinical trials showed that patients receiving ICS were among those particularly likely to experience a reduction of exacerbations in roflumilast treatment.42 A subsequent 1-year trial showed that roflumilast treatment reduced exacerbations in patients also receiving ICS and LABA.13
Proinflammatory cytokines and chemokines
Numerous proinflammatory cytokines and chemokines are significantly higher in COPD patients than in healthy controls,43 and multiple biologics and small molecules that target these mediators are under investigation (Tables 1 and 2). Chemokines significantly contribute to inflammation by attracting neutrophils and other inflammation-related cells, and an antagonist of the CXCR2 receptor for interleukin (IL)-8 and other chemokines44 has been found beneficial in COPD patients. A different CXCR2 inhibitor reduced blood neutrophil counts with no increased rate of infection,45 but compounds blocking CCR146 and CCR247,48 (NCT01215279) had no effect.
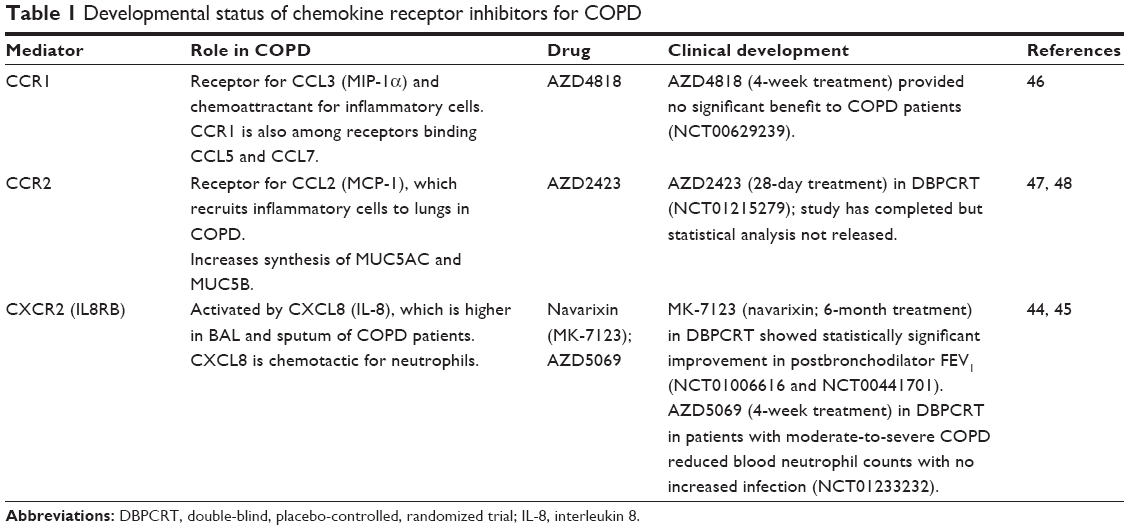 | Table 1 Developmental status of chemokine receptor inhibitors for COPD |
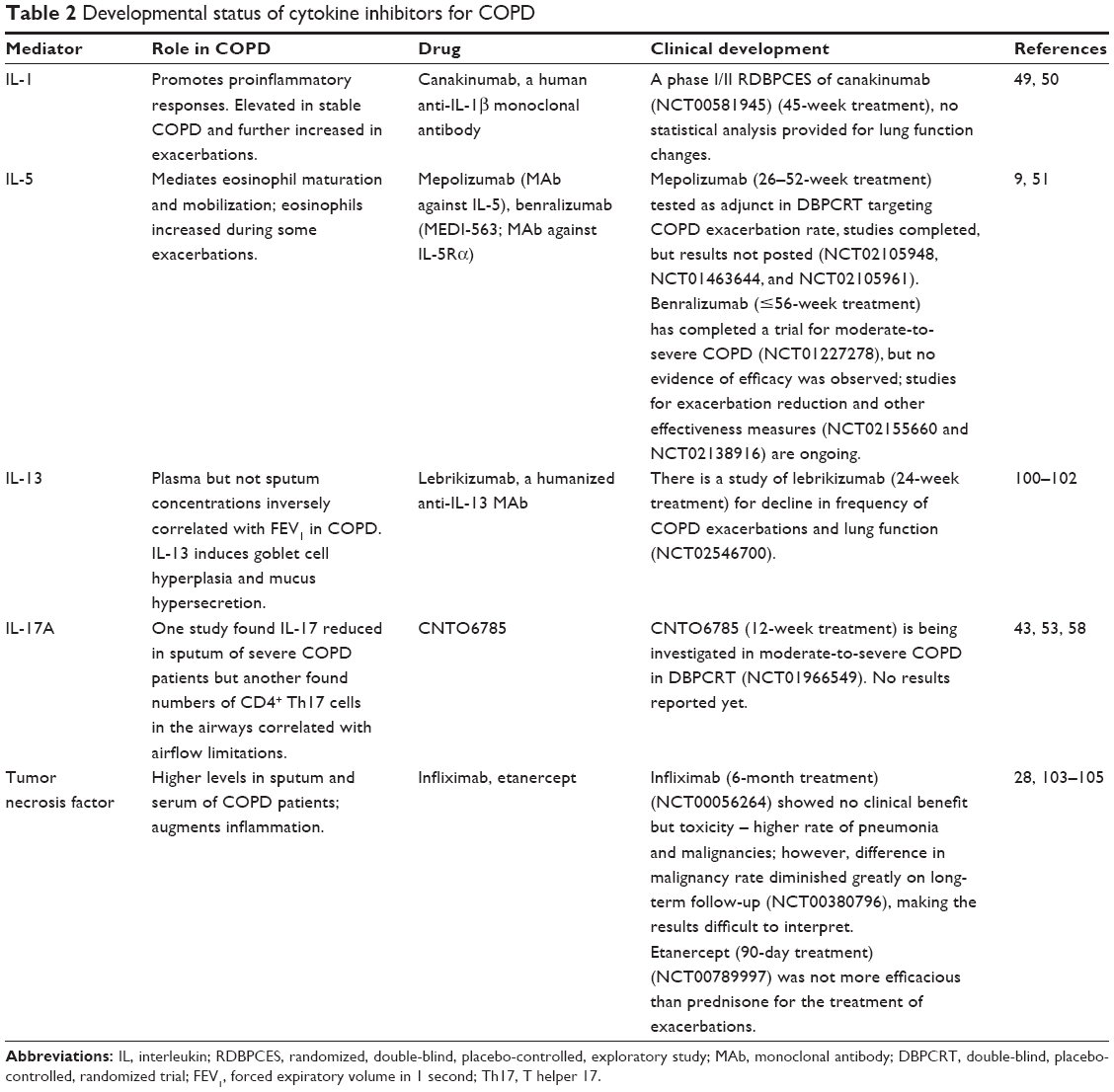 | Table 2 Developmental status of cytokine inhibitors for COPD |
IL-1 expression was significantly higher in lung and sputum from COPD patients compared to non-COPD controls.49 However, treatment with a monoclonal antibody (MAb) inhibiting IL-1b (canakinumab)50 remains an open question in COPD patients (Table 2).
IL-5 is largely produced by eosinophils and is therefore low in typical COPD. A subset of COPD patients have elevated eosinophil numbers and IL-5 levels in their blood and airways; these patients are at increased risk for acute exacerbations.9,51 Two MAbs targeted to IL-5 have been investigated for their ability to reduce exacerbation rate in COPD patients, but one showed no evidence of efficacy and results for the other are not yet available (Table 2).
Secretion of IL-17A is a canonical marker of T helper 17 (Th17) lymphocytes, which are distinct from Th1 and Th2 cells. However, most of the cells secreting IL-17 in patients with very severe COPD were identified as mast cells.52 This study also found that levels of IL-17A in lungs of COPD patients correlated with functional decline, with elevations becoming significant in patients with severe and very severe disease. Another study found that the numbers of CD4+IL-17+ cells in the alveolar walls and small airways of COPD patients and CD4+IL-17+ numbers in small airways positively correlated with airflow limitations.53 These results are compatible with the finding that Th17 cells were present in the lungs of patients with emphysema but not normal controls and that IL-17A-induced secretion of the elastin-degrading enzyme matrix metalloprotease 12 by lung macrophages.54 The levels of IL-17A were also elevated in the sputum of patients with acute COPD exacerbations associated with Haemophilus influenzae infection but not other acute exacerbations.55 Other studies have not similarly distinguished among causes of acute exacerbations, likely accounting for findings that the ratio of regulatory T cells to IL-17 levels in peripheral blood was similar in COPD patients with and without current acute exacerbations, although exacerbations significantly increased levels of transforming growth factor β (TGF-β).56 Indeed, one study was unable to detect IL-17 in sputum or serum of COPD patients with or without exacerbations.57 Another study found that the sputum of patients with severe COPD had significantly higher levels of IL-8 but 4.8-fold lower levels of IL-17 compared to that of patients with mild COPD and healthy controls.58 T cells from many COPD patients have also been reported to produce less IL-17A and IL-22 (also a signature cytokine of Th17 cells) than those of most normal smokers.59 These complex and apparently contradictory findings underline the uncertainty of the role of IL-17 in COPD. Nevertheless, an IL-17 modulator is currently in clinical trials for COPD (Table 2).
Based on an IL-18-overexpressing transgenic mouse model that develops emphysema and airway remodeling,60 Kang et al60 and Nakajima and Owen61 proposed that IL-18 is a master regulator of lung pathology in COPD. A phase I safety and tolerability clinical trial (NCT01322594) of the MAb MEDI2338 (targeted to IL-18) in COPD patients found no serious adverse events, but there have been no efficacy studies.62
Tumor necrosis factor α (TNF-α) plays multiple roles in COPD inflammatory pathology, and the levels of interferon γ (IFNγ) and TNF-α in the intraepithelial T cells from bronchi of COPD patients with GOLD II–III disease showed a significant negative correlation with FEV1.63 Nevertheless, studies with infliximab showed no clinical benefits on FEV1, dyspnea, or exacerbations and were associated with higher rates of pneumonia and malignancy (Table 2). Similarly, treatment with etanercept, a fusion protein that competitively binds TNF-α, was not superior to prednisone in COPD exacerbations and in fact was less effective among patients with eosinophilia (Table 2).
Taken together, these data show that an increased level of a specific cytokine or chemokine during COPD exacerbations or stable COPD does not necessarily predict the efficacy of its specific inhibitor in COPD patients. Whether modulators of specific cytokines or chemokines can provide improved efficacy in a subgroup of patients is a possibility and warrants further investigation.6
Signaling molecules
Multiple signaling molecules help regulate inflammation and airway remodeling and represent plausible targets for the development of therapeutic candidates. Candidate drugs include inhibitors of p38 MAPK and related kinases, phosphoinositide kinase δ (PI3Kδ), leukotriene B4, selectins, and vasoactive intestinal peptide (Table 3). Although several oral and inhaled p38 MAPK inhibitors have been discontinued, the inhaled narrow spectrum kinase (p38α + Src family) inhibitor JNJ49095397 (previously RV568) shows promising activity in COPD patients; conference presentations have indicated that RV568 significantly increased FEV1 and inhibited IL-1β (90% at 800 μg dose) and CXCL8 expression (73%).64 However, a recent conference report performed in over 200 COPD patients (half placebo, half 400 mg dose) showed no benefit with RV568 on lung function or EXACT-PRO.65 PI3Kδ participates in many functions of lymphoid and myeloid cells: B-cell development, migration and activation of natural killer (NK) cells and T cells, neutrophil oxidative burst, macrophage activation triggered by immune complexes, and degranulation and maturation of mast cells.66 Specific PI3Kδ inhibitors are being developed,67 and studies on the effects of such inhibitors in COPD are in progress. Efficacy data remain limited, however (Table 3). Selectins are essential for migration of inflammatory cells from the bloodstream into pulmonary tissue; the selectin modulator bimosiamose reduced inflammation in COPD patients and thus warrants further testing (Table 3).68
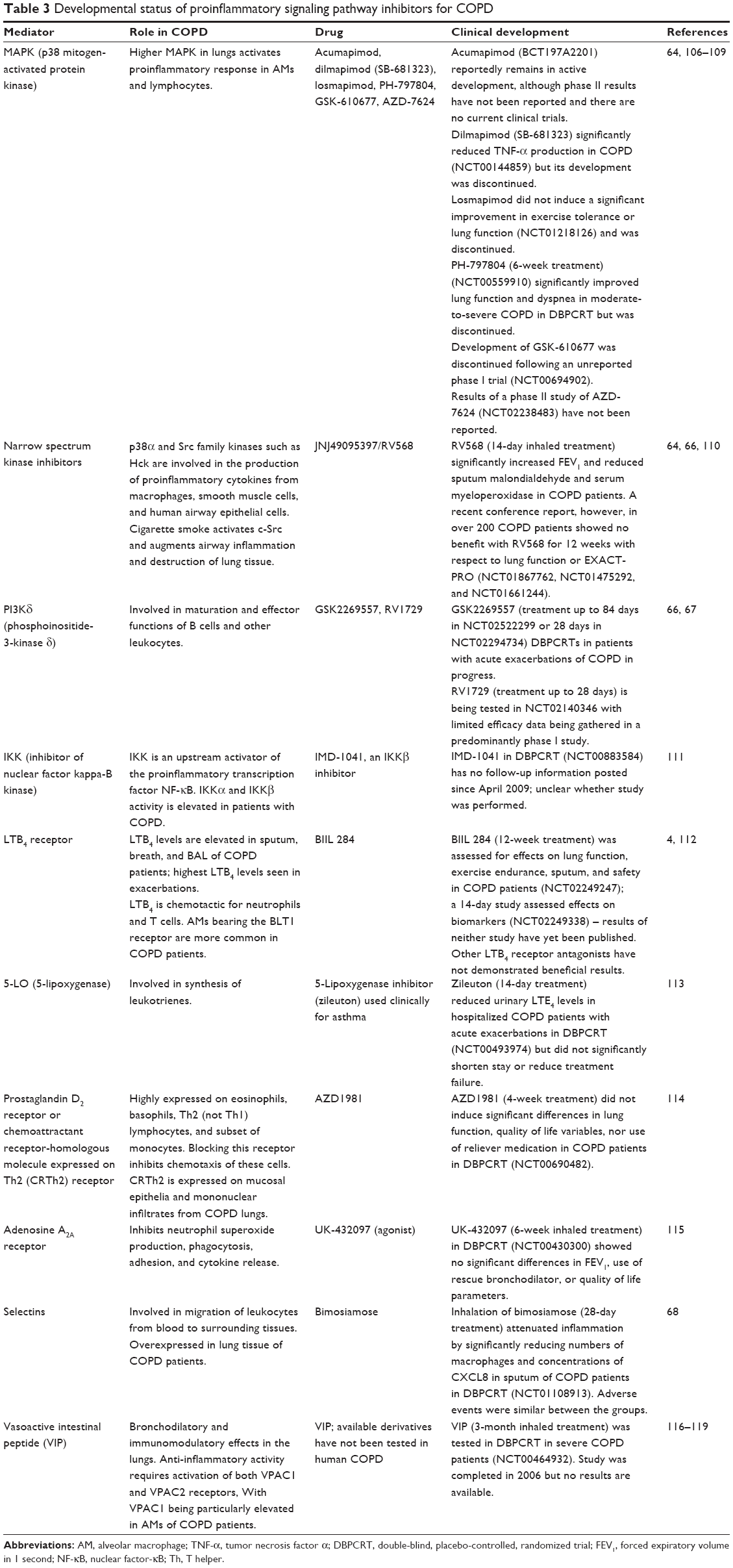 | Table 3 Developmental status of proinflammatory signaling pathway inhibitors for COPD |
Similar to the glucocorticoid receptor, peroxisome proliferator-activated receptor γ (PPARγ), also a member of the nuclear hormone receptor superfamily, exerts potent antioxidant and anti-inflammatory effects via multiple mechanisms, including downregulation of NF-κB and other proinflammatory transcription factors. Lung tissue and bronchial epithelial cells from COPD patients express significantly lower levels of PPARγ than those of nonsmoking controls.69 In vitro treatment of COPD patient and control bronchial epithelial cells with PPARγ agonists including 10-nitro-oleic acid (a possible endogenous ligand) or rosiglitazone (a thiazolidinedione) blocked cigarette smoke-induced production of cytokines, chemokines, and ROS, and accompanying suppression of HDAC2 levels.69 The PPARγ agonist rosiglitazone dose dependently inhibited LPS-induced production of TNF-α and CCL5 by AMs of COPD patients, smokers, and never-smokers, shifting them toward an anti-inflammatory M2 phenotype, while both rosiglitazone and pioglitazone attenuated pulmonary inflammation in a tobacco smoke mouse model.70 PPARγ also appears to play a central role in the development of emphysema. PPARγ is downregulated in lung myeloid dendritic cells of smokers with emphysema and endogenous PPARγ agonist activity is reduced in plasma of those with emphysema, while treatment with the thiazolidinedione ciglitazone reverses emphysema in a mouse model.71 A retrospective epidemiological study of veterans with both diabetes and COPD indicated that the 7,887 veterans treated with a PPARγ agonist for their diabetes (97.1% rosiglitazone) significantly reduced the risk of COPD exacerbations in comparison to those receiving other diabetes medications (n=42,347; incidence rate ratio [IRR]=0.85; CI: 0.80–0.91) (Table 4).72 Taken together, these data support further studies of PPARγ agonists in COPD patients with early signs of emphysema.
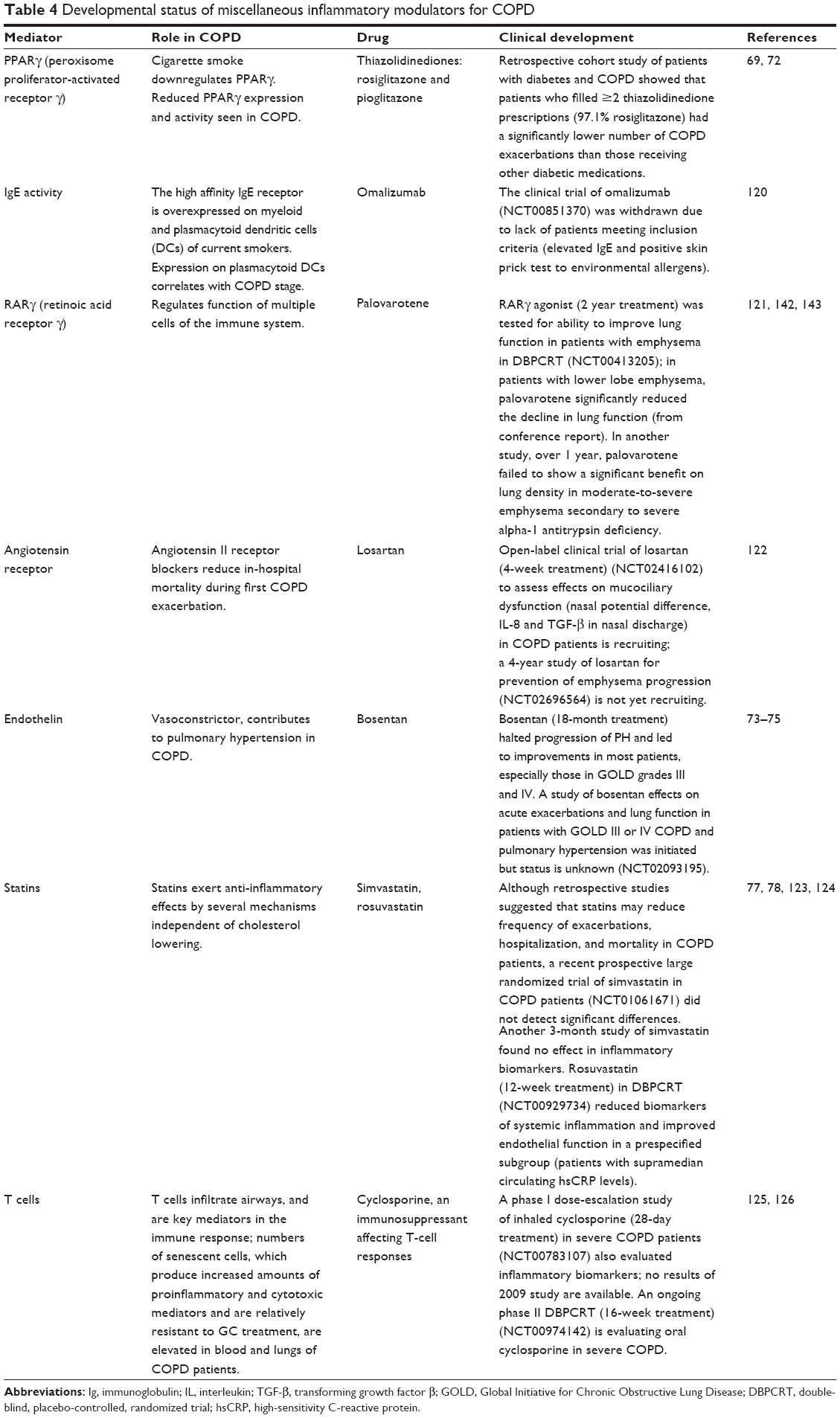 | Table 4 Developmental status of miscellaneous inflammatory modulators for COPD |
COPD also involves progressive increase in pulmonary arterial pressure, and 20%–91% (depending on definition, COPD severity, and method of measurement) of COPD patients have progressed to pulmonary hypertension.73 Endothelin signaling plays a major role in vascular remodeling and hence in development of pulmonary hypertension.74,75 An 18-month treatment with the endothelin antagonist bosentan improved measures of pulmonary hypertension compared to those at baseline, especially in GOLD grade III and IV COPD patients, while pulmonary hypertension worsened in placebo-treated patients (Table 4).74 However, bosentan can actually worsen hypoxemia in COPD patients without pulmonary hypertension.76
Although statins are known to have anti-inflammatory properties, simvastatin treatment (40 mg/day) of COPD patients at high risk for exacerbation did not increase the time to first exacerbation nor reduce the number of exacerbations.77 However, rosuvastatin (12-week treatment) improved endothelium-dependent vascular function in a prespecified subgroup (patients with supramedian circulating high-sensitivity C-reactive protein [hsCRP]) but not in the total COPD population in a double-blind, placebo-controlled trial.78 This study found no statistically significant effect on pulmonary function parameters, however.
PDE inhibitors
The 11-membered phosphodiesterase enzyme family (PDE1-11) differentially hydrolyzes cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), which regulate many cellular processes including release of inflammatory mediators and relaxation of smooth muscles.10 PDE3 appears to be involved in bronchoconstriction since its inhibitors induce bronchodilation in humans.10 PDE4 is expressed in most inflammatory cell types and is a main target for emerging COPD therapies (Table 5), with the PDE4 inhibitor roflumilast having been approved by the FDA as a COPD treatment. In vitro studies of low-dose combinations of PDE4 and PI3Kδ inhibitors significantly reduced cigarette smoke extract-induced apoptosis of lung epithelial cells, neutrophil elastase production, and macrophage secretion of TNF-α, phosphorylated protein kinase B, and matrix metalloproteinase 9 (MMP-9).79
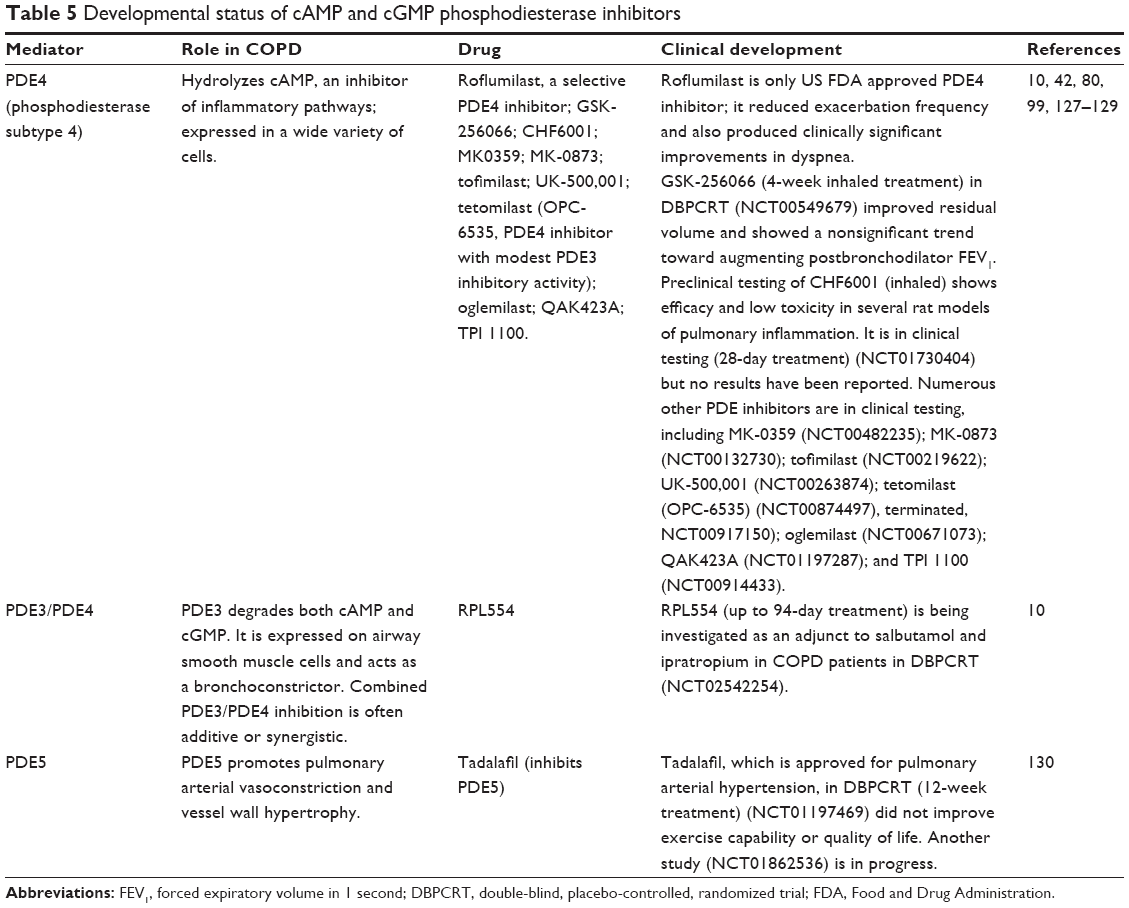 | Table 5 Developmental status of cAMP and cGMP phosphodiesterase inhibitors |
Gastrointestinal adverse effects can be a significant problem with roflumilast, but PDE4 inhibitors with inhaled or nebulized (eg, RPL554, GSK-256066) formulations appear to have more tolerable side effect profiles than orally administered roflumilast.80,81
The severity of emphysema and small airways disease is associated with higher expression of multiple MMPs.82 However, despite promising results in an animal model,83 an inhibitor of MMP-9 and -12 has not shown significant benefit for COPD patients in a clinical trial (Table 6).
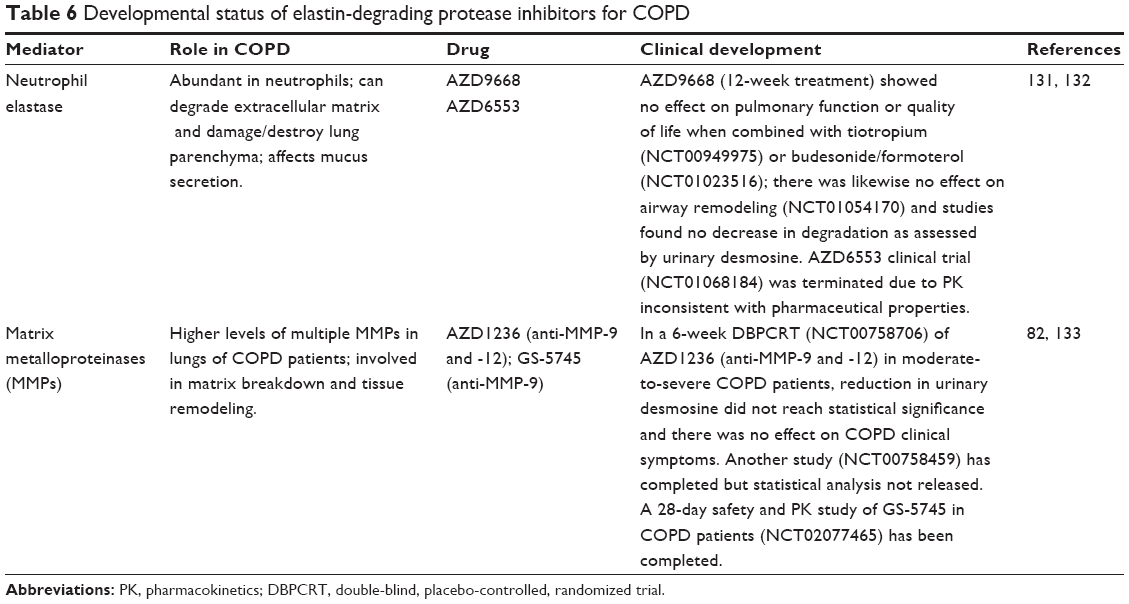 | Table 6 Developmental status of elastin-degrading protease inhibitors for COPD |
Oxidative stress and antioxidants
Oxidants, both from inhaled pollutants and produced as part of the inflammatory response, are major contributors to COPD pathophysiology. Furthermore, inflammation-associated production of ROS and reactive nitrogen species (RNS) is accompanied by downregulation of the antioxidant transcription factor Nrf2. ROS activates the proinflammatory transcription factors activator protein 1 (AP-1) and NF-κB, with consequent production of inflammatory proteins and mediators. Excess ROS in COPD patients also directly contributes to airspace epithelial injury and inactivates antiproteases that help prevent emphysema.84 Therapeutic agents that activate Nrf2 and thus neutralize the excess oxidants may prove beneficial to COPD treatment.85
The antioxidant transcription factor Nrf2, the primary mechanism for limiting oxidative stress, is reduced in COPD patients.36 Kelch-like ECH-associated protein 1 (Keap1) sequesters Nrf2 in the cytoplasm and under healthy conditions targets Nrf2 to Cullin-3 for ubiquitination and degradation.86 Keap1 monitors oxidative stress through its multiple cysteines with distinct stressors binding one or more specific cysteines. Following binding of these stress-related compounds, release of Nrf2 from Keap1 occurs, which transfers to the nucleus and activates multiple antioxidant enzymes and phase II proteins that counteract oxidative stress.86 This makes the Nrf2/Keap1 system an attractive therapeutic target in COPD and other inflammatory diseases. The natural product sulforaphane activated Nrf2 in AMs isolated from COPD patients, denitrosylated HDAC2, and restored sensitivity to the glucocorticoid dexamethasone in a glutathione-dependent manner.37 A study of the effect of two doses of sulforaphane on Nrf2 expression in 89 COPD patients was recently completed, and sulforaphane administered for four weeks to patients with COPD did not induce the expression of Nrf2 target genes or have an effect on oxidative stress, airway inflammation, or lung function. (NCT01335971).87
Erdosteine and N-acetylcysteine directly scavenge ROS via their thiol groups (of the metabolite in the case of erdosteine) and also have mucolytic activity. High-dose (900 mg/day) erdosteine increased the ability of salbutamol to improve %FEV1 reversibility,88 and long-term treatment reduces exacerbations and improves quality of life.89 High-dose (600 mg bid) N-acetylcysteine reduces the number of exacerbations patients experience;12,90,91 reported effects with a lower dose (600 mg/day) have been inconsistent.92–94
Mucus hypersecretion
Mucus hypersecretion can be modulated by blocking its overproduction and/or by inhibiting its secretion (Table 7). Hypothetically, a rebound effect after cessation of an inhibitor of mucin secretion may involve a rapid release of produced but unsecreted mucin. In contrast, the rebound effect after cessation of an inhibitor of mucin production may involve a lag phase for mucin production and a more gradual increase of mucin secretion. Multiple mucins comprise the gel-forming layer of normal airway mucus. Numerous signals can promote mucus secretion: bacterial products, cytokines, cholinergic agonists, elastases, matrix metalloproteases, and activation of epidermal growth factor receptor (EGFR).95,96 Cigarette smoke induces EGFR- and hypoxia inducible factor-1 (HIF-1)-mediated signaling95 and thus can promote hyperplasia of mucin-producing goblet cells.97 Supporting this concept, nuclear HIF-1α was expressed in the majority of goblet cells in areas of remodeled airway tissues showing goblet cell hyperplasia from COPD patients but not in subjects without COPD.97 IL-13, an essential component of COPD-associated inflammation, promotes goblet cell production of mucins.98 After the mucins are expressed, glycosylated, and packaged in mucin granules, myristoylated alanine-rich C kinase substrate (MARCKS) mediates movement of intracellular mucin granules to the goblet cell apical membrane and is therefore essential for mucin exocytosis and secretion.98
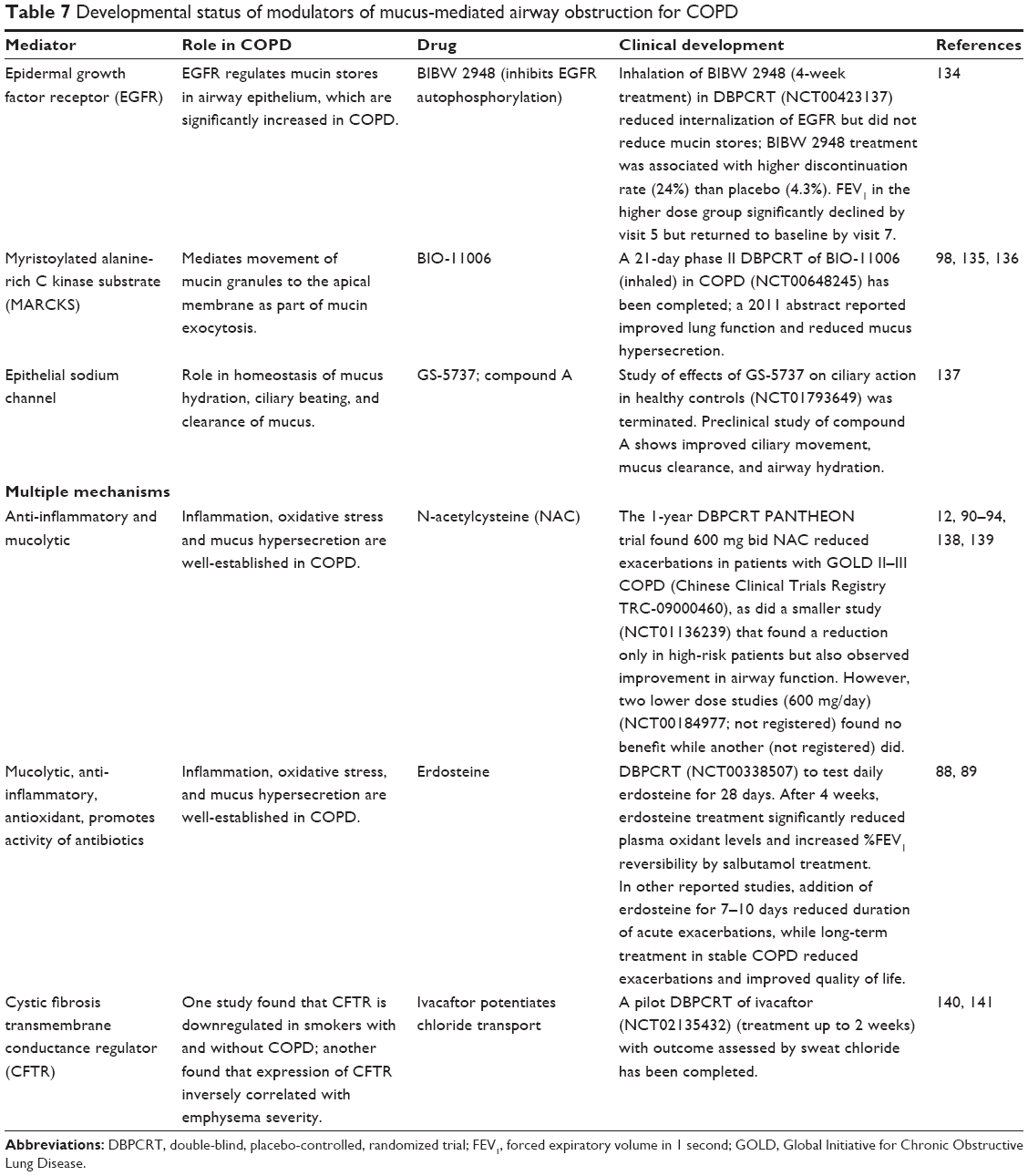 | Table 7 Developmental status of modulators of mucus-mediated airway obstruction for COPD |
Summary
The heterogeneity of COPD presentation augments the challenges in identifying and developing therapeutic compounds for the treatment of COPD patients. It also emphasizes the importance of tailoring therapy to individual patients and their disease status, which may involve considerations beyond the standard GOLD categories. Several distinct emerging therapies have shown efficacy in at least some COPD patients. The length of treatment (>3 months) and the inhaled route of administration can be associated with a higher probability of observing a positive effect on COPD variables and a reduced side effect profile, respectively. Although some of the emerging compounds showed significant activity in the total COPD population tested,45,68,74,88,99 sometimes only in connection with assessed biomarkers rather than clinically significant outcomes, subgroup analysis showed that most compounds were significantly affected in one or more of the following subgroups: smokers or ex-smokers,44,99 patients with chronic bronchitis or emphysema,80,99 use of standard COPD therapies,99 patients with alteration of relevant biomarkers,78 or at different COPD grades.74 These data suggest that subgroup analyses and the possibility of individualized therapy can benefit developers of emerging therapies by identification of the patients most likely to benefit from a new therapy.
Acknowledgments
This work was supported by a Merit Review award from the US Department of Veterans Affairs and the National Institutes of Health grants HL093196 and AI125338 (RCR). We gratefully acknowledge the scientific and writing expertise of KL Molnar-Kimber, PhD. The contents in this article do not represent the views of the US Department of Veterans Affairs or the United States Government.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferkol T, Schraufnagel D. The global burden of respiratory disease. Ann Am Thorac Soc. 2014;11(3):404–406. | ||
Heron M. Deaths: leading causes for 2013. Natl Vital Stat Rep. 2015;65(2):95. | ||
Centers for Disease Control and Prevention (CDC). Chronic obstructive pulmonary disease among adults – United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61(46):938–943. | ||
Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12(7):543–559. | ||
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Woodruff PG, Agusti A, Roche N, Singh D, Martinez FJ. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: making progress towards personalised management. Lancet. 2015;385(9979):1789–1798. | ||
Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38(4):497–515. | ||
Wiegman CH, Michaeloudes C, Haji G, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769–780. | ||
Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. The Copenhagen General Population Study. Am J Respir Crit Care Med. 2016;193(9):965–974. | ||
Abbott-Banner KH, Page CP. Dual PDE3/4 and PDE4 inhibitors: novel treatments for COPD and other inflammatory airway diseases. Basic Clin Pharmacol Toxicol. 2014;114(5):365–376. | ||
Pavord ID, Jones PW, Burgel PR, Rabe KF. Exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2016;11(Spec Iss):21–30. | ||
Tse HN, Raiteri L, Wong KY, Ng LY, Yee KS, Tseng CZ. Benefits of high-dose N-acetylcysteine to exacerbation-prone patients with COPD. Chest. 2014;146(3):611–623. | ||
Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet. 2015;385(9971):857–866. | ||
Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Celli BR, Thomas NE, Anderson JA, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med. 2008;178(4):332–338. | ||
Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk (SUMMIT): a double-blind randomised controlled trial. Lancet. 2016;387(10030):1817–1826. | ||
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671. | ||
Soter S, Barta I, Antus B. Predicting sputum eosinophilia in exacerbations of COPD using exhaled nitric oxide. Inflammation. 2013;36(5):1178–1185. | ||
Brightling CE, Monteiro W, Ward R, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356(9240):1480–1485. | ||
Chanez P, Vignola AM, O’Shaugnessy T, et al. Corticosteroid reversibility in COPD is related to features of asthma. Am J Respir Crit Care Med. 1997;155(5):1529–1534. | ||
Pizzichini E, Pizzichini MM, Gibson P, et al. Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am J Respir Crit Care Med. 1998;158(5 pt 1):1511–1517. | ||
Pace E, Giarratano A, Ferraro M, et al. TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum Immunol. 2011;72(1):54–62. | ||
Mortaz E, Adcock IM, Ito K, Kraneveld AD, Nijkamp FP, Folkerts G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur Respir J. 2010;36(5):1143–1154. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Burgel PR, Bourdin A, Chanez P, et al. Update on the roles of distal airways in COPD. Eur Respir Rev. 2011;20(119):7–22. | ||
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–1356. | ||
McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. | ||
Loza MJ, Watt R, Baribaud F, Barnathan ES, Rennard SI. Systemic inflammatory profile and response to anti-tumor necrosis factor therapy in chronic obstructive pulmonary disease. Respir Res. 2012;13:12. | ||
Hogg JC, Chu FS, Tan WC, et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am J Respir Crit Care Med. 2007;176(5):454–459. | ||
Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 2010;120(2–3):76–85. | ||
Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. | ||
Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. | ||
Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200(5):689–695. | ||
Subramanian, Ragulan, Jindal A, Viswambhar V, Arun Babu V. The study of efficacy, tolerability and safety of theophylline given along with formoterol plus budesonide in COPD. J Clin Diagn Res. 2015;9(2):OC10–OC13. | ||
Cosio BG, Shafiek H, Iglesias A, et al. Oral low-dose theophylline on top of inhaled fluticasone-salmeterol does not reduce exacerbations in patients with severe COPD: a pilot clinical trial. Chest. 2016;150(1):123–130. | ||
Suzuki M, Betsuyaku T, Ito Y, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2008;39(6):673–682. | ||
Malhotra D, Thimmulappa RK, Mercado N, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121(11):4289–4302. | ||
Marwick JA, Caramori G, Stevenson CS, et al. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 2009;179(7):542–548. | ||
Boardman C, Chachi L, Gavrila A, et al. Mechanisms of glucocorticoid action and insensitivity in airways disease. Pulm Pharmacol Ther. 2014;29(2):129–143. | ||
Mercado N, Hakim A, Kobayashi Y, et al. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS One. 2012;7(7):e41582. | ||
Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2014;134(2):314–322. | ||
Rennard SI, Calverley PM, Goehring UM, Bredenbroker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast – the importance of defining different subsets of patients with COPD. Respir Res. 2011;12:18. | ||
Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412. | ||
Rennard SI, Dale DC, Donohue JF, et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–1011. | ||
Kirsten AM, Forster K, Radeczky E, et al. The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate-to-severe COPD. Pulm Pharmacol Ther. 2015;31:36–41. | ||
Kerstjens HA, Bjermer L, Eriksson L, Dahlstrom K, Vestbo J. Tolerability and efficacy of inhaled AZD4818, a CCR1 antagonist, in moderate to severe COPD patients. Respir Med. 2010;104(9):1297–1303. | ||
Bai J, Song H, Cai C, Zhang M, Xu S, Tan J. The association of monocyte chemotactic protein-1 and CC chemokine receptor 2 gene variants with chronic obstructive pulmonary disease. DNA Cell Biol. 2012;31(6):1058–1063. | ||
Monzon ME, Forteza RM, Casalino-Matsuda SM. MCP-1/CCR2B-dependent loop upregulates MUC5AC and MUC5B in human airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L204–L215. | ||
Botelho FM, Bauer CM, Finch D, et al. IL-1alpha/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smoke-induced neutrophilia in mice. PLoS One. 2011;6(12):e28457. | ||
Rogliani P, Calzetta L, Ora J, Matera MG. Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2015;31:15–27. | ||
Varricchi G, Bagnasco D, Borriello F, Heffler E, Canonica GW. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr Opin Allergy Clin Immunol. 2016;16(2):186–200. | ||
Roos AB, Sanden C, Mori M, Bjermer L, Stampfli MR, Erjefalt JS. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191(11):1232–1241. | ||
Zhang J, Chu S, Zhong X, Lao Q, He Z, Liang Y. Increased expression of CD4+IL-17+ cells in the lung tissue of patients with stable chronic obstructive pulmonary disease (COPD) and smokers. Int Immunopharmacol. 2013;15(1):58–66. | ||
Shan M, Cheng HF, Song LZ, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1(4):4ra10. | ||
Roos AB, Sethi S, Nikota J, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(4):428–437. | ||
Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and Treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. | ||
Freeman CM, Martinez CH, Todt JC, et al. Acute exacerbations of chronic obstructive pulmonary disease are associated with decreased CD4+ & CD8+ T cells and increased growth & differentiation factor-15 (GDF-15) in peripheral blood. Respir Res. 2015;16:94. | ||
Zhang X, Zheng H, Zhang H, et al. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine. 2011;56(3):717–725. | ||
Freeman CM, McCubbrey AL, Crudgington S, et al. Basal gene expression by lung CD4+ T cells in chronic obstructive pulmonary disease identifies independent molecular correlates of airflow obstruction and emphysema extent. PLoS One. 2014;9(5):e96421. | ||
Kang MJ, Choi JM, Kim BH, et al. IL-18 induces emphysema and airway and vascular remodeling via IFN-gamma, IL-17A, and IL-13. Am J Respir Crit Care Med. 2012;185(11):1205–1217. | ||
Nakajima T, Owen C. Interleukin-18: the master regulator driving destructive and remodeling processes in the lungs of patients with chronic obstructive pulmonary disease? Am J Respir Crit Care Med. 2012;185(11):1137–1139. | ||
MedImmune LLC. A study to evaluate the safety of MEDI2338 in subjects with chronic obstructive pulmonary disease; 2013. Available from: https://clinicaltrials.gov/ct2/show/results/NCT01322594?term=MEDI2338&rank=1. NLM identifier: NCT01322594. Accessed January 15, 2016. | ||
Hodge G, Reynolds PN, Holmes M, Hodge S. Differential expression of pro-inflammatory cytokines in intra-epithelial T cells between trachea and bronchi distinguishes severity of COPD. Cytokine. 2012;60(3):843–848. | ||
Norman P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin Investig Drugs. 2015;24(3):383–392. | ||
Robinson C, Xia K, Russell P, et al. A 12 week, randomized, double-blind, placebo controlled study of the efficacy of RV568 (JNJ49095397), a narrow spectrum kinase inhibitor, in COPD patients. Am J Respir Crit Care Med. 2016;193:A6843. | ||
Fung-Leung WP. Phosphoinositide 3-kinase delta (PI3Kdelta) in leukocyte signaling and function. Cell Signal. 2011;23(4):603–608. | ||
Down K, Amour A, Baldwin IR, et al. Optimization of novel indazoles as highly potent and selective inhibitors of phosphoinositide 3-kinase delta for the treatment of respiratory disease. J Med Chem. 2015;58(18):7381–7399. | ||
Watz H, Bock D, Meyer M, et al. Inhaled pan-selectin antagonist bimosiamose attenuates airway inflammation in COPD. Pulm Pharmacol Ther. 2013;26(2):265–270. | ||
Lakshmi SP, Reddy AT, Zhang Y, et al. Down-regulated peroxisome proliferator-activated receptor gamma (PPARgamma) in lung epithelial cells promotes a PPARgamma agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. 2014;289(10):6383–6393. | ||
Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-gamma ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. | ||
Shan M, You R, Yuan X, et al. Agonistic induction of PPARgamma reverses cigarette smoke-induced emphysema. J Clin Invest. 2014;124(3):1371–1381. | ||
Rinne ST, Liu CF, Feemster LC, et al. Thiazolidinediones are associated with a reduced risk of COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2015;10:1591–1597. | ||
Shujaat A, Bajwa AA, Cury JD. Pulmonary hypertension secondary to COPD. Pulm Med. 2012;2012:203952. | ||
Valerio G, Bracciale P, Grazia D’Agostino A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2009;3(1):15–21. | ||
Zakynthinos E, Daniil Z, Papanikolaou J, Makris D. Pulmonary hypertension in COPD: pathophysiology and therapeutic targets. Curr Drug Targets. 2011;12(4):501–513. | ||
Stolz D, Rasch H, Linka A, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. 2008;32(3):619–628. | ||
Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. 2014;370(23):2201–2210. | ||
Neukamm A, Hoiseth AD, Einvik G, et al. Rosuvastatin treatment in stable chronic obstructive pulmonary disease (RODEO): a randomized controlled trial. J Intern Med. 2015;278(1):59–67. | ||
Dinavahi SS, Nyayapathy S, Perumal Y, Dharmarajan S, Viswanadha S. Combined inhibition of PDE4 and PI3Kdelta modulates the inflammatory component involved in the progression of chronic obstructive pulmonary disease. Drug Res (Stuttg). 2014;64(4):214–219. | ||
Mulhall AM, Droege CA, Ernst NE, Panos RJ, Zafar MA. Phosphodiesterase 4 inhibitors for the treatment of chronic obstructive pulmonary disease: a review of current and developing drugs. Expert Opin Investig Drugs. 2015;24(12):1597–1611. | ||
De Savi C, Cox RJ, Warner DJ, et al. Efficacious inhaled PDE4 inhibitors with low emetic potential and long duration of action for the treatment of COPD. J Med Chem. 2014;57(11):4661–4676. | ||
Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax. 2016;71(2):126–132. | ||
Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. 2007;62(8):706–713. | ||
MacNee W. Oxidants/antioxidants and COPD. Chest. 2000;117(5 suppl 1):303S–317S. | ||
Bernardo I, Bozinovski S, Vlahos R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol Ther. 2015;155:60–79. | ||
Bocci V, Valacchi G. Nrf2 activation as target to implement therapeutic treatments. Front Chem. 2015;3:4. | ||
Wise RA, Holbrook JT, Criner G, et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: a randomized, double-blind, placebo controlled trial. PLoS One. 2016;11(11):e0163716. | ||
Dal Negro RW, Visconti M, Turco P. Efficacy of erdosteine 900 versus 600 mg/day in reducing oxidative stress in patients with COPD exacerbations: results of a double blind, placebo-controlled trial. Pulm Pharmacol Ther. 2015;33:47–51. | ||
Moretti M. Erdosteine: its relevance in COPD treatment. Expert Opin Drug Metab Toxicol. 2009;5(3):333–343. | ||
Tse HN, Raiteri L, Wong KY, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest. 2013;144(1):106–118. | ||
Zheng JP, Wen FQ, Bai CX, et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med. 2014;2(3):187–194. | ||
Decramer M, Rutten-van Molken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet. 2005;365(9470):1552–1560. | ||
Pela R, Calcagni AM, Subiaco S, Isidori P, Tubaldi A, Sanguinetti CM. N-acetylcysteine reduces the exacerbation rate in patients with moderate to severe COPD. Respiration. 1999;66(6):495–500. | ||
Schermer T, Chavannes N, Dekhuijzen R, et al. Fluticasone and N-acetylcysteine in primary care patients with COPD or chronic bronchitis. Respir Med. 2009;103(4):542–551. | ||
Yu H, Li Q, Kolosov VP, Perelman JM, Zhou X. Regulation of cigarette smoke-mediated mucin expression by hypoxia-inducible factor-1alpha via epidermal growth factor receptor-mediated signaling pathways. J Appl Toxicol. 2012;32(4):282–292. | ||
Rubin BK, Priftis KN, Schmidt HJ, Henke MO. Secretory hyperresponsiveness and pulmonary mucus hypersecretion. Chest. 2014;146(2):496–507. | ||
Polosukhin VV, Cates JM, Lawson WE, et al. Hypoxia-inducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J Pathol. 2011;224(2):203–211. | ||
Curran DR, Cohn L. Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol. 2010;42(3):268–275. | ||
Rennard SI, Sun SX, Tourkodimitris S, et al. Roflumilast and dyspnea in patients with moderate to very severe chronic obstructive pulmonary disease: a pooled analysis of four clinical trials. Int J Chron Obstruct Pulmon Dis. 2014;9:657–673. | ||
Grubek-Jaworska H, Paplinska M, Hermanowicz-Salamon J, et al. IL-6 and IL-13 in induced sputum of COPD and asthma patients: correlation with respiratory tests. Respiration. 2012;84(2):101–107. | ||
Lee JS, Rosengart MR, Kondragunta V, et al. Inverse association of plasma IL-13 and inflammatory chemokines with lung function impairment in stable COPD: a cross-sectional cohort study. Respir Res. 2007;8:64. | ||
Tanabe T, Kanoh S, Tsushima K, Yamazaki Y, Kubo K, Rubin BK. Clarithromycin inhibits interleukin-13-induced goblet cell hyperplasia in human airway cells. Am J Respir Cell Mol Biol. 2011;45(5):1075–1083. | ||
Rennard SI, Flavin SK, Agarwal PK, Lo KH, Barnathan ES. Long-term safety study of infliximab in moderate-to-severe chronic obstructive pulmonary disease. Respir Med. 2013;107(3):424–432. | ||
Aaron SD, Vandemheen KL, Maltais F, et al. TNFalpha antagonists for acute exacerbations of COPD: a randomised double-blind controlled trial. Thorax. 2013;68(2):142–148. | ||
Chung KF. Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy. 2005;4(6):619–625. | ||
Betts JC, Mayer RJ, Tal-Singer R, et al. Gene expression changes caused by the p38 MAPK inhibitor dilmapimod in COPD patients: analysis of blood and sputum samples from a randomized, placebo-controlled clinical trial. Pharmacol Res Perspect. 2015;3(1):e00094. | ||
MacNee W, Allan RJ, Jones I, De Salvo MC, Tan LF. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax. 2013;68(8):738–745. | ||
Singh D, Smyth L, Borrill Z, Sweeney L, Tal-Singer R. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J Clin Pharmacol. 2010;50(1):94–100. | ||
Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. | ||
Geraghty P, Hardigan A, Foronjy RF. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am J Respir Cell Mol Biol. 2014;50(3):559–570. | ||
Gagliardo R, Chanez P, Profita M, et al. IkappaB kinase-driven nuclear factor-kappaB activation in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2011;128(3):635–645.e631–e632. | ||
Marian E, Baraldo S, Visentin A, et al. Up-regulated membrane and nuclear leukotriene B4 receptors in COPD. Chest. 2006;129(6):1523–1530. | ||
Woodruff PG, Albert RK, Bailey WC, et al. Randomized trial of zileuton for treatment of COPD exacerbations requiring hospitalization. COPD. 2011;8(1):21–29. | ||
Snell N, Foster M, Vestbo J. Efficacy and safety of AZD1981, a CRTH2 receptor antagonist, in patients with moderate to severe COPD. Respir Med. 2013;107(11):1722–1730. | ||
Mantell S, Jones R, Trevethick M. Design and application of locally delivered agonists of the adenosine A(2A) receptor. Expert Rev Clin Pharmacol. 2010;3(1):55–72. | ||
Mathioudakis A, Chatzimavridou-Grigoriadou V, Evangelopoulou E, Mathioudakis G. Vasoactive intestinal Peptide inhaled agonists: potential role in respiratory therapeutics. Hippokratia. 2013;17(1):12–16. | ||
Burian B, Storka A, Marzluf BA, et al. Vasoactive intestinal peptide (VIP) receptor expression in monocyte-derived macrophages from COPD patients. Peptides. 2010;31(4):603–608. | ||
Onoue S, Aoki Y, Matsui T, et al. Formulation design and in vivo evaluation of dry powder inhalation system of new vasoactive intestinal peptide derivative ([R(15, 20, 21), L(17), A(24, 25), des-N(28)]-VIP-GRR) in experimental asthma/COPD model rats. Int J Pharm. 2011;410(1–2):54–60. | ||
Wu D, Lee D, Sung YK. Prospect of vasoactive intestinal peptide therapy for COPD/PAH and asthma: a review. Respir Res. 2011;12:45. | ||
Stoll P, Bahker A, Ulrich M, et al. The dendritic cell high-affinity IgE receptor is overexpressed in both asthma and severe COPD. Clin Exp Allergy. 2016;46(4):575–583. | ||
Raverdeau M, Mills KH. Modulation of T cell and innate immune responses by retinoic Acid. J Immunol. 2014;192(7):2953–2958. | ||
Ho TW, Tsai YJ, Ruan SY, et al. In-hospital and one-year mortality and their predictors in patients hospitalized for first-ever chronic obstructive pulmonary disease exacerbations: a nationwide population-based study. PLoS One. 2014;9(12):e114866. | ||
Kaczmarek P, Sladek K, Skucha W, et al. The influence of simvastatin on selected inflammatory markers in patients with chronic obstructive pulmonary disease. Pol Arch Med Wewn. 2010;120(1–2):11–17. | ||
Lawes CM, Thornley S, Young R, et al. Statin use in COPD patients is associated with a reduction in mortality: a national cohort study. Prim Care Respir J. 2012;21(1):35–40. | ||
Hodge G, Jersmann H, Tran HB, et al. Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes. Respir Res. 2015;16:130. | ||
Podolin PL, Foley JP, Carpenter DC, et al. T cell depletion protects against alveolar destruction due to chronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L312–L323. | ||
Moretto N, Caruso P, Bosco R, et al. CHF6001 I: a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration. J Pharmacol Exp Ther. 2015;352(3):559–567. | ||
Villetti G, Carnini C, Battipaglia L, et al. CHF6001 II: a novel phosphodiesterase 4 inhibitor, suitable for topical pulmonary administration – in vivo preclinical pharmacology profile defines a potent anti-inflammatory compound with a wide therapeutic window. J Pharmacol Exp Ther. 2015;352(3):568–578. | ||
Watz H, Mistry SJ, Lazaar AL; Investigators IPC. Safety and tolerability of the inhaled phosphodiesterase 4 inhibitor GSK256066 in moderate COPD. Pulm Pharmacol Ther. 2013;26(5):588–595. | ||
Goudie AR, Lipworth BJ, Hopkinson PJ, Wei L, Struthers AD. Tadalafil in patients with chronic obstructive pulmonary disease: a randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2014;2(4):293–300. | ||
Kuna P, Jenkins M, O’Brien CD, Fahy WA. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir Med. 2012;106(4):531–539. | ||
Henriksen PA. The potential of neutrophil elastase inhibitors as anti-inflammatory therapies. Curr Opin Hematol. 2014;21(1):23–28. | ||
Dahl R, Titlestad I, Lindqvist A, et al. Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial. Pulm Pharmacol Ther. 2012;25(2):169–177. | ||
Woodruff PG, Wolff M, Hohlfeld JM, et al. Safety and efficacy of an inhaled epidermal growth factor receptor inhibitor (BIBW 2948 BS) in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(5):438–445. | ||
Damera G, Jester WF, Jiang M, et al. Inhibition of myristoylated alanine-rich C kinase substrate (MARCKS) protein inhibits ozone-induced airway neutrophilia and inflammation. Exp Lung Res. 2010;36(2):75–84. | ||
Ha EV, Rogers DF. Novel therapies to inhibit mucus synthesis and secretion in airway hypersecretory diseases. Pharmacology. 2016;97(1–2):84–100. | ||
Astrand AB, Hemmerling M, Root J, et al. Linking increased airway hydration, ciliary beating, and mucociliary clearance through ENaC inhibition. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L22–L32. | ||
Black PN, Morgan-Day A, McMillan TE, Poole PJ, Young RP. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344]. BMC Pulm Med. 2004;4:13. | ||
Ayfer Aytemur Z, Baysak A, Ozdemir O, Kose T, Sayiner A. N-acetylcysteine in patients with COPD exacerbations associated with increased sputum. Wien Klin Wochenschr. 2015;127(7–8):256–261. | ||
Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest. 2013;144(2):498–506. | ||
Bodas M, Min T, Mazur S, Vij N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J Immunol. 2011;186(1):602–613. | ||
Jones PW, Rames AD. TESRA (treatment of emphysema with a selective retinoid agonist) study results. Am J Respir Crit Care Med. 2011;183:A6418. | ||
Stolk J, Stockley RA, Stoel BC, et al. Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor. Eur Respir J. 2012;40(2):306–312. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.