")
Back to Journals » OncoTargets and Therapy » Volume 7
Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma
Authors Neesse A, Krug S, Gress TM, Tuveson DA, Michl P
Received 24 September 2013
Accepted for publication 2 November 2013
Published 18 December 2013 Volume 2014:7 Pages 33—43
DOI https://doi.org/10.2147/OTT.S38111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Albrecht Neesse,1 Sebastian Krug,1 Thomas M Gress,1 David A Tuveson,2 Patrick Michl1
1Department of Gastroenterology, Endocrinology, Infectiology and Metabolism, Philipps University Marburg, Marburg, Germany; 2Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
Abstract: Pancreatic ductal adenocarcinoma is a stroma-rich and highly challenging cancer to treat. Over recent years, it has become increasingly evident that the complex network of soluble cytokines, growth factors, proteases, and components of the extracellular matrix collaboratively interact within the tumor microenvironment, sustaining and driving cancer cell proliferation, invasion, and early metastasis. More recently, the tumor microenvironment has also been appreciated to mediate therapeutic resistance in pancreatic ductal adenocarcinoma, thus opening numerous avenues for novel therapeutic explorations. Inert and soluble components of the tumor stroma have been targeted in order to break down the extracellular matrix scaffold, relieve vessel compression, and increase drug delivery to hypovascular tumors. Moreover, targeting of antiapoptotic, immunosuppressive, and pro-proliferative effects of the tumor stroma provides novel vantage points of attack. This review focuses on current and future developments in pancreatic cancer medicine, with a particular emphasis on biophysical and biochemical approaches that target the tumor microenvironment.
Keywords: pancreatic cancer, chemoresistance, tumor stroma, drug delivery
Introduction to pancreatic cancer
Pancreatic ductal adenocarcinoma (PDA) is an extremely stroma-rich and highly aggressive solid tumor within the exocrine compartment of the pancreatic gland, and its incidence rates are steadily increasing in the Western world.1 In the US, an estimated 43,920 new cases are diagnosed each year, and many diagnosed patients succumb to the disease after only a few months.2 The inability of clinicians to substantially improve the prognosis of PDA patients over the last few decades is reflected in a virtually unchanged 5-year survival rate of 5%–6% and a median survival of less than 12 months.2 Potential reasons for such a poor clinical outcome reflect both the clinical and biological characteristics of pancreatic cancer. First, PDA is chiefly a disease of the elderly, and patients often initially note relatively nonspecific symptoms, including back pain and dyspepsia.3 These symptoms often point towards degenerative and relatively harmless conditions that may not immediately raise concerns on the part of patients and general practitioners. More alarming symptoms include new onset of diabetes,4–6 painless jaundice, weight loss, or spontaneous deep vein thrombosis.7,8 As a result, a timely diagnostic work-up, including use of specialized imaging modalities such as abdominal ultrasound scans, computed tomography (CT), or magnetic resonance imaging is often delayed for several months after initial symptom onset.9 Second, no specific blood or urine biomarkers are presently available that would help to identify subgroups of patients with increased risk of developing PDA. Due to these issues and the lack of early detection methods, 80% of PDA patients are diagnosed with unresectable locally advanced or metastatic disease.
Families having at least two first-degree relatives with confirmed PDA that does not meet the criteria of other inherited tumor syndromes, such as Peutz–Jeghers syndrome or hereditary chronic pancreatitis, comprise a small subset of the overall population of PDA patients (5%–10%).10 Increased surveillance by high resolution imaging and various chemopreventive strategies are under investigation as potential modalities to detect PDA early or prevent its onset.11–16 While the optimal treatment of locally advanced PDA without detectable distant metastases remains to be determined, patients with distant metastases are candidates for systemic palliative chemotherapy dependent on comorbidities and general performance status. A relative minority of patients (15%–20%) who qualify for pancreatic resection receive a 6-month course of adjuvant gemcitabine or 5-fluorouracil plus folinic acid chemotherapy (Figure 1).17 Perioperative morbidity and mortality have improved in high-volume centers owing to improved surgical resection techniques, postoperative care, and multidisciplinary approaches;18–20 however, tumor relapse is unfortunately common, and the median survival rate for patients with R0 resection is approximately 2 years, with a 5-year survival of 15%–20%.21
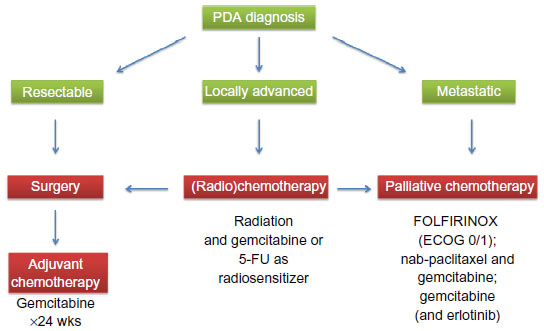 | Figure 1 Schematic overview of current clinical treatment strategies for pancreatic cancer patients according to whether they have resectable, locally advanced, or metastatic pancreatic cancer, and ECOG performance status 0–5. |
This review focuses on current and future developments in pancreatic cancer medicine, with a particular emphasis on novel treatment options that target the tumor microenvironment, in particular the tumor stroma.
Patient outcomes and current treatment options
Despite intensive clinical research activities aiming to identify effective chemotherapies, PDA has remained virtually unresponsive to conventional and targeted therapies.22 A large number of randomized clinical trials have been conducted in an attempt to improve overall survival in PDA patients. To this end, the nucleoside analogue gemcitabine was combined with a second cytotoxic agent, eg, 5-fluorouracil, capecitabine, cisplatin, oxaliplatin, irinotecan, pemetrexed, or exatecan, but all combinations failed to achieve significant improvement in overall survival.23–29 Although much hope lay in novel targeted therapies such as the vascular endothelial growth factor inhibitor bevacizumab,30 the matrix metalloproteinase inhibitor marimastat,31,32 and the antiepidermal growth factor receptor agent cetuximab,33,34 none of these drugs alone or in combination with standard chemotherapies had a significant impact on patient outcome. Therefore, the relatively well tolerated nucleoside analogue gemcitabine remained the standard of care chemotherapy in most countries, mainly on the basis of modest patient benefit with only a marginal increase in median survival.35
The only targeted agent that has been approved for PDA patients is the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib (Tarceva®, Genentech, CA, USA), since the combination of gemcitabine and erlotinib conferred a marginal survival benefit over gemcitabine alone (6.24 months versus 5.91 months).36 Although statistically significant, the clinical relevance remains questionable. More recently, the gemcitabine-free FOLFIRINOX protocol (folinic acid, fluorouracil, irinotecan, and oxaliplatin) was reported, and achieved a significant survival benefit for patients with metastatic PDA compared with gemcitabine monotherapy (11.1 months versus 6.8 months).37 Although FOLFIRINOX significantly improves quality of life compared with gemcitabine,38 severe side effects such as grade 3 and 4 neutropenia and dehydration limit the use of this aggressive combination chemotherapy to carefully selected patients with good performance status. More recently, results from the Phase III Metastatic Pancreatic Adenocarcinoma Trial (MPACT) in 861 patients comparing nanoformulated albumin-bound paclitaxel (nab-paclitaxel, Celgene, NY, USA) with gemcitabine and gemcitabine monotherapy were presented. The data show a significant survival benefit for the combination of nab-paclitaxel and gemcitabine (8.5 versus 6.7 months), with a reasonable toxicity profile.39 Although the mechanism of action of nab-paclitaxel remains unclear, its tolerability profile is superior to that of conventional paclitaxel dissolved in cremophor, and the MPACT regimen will likely become a new standard of care treatment and new backbone for novel targeted therapies in the future. Furthermore, the nab-paclitaxel and gemcitabine combination is undergoing investigation in localized resectable and unresectable PDA for its potential clinical benefit in these disease stages. Figure 1 shows the different clinical treatment algorithms for resectable, locally advanced, and metastasized PDA.
Cellular and molecular evolution of pancreatic cancer
Pancreatic carcinogenesis occurs through an accumulation of genetic alterations that result in deregulation of tumor cell-autonomous and nontumor cell-autonomous pathways. These genetic changes are accompanied by typical morphologic and histologic alterations in epithelial, stromal, and inflammatory cells within the pancreas that eventually culminate in desmoplastic, highly invasive, and metastatic ductal adenocarcinoma. Activating mutations in the K-ras gene represent a signature event in almost all pancreatic cancers (>90%), followed by subsequent somatic mutations involving the tumor suppressor genes p16, p53, and DPC4/SMAD4.40–45 Interestingly, the number of actual mutations of these key drivers of pancreatic carcinogenesis correlates positively with a poor prognosis and shortened survival for patients.46 Mechanistically, oncogenic K-ras activation governs a multitude of mitogenic signals with profound cell-autonomous and noncell-autonomous effects that initiate epithelial transformation, dynamic rearrangement of a proinflammatory and immunosuppressive microenvironment, metabolic requirements, and finally drive frank malignancy.47–54 Recent evidence from genetically engineered mouse models (GEMMs) with pancreas-specific and doxycycline-inducible expression of oncogenic K-rasG12D indicates that abrogation of mutant K-ras in established tumors led to dramatic tumor shrinkage and depletion of the surrounding stroma after only a few days,55,56 further highlighting the key function of mutant K-ras in shaping tumor biology in PDA.
Global sequencing analysis and transposon-mediated insertional mutagenesis screens have also discovered genetic alterations at low frequency and provide multiple examples for the genomic instability and heterogeneity of PDA.57–59 For the small subset of patients with inherited predisposition to PDA, several germline mutations, including BRCA2, STK11/LKB1, p16/CDKN2A, ATM,60 and PRSS1, have been reported.10
Alongside these molecular discoveries, a unique histopathologic progression model similar to that of the adenoma-carcinoma sequence in the development of colon cancer61 was proposed to describe the progression from a normal pancreas via preneoplastic lesions to invasive cancer.62,63 Preneoplastic lesions are classified as pancreatic intraepithelial neoplasms (PanINs) 1a/b, 2, and 3, according to their stepwise accumulation of histopathologic and molecular alterations. The discovery that high-grade PanIN lesions increase the risk of developing PDA has sparked attempts to detect these lesions early by cross-sectional and endoscopic imaging techniques.16,64,65 Further, early (partial) pancreatectomy in patients with high-grade PanIN lesions is considered in high-risk individuals (eg, familial PDA), but the ideal timing is still debated and data on overall survival from randomized clinical trials are currently not available.10
The traditional PanIN-PDA sequence (“ductal carcinogenesis”) has recently been challenged by the description of an alternative route of pancreatic carcinogenesis known as “acinoductal carcinogenesis”. Careful histopathologic investigations by Esposito et al identified tubular complexes within areas of acinar ductal metaplasia that form atypical flat lesions and may bypass the common PanIN precursor stages and directly evolve to invasive cancer.66–68 Histologic analysis of sporadic PDA cases confirmed the presence of tubular complexes in almost 80% of cases, and atypical flat lesions were also detected in cases of familial pancreatic cancer. These exciting findings need to be confirmed in larger series of sporadic pancreatic cancer cases and may provide novel insights into the development of PDA, with potentially profound implications in future diagnostic and preventive algorithms. Interestingly, earlier studies in GEMMs of pancreas cancer had also pointed towards a putative role of the acinar cell compartment in driving carcinogenesis with and without concomitant inflammation.69–72
Role of the stroma in pancreatic cancer
Histologically, PDA is an extremely stroma-rich and hypovascular tumor, and indeed, most of the pancreatic tumor mass consists of activated (myo)fibroblasts, immune cells, and extracellular matrix components, such as collagen, desmin, fibronectin, and hyaluronic acid.9,73,74 Over recent years, it has become increasingly evident that the complex network of soluble cytokines, growth factors, proteases, and extracellular matrix components collaboratively interact within the tumor microenvironment, sustaining and driving cancer cell proliferation, invasion, early metastasis, and therapeutic resistance.75–80 An important subtype within the stromal population is pancreatic stellate cells, which have emerged as pancreas-specific myofibroblasts and share morphologic and functional characteristics with hepatic stellate cells.81–83 Activated pancreatic stellate cells secrete profibrotic proteins abundantly and interact with tumor cells in multiple ways to establish and maintain the pronounced desmoplastic reaction in PDA (Figure 2).84–86 The clinical relevance is highlighted by histologic characteristics of patient samples, suggesting that the extent of the stromal reaction correlates with shortened survival in patients undergoing surgery.87 The complex cellular and biochemical interactions of pancreatic stellate cells and cancer cells have limited faithful in vitro investigation to traditional two-dimensional coculture assays in the laboratory. Therefore, intensive efforts are currently being made to establish three-dimensional or organotypic culture systems where cancer cells and stromal cells can be grown within a reconstituted extracellular matrix gel to study the tumor-stromal crosstalk and test novel compounds.88,89
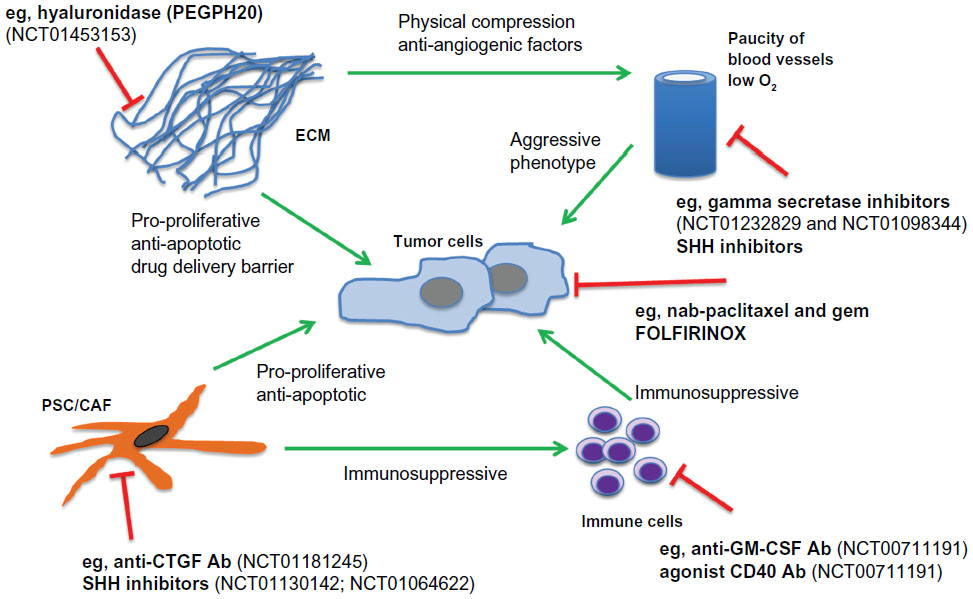 | Figure 2 Schematic of tumor microenvironment crosstalk and interdependence in PDA, with a particular focus on novel experimental therapeutic interventions and clinical trials. |
The advent of various GEMMs of pancreas cancer has marked a milestone for the scientific community in understanding the biological implications of the tumor stroma and provides ample opportunities for preclinical testing of novel agents directed against cell-autonomous and noncell-autonomous targets.90–93 The most commonly used pancreatic cancer GEMM bears an activating K-rasG12D allele that is conditionally activated in pancreatic progenitor cells by crossing mice with transgenic strains that express Cre recombinase in pancreatic lineages (PdxCre or p48Cre). These mice are referred to as “KC” mice and develop murine PanIN lesions with 100% penetrance, and progress to PDA with a long latency.94 The addition of a dominant negative mutation in the tumor suppressor gene p53 (Trp53R172H/+) greatly accelerates pancreatic tumor development and penetrance at an early age, and these mice are accordingly termed “KPC” mice.95 In contrast with traditional xenograft tumors, GEMMs faithfully recapitulate human PDA, including the presence of abundant tumor stroma and comorbidities such as cachexia, jaundice, metastasis to distant sites, and activation of biochemical pathways. Although the KPC is a faithful mouse model of PDA, the resources and staffing required to maintain, image, and treat a large cohort of animals is substantial and beyond the practical abilities of a single laboratory.
The first ultrasound-guided and controlled preclinical study in KPC mice was performed by Ken Olive in the Tuveson laboratory and targeted the profibrotic Sonic Hedgehog (SHH) pathway. SHH plays important roles during pancreas organ development, and is re-expressed during malignant transformation to activate and expand stromal rather than epithelial cells, thus promoting desmoplasia in pancreatic carcinogenesis.96–98 Strikingly, pharmacologic inhibition of SHH by the smoothened inhibitor IPI-926 resulted in marked stromal depletion and increased microvessel density and patency, paralleled by significantly improved delivery of several chemotherapeutics in the KPC mouse model.99 Although the intrinsic effects of SHH inhibition on chemosensitivity could not be excluded, this study identified the tumor stroma as a biophysical barrier for drug delivery. Thus, the tumor microenvironment, in particular the dense tumor stroma, is now considered to be a potential reason for the failure of most systemic therapies in PDA. Indeed, hypoenhancing masses are visualized in PDA patients whenever contrast-enhanced imaging is used, and poor perfusion has been associated with an aggressive phenotype.100
Novel targets in the microenvironment of pancreatic cancer
The biophysical role of the pancreatic tumor stroma as a barrier to drug delivery has been the focus of intensive clinical and preclinical research over the last 3 years, and also attracted attention in other tumor entities.101,102 Surprisingly, an SHH inhibitor IPI-926 (saridegib, Infinity, MA, USA) and GDC-0449 (vismodegib, Genentech, CA, USA) both failed in Phase II clinical trials, and investigations are still ongoing to comprehend the discrepancy between the clinical and preclinical data. However, the SHH signaling cascade remains an intriguing and widely investigated pathway in PDA, and pharmacologic inhibition may still be a beneficial therapeutic option in the future, depending on the specific compound and cotreatments.103
Other solid and soluble components of the tumor microenvironment have been targeted in order to break down the extracellular matrix scaffold, relieve vessel compression, and increase drug accumulation within the tumor (Figure 2). One prominent example is the enzymatic depletion of hyaluronic acid, a glycosaminoglycan, by human recombinant PEGylated hyaluronidase (PEGPH20). Pancreatic cancers are extremely rich in the megadalton form of hyaluronic acid, and the solvation of water by hyaluronic acid is thought to be responsible for the high interstitial fluid pressure in PDA that results in compression of intratumoral blood vessels.104,105 The Hingorani group has shown that treatment with PEGPH20 in murine pancreas tumors results in decreased levels of intratumoral fluid pressure in PDA, and this group and ours have noted that treatment with PEGPH20 results in the re-expansion of blood vessels and improved drug delivery, accompanied by slowing of tumor growth and prolonged survival in KPC mice.106,107 The results of a Phase I/II dose-escalation study (NCT01453153) with PEGPH20 in combination with gemcitabine in 28 patients with previously untreated stage IV pancreatic cancer were presented at the 2013 annual scientific meeting of the American Society of Clinical Oncology, and suggested promising efficacy, particularly in patients with a high intratumoral content of hyaluronic acid.108
Connective tissue growth factor (CTGF/CCN2) is a pleiotropic growth factor that is overexpressed in human and murine pancreas tumors. Therapeutic inhibition of CTGF using a monoclonal human antibody (FG-3019, Fibrogen, CA, USA) resulted in significantly increased induction of tumor cell apoptosis in KPC mice when combined with gemcitabine.78 Notably, neither stromal depletion nor increased drug delivery was observed. Rather, stromal-derived CTGF impinged on the antiapoptotic machinery in tumor cells, and the X-linked inhibitor of apoptosis protein was downregulated upon treatment with FG-3019. Finally, cotreatment with FG-3019 and gemcitabine resulted in slowing of murine tumors and prolonged survival in KPC mice.78 FG-3019 in combination with gemcitabine and erlotinib (an epidermal growth factor receptor tyrosine kinase inhibitor) is currently being investigated in a Phase I safety and bioactivity study in patients with locally advanced and metastasized PDA (NCT01181245).
Secreted protein acidic and rich in cysteine (SPARC) is overexpressed by cancer-associated fibroblasts and represents another intriguing target in PDA. Results from a Phase I/II clinical study showed that patients with high stromal SPARC levels responded better to nab-paclitaxel and gemcitabine (median survival 17.8 months versus 8.1 months for low SPARC),109 suggesting that the albumin-binding protein SPARC may act as a novel biomarker for PDA that retains nab-paclitaxel to accumulate the drug intratumorally. In contrast, two independent studies identified high expression of SPARC as a negative prognostic marker for patients with resectable and unresectable PDA.110,111 Our group has undertaken several preclinical studies regarding SPARC and the efficacy and mechanism of action of nab-paclitaxel in combination with gemcitabine. We reported remarkable therapeutic efficacy for nab-paclitaxel in highly treatment-resistant KPC tumors. Importantly, nab-paclitaxel was much better tolerated than cremophor-paclitaxel and could be administered in more than four-fold higher concentrations. Moreover, we identified a synergistic effect of nab-paclitaxel through reactive oxygen species-mediated degradation of the primary gemcitabine-metabolizing enzyme, cytidine deaminase. Therefore, combinations of nab-paclitaxel and gemcitabine resulted in increased intratumoral gemcitabine levels.112 However, genetic ablation of SPARC in the KPC model neither resulted in decreased intratumoral (nab-)paclitaxel levels nor altered the response to treatment.113 Nonetheless, elevated serum SPARC levels may play a role in paclitaxel uptake in certain PDA patients and could be investigated noninvasively in PDA patients prior to starting nab-paclitaxel-based chemotherapies.113 The exact function of SPARC and the mechanism of action of nab-paclitaxel remains a subject of intense clinical114 and preclinical investigation,115 and will help to systematically evaluate the predictive power of different in vivo models by rigorous comparison with the human data. To this end, tissue analysis of the MPACT trial is anxiously awaited by the field and should provide more answers on this exciting topic.
Partly owing to the hypovascular state of PDA, hypoxia is considered a hallmark feature that may predict more aggressive behavior and impair the response to therapies by providing a niche for slow-cycling, highly drug-resistant cells.116–118
Chemotherapeutic agents such as TH-302 are selectively activated in the hypoxic tumor microenvironment and are currently being investigated in preclinical119 and several clinical trials in PDA patients (NCT01746979, NCT00743379, NCT01144455, NCT01833546). Hypoxia is also known to stimulate the Notch signaling pathway, and gamma secretase inhibitors are currently under early clinical investigation in PDA patients (NCT01232829, NCT01098344). Experimental data in human cell lines and GEMMs underscore the therapeutic potential of Notch inhibitors in inducing treatment responses.120,121
The field of cancer immunotherapy is rapidly evolving and has recently provided fascinating insights into pancreatic carcinogenesis with potential therapeutic implications for patients. For instance, tumor-derived granulocyte-macrophage colony-stimulating factor has been ascribed a central role in mediating a proinflammatory and immunosuppressive tumor microenvironment in PDA, and abrogation of granulocyte-macrophage colony-stimulating factor blocked tumor development by inhibition of Gr-1+ CD11b+ and recruitment of cytotoxic CD8+ T-cells into the tumor microenvironment.122,123 However, immune surveillance does not inevitably depend on therapy-induced T-cells. A combined Phase I preclinical-clinical study (NCT00711191) investigated the effects of a CD40 agonist antibody in 21 patients with metastatic PDA, and showed promising clinical activity, with tumor regression in some patients.124,125 Mechanistically, activated macrophages, but not activated T-cell-infiltrated tumors, induced tumor cell death and depleted the tumor stroma.124 This is the first clear example that shows how closely interconnected the immune cell and stromal cell compartment is in PDA, and suggests that critical cross points between immune and stromal cells must be interrupted in order to achieve robust treatment responses.
Conclusion and future perspectives
Tumor-stromal interactions are highly complex and contribute to the key hallmarks of cancer, such as sustained proliferative signaling, angiogenesis, activation of invasion, and metastasis, as well as extracellular matrix remodeling.126 More recently, the tumor microenvironment has been increasingly appreciated as being instrumental in mediating resistance to therapy in PDA and other cancers, thus opening up numerous avenues for therapeutic exploration, for both the biophysical and biochemical approaches described above.
Emerging evidence of metabolic reprogramming driven by oncogenic K-ras,56 and the dependency on autophagy,51 a catabolic pathway degrading cellular organelles and macromolecules, highlight additional metabolic targets that will be investigated in the near future. Further, exciting data have been reported recently by the Fearon group in Cambridge, UK, showing that a subtype of stromal cells expressing fibroblast activation protein-α not only cause failure of immune surveillance in murine tumors but may also contribute to tumor syndromes, such as cachexia and anemia,127 symptoms that are most relevant for patient well-being and survival.
A critical and clinically relevant issue remains the dynamics of tumor cell dissemination, and whether PDA metastasizes early or late during disease progression. Whereas deep sequencing data for human PDA suggested a long latency (on average 17 years) for the occurrence of distant metastases,71 a computational analysis of 228 PDA patients supported the notion that spread of malignant cells represents an early event during carcinogenesis, and most patients may harbor distant metastases at earlier disease stages than previously anticipated.128 Provocative lineage tracing experiments in GEMMs of pancreatic cancer proposed a mechanism by which single mutant cells detach from the basement membrane by epithelial-to-mesenchymal transition to enter the blood circulation prior to the development of frank malignancy. The disseminated cells seeded the liver and showed stem cell properties, a process that was further promoted by pancreatic inflammation.129
These studies must be confirmed independently but may transform our understanding of the evolution of pancreatic cancer and prioritize our efforts toward investigational clinical trials that compare neoadjuvant cytotoxic and antimetastatic therapies with upfront surgery. Also, patients at risk for developing pancreatic cancer (eg, Peutz–Jeghers syndrome, hereditary pancreatitis, familial pancreatic cancer) should be considered for evaluation of anti-inflammatory and antimetastatic therapeutic approaches. Moreover, these studies may open new avenues to understand the genetic, epigenetic, and microenvironmental determinants that may explain long-term survivors and those who never develop metastases.130
GEMMs of PDA that recapitulate important aspects of the tumor microenvironment are critical tools for investigating tumor-stromal interactions in the laboratory and for testing novel compounds that target components of the microenvironment prior to clinical testing. Given the long list of failed clinical trials in the past, it remains speculative which compounds will make the difference for patients with pancreatic cancer. Therefore, it is timely for the field to consider including these GEMMs prior to evaluating therapies in the clinic.
To enhance our molecular understanding of treatment success or failure, clinicians should seek to obtain pretreatment biopsies from patients via endoscopic ultrasound-guided fine needle aspiration biopsy or endoscopic ultrasound-guided core biopsy. Limitations in the quantity and quality of biopsy samples requires optimized approaches to obtain tissue specimens for histologic and immunohistochemical analysis of stromal, inflammatory, and parenchymal tissue components,131 and post-treatment biopsies are highly desirable for monitoring the effects of treatment on tumor biology and to prospectively explore potential biomarkers. Contrast-enhanced endoscopic ultrasound combined with elastography is an additional noninvasive technique that may provide useful information before and during therapies.114,132,133 Further, experimental molecular imaging approaches in various mouse models of pancreas cancer have recently identified potentially promising candidates, such as plectin-1, cathepsins, and the tight-junction protein claudin-4, that could be deployed for early detection,134–137 and should now be rigorously evaluated in the clinical setting to improve early diagnosis of PDA.
To conclude, the last few years have seen a virtual explosion of knowledge in the field of basic and translational pancreatic cancer research, and we are hopeful that the continuing effort to translate these findings to the clinic will eventually benefit our patients. Targeting the tumor microenvironment provides a novel and much needed vantage point of attack, and we anticipate that several of the components within the tumor microenvironment described here will be exploited to achieve robust treatment responses for this recalcitrant tumor.
Acknowledgments
AN, PM, TMG, and DAT were supported by a European Community grant (EPC-TM-Net 256974). DAT was also supported by the Lustgarten Foundation for Pancreatic Cancer Research, and by the Cold Spring Harbor Laboratory Association.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | |
Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol. 2005;7(5):189–197. | |
Sah RP, Nagpal SJ, Mukhopadhyay D, Chari ST. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat Rev Gastroenterol Hepatol. 2013;10(7):423–433. | |
Aggarwal G, Ramachandran V, Javeed N, et al. Adrenomedullin is up-regulated in patients with pancreatic cancer and causes insulin resistance in beta cells and mice. Gastroenterology. 2012;143(6):1510–1517. e1511. | |
Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology. 2012;12(2):156–161. | |
Watanabe I, Sasaki S, Konishi M, et al. Onset symptoms and tumor locations as prognostic factors of pancreatic cancer. Pancreas. 2004;28(2):160–165. | |
Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol. 2004;5(11):655–663. | |
Erkan M, Hausmann S, Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9(8):454–467. | |
Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer – current knowledge. Nat Rev Gastroenterol Hepatol. 2012;9(8):445–453. | |
Fendrich V, Chen NM, Neef M, et al. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut. 2010;59(5):630–637. | |
Fendrich V, Schneider R, Maitra A, Jacobsen ID, Opfermann T, Bartsch DK. Detection of precursor lesions of pancreatic adenocarcinoma in PET-CT in a genetically engineered mouse model of pancreatic cancer. Neoplasia. 2011;13(2):180–186. | |
Fendrich V. Chemoprevention of pancreatic cancer-one step closer. Langenbecks Arch Surg. 2012;397(4):495–505. | |
Husain K, Centeno BA, Chen DT, et al. Prolonged survival and delayed progression of pancreatic intraepithelial neoplasia in LSL-KrasG12D/+;Pdx-1-Cre mice by vitamin E delta-tocotrienol. Carcinogenesis. 2013;34(4):858–863. | |
Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62(3):339–347. | |
Canto MI, Hruban RH, Fishman EK, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142(4):796–804. | |
Sultana A, Cox T, Ghaneh P, Neoptolemos JP. Adjuvant therapy for pancreatic cancer. Recent Results Cancer Res. 2012;196:65–88. | |
Crist DW, Sitzmann JV, Cameron JL. Improved hospital morbidity, mortality, and survival after the Whipple procedure. Ann Surg. 1987;206(3):358–365. | |
Lieberman MD, Kilburn H, Lindsey M, Brennan MF. Relation of perioperative deaths to hospital volume among patients undergoing pancreatic resection for malignancy. Ann Surg. 1995;222(5):638–645. | |
Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346(15):1128–1137. | |
Neoptolemos JP. Adjuvant treatment of pancreatic cancer. Eur J Cancer. 2011;47 Suppl 3:S378–S380. | |
Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol. 2013;6(4):321–337. | |
Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB 3rd. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J Clin Oncol. 2002;20(15):3270–3275. | |
Herrmann R, Bodoky G, Ruhstaller T, et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol. 2007;25(16):2212–2217. | |
Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD Phase III trial. J Clin Oncol. 2005;23(15):3509–3516. | |
Poplin E, Feng Y, Berlin J, et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2009;27(23):3778–3785. | |
Heinemann V, Quietzsch D, Gieseler F, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24(24):3946–3952. | |
Colucci G, Giuliani F, Gebbia V, et al. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer. 2002;94(4):902–910. | |
Rocha Lima CM, Green MR, Rotche R, et al. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol. 2004;22(18):3776–3783. | |
Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. | |
Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: a randomized trial. J Clin Oncol. 2001;19(15):3447–3455. | |
Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JA. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer. 2002;87(2):161–167. | |
Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol. 2010;28(22):3605–3610. | |
Fensterer H, Schade-Brittinger C, Muller HH, et al. Multicenter phase II trial to investigate safety and efficacy of gemcitabine combined with cetuximab as adjuvant therapy in pancreatic cancer (ATIP). Ann Oncol. 2013;24(10):2576–2581. | |
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | |
Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | |
Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. | |
Gourgou-Bourgade S, Bascoul-Mollevi C, Desseigne F, et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: results from the PRODIGE 4/ACCORD 11 randomized trial. J Clin Oncol. 2013;31(1):23–29. | |
Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691–1703. | |
Hahn SA, Hoque AT, Moskaluk CA, et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996;56(3):490–494. | |
Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–353. | |
Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20(10):1218–1249. | |
Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. | |
Pellegata NS, Sessa F, Renault B, et al. K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994;54(6):1556–1560. | |
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549–554. | |
Yachida S, White CM, Naito Y, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res. 2012;18(22):6339–6347. | |
Ardito CM, Gruner BM, Takeuchi KK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22(3):304–317. | |
Heid I, Lubeseder-Martellato C, Sipos B, et al. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011;141(2):719–730. e711–e717. | |
Morris JP 4th, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120(2):508–520. | |
Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. | |
Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. | |
Hamidi T, Algul H, Cano CE, et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J Clin Invest. 2012;122(6):2092–2103. | |
Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–469. | |
Fukuda A, Wang SC, Morris JP 4th, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):441–455. | |
Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–653. | |
Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–670. | |
Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. | |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | |
Perez-Mancera PA, Rust AG, van der Weyden L, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486(7402):266–270. | |
Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–46. | |
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–532. | |
Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–2972. | |
Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156(6):1821–1825. | |
Verna EC, Hwang C, Stevens PD, et al. Pancreatic cancer screening in a prospective cohort of high-risk patients: a comprehensive strategy of imaging and genetics. Clin Cancer Res. 2010;16(20):5028–5037. | |
Langer P, Kann PH, Fendrich V, et al. Five years of prospective screening of high-risk individuals from families with familial pancreatic cancer. Gut. 2009;58(10):1410–1418. | |
Aichler M, Seiler C, Tost M, et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol. 2012;226(5):723–734. | |
Esposito I, Seiler C, Bergmann F, Kleeff J, Friess H, Schirmacher P. Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas. 2007;35(3):212–217. | |
Esposito I, Konukiewitz B, Schlitter AM, Kloppel G. [New insights into the origin of pancreatic cancer. Role of atypical flat lesions in pancreatic carcinogenesis]. Pathologe. 2012;33 Suppl 2:189–193. German. | |
Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105(48):18913–18918. | |
Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. | |
Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. | |
Gidekel Friedlander SY, Chu GC, Snyder EL, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16(5):379–389. | |
Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60(6):861–868. | |
Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene. July 15, 2013. [Epub ahead of print.] | |
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. | |
Buchholz M, Biebl A, Neesse A, et al. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 2003;63(16):4945–4951. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Neesse A, Frese KK, Bapiro TE, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci U S A. 2013;110(30):12325–12330. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Dangi-Garimella S, Krantz SB, Barron MR, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011;71(3):1019–1028. | |
Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. | |
Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43(1):128–133. | |
Buchholz M, Kestler HA, Holzmann K, et al. Transcriptome analysis of human hepatic and pancreatic stellate cells: organ-specific variations of a common transcriptional phenotype. J Mol Med (Berl). 2005;83(10):795–805. | |
Ene-Obong A, Clear AJ, Watt J, et al. Activated pancreatic stellate cells sequester CD8+ T-cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–1132. | |
Froeling FE, Feig C, Chelala C, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141(4):1486–1497. | |
Erkan M, Reiser-Erkan C, Michalski CW, et al. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11(5):497–508. | |
Erkan M, Michalski CW, Rieder S, et al. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6(10):1155–1161. | |
Longati P, Jia X, Eimer J, et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:95. | |
Kadaba R, Birke H, Wang J, et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J Pathol. 2013;230(1):107–117. | |
Cook N, Jodrell DI, Tuveson DA. Predictive in vivo animal models and translation to clinical trials. Drug Discov Today. 2012;17(5–6):253–260. | |
Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7(9):645–658. | |
Perez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142(5):1079–1092. | |
Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2012;61(10):1488–1500. | |
Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. | |
Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. | |
Bailey JM, Swanson BJ, Hamada T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14(19):5995–6004. | |
Tian H, Callahan CA, DuPree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106(11):4254–4259. | |
Nolan-Stevaux O, Lau J, Truitt ML, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. | |
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | |
Komar G, Kauhanen S, Liukko K, et al. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin Cancer Res. 2009;15(17):5511–5517. | |
Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011;108(7):2909–2914. | |
Chauhan VP, Stylianopoulos T, Martin JD, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383–388. | |
Rosow DE, Liss AS, Strobel O, et al. Sonic Hedgehog in pancreatic cancer: from bench to bedside, then back to the bench. Surgery. 2012;152(3 Suppl 1):S19–S32. | |
Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008;18(4):288–295. | |
Thompson CB, Shepard HM, O’Connor PM, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9(11):3052–3064. | |
Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–120. | |
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. | |
Hingorani SR, Harris WP, Beck JT, et al. A phase Ib study of gemcitabine plus PEGPH20 (pegylated recombinant human hyaluronidase) in patients with stage IV previously untreated pancreatic cancer. J Clin Oncol. 2013;31 Suppl:Abstr 4010. | |
Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29(34):4548–4554. | |
Infante JR, Matsubayashi H, Sato N, et al. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25(3):319–325. | |
Mantoni TS, Schendel RR, Rodel F, et al. Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7(11):1806–1815. | |
Frese KK, Neesse A, Cook N, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. | |
Neesse A, Frese KK, Chan DS, Bapiro TE, Howat WJ, Richards FM. SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut. September 25, 2013. [Epub ahead of print.] | |
Alvarez R, Musteanu M, Garcia-Garcia E, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer. 2013;109(4):926–933. | |
Awasthi N, Zhang C, Schwarz AM, et al. Comparative benefits of nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis. 2013;34(10):2361–2369. | |
Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48(4):919–922. | |
Hiraoka N, Ino Y, Sekine S, et al. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br J Cancer. 2010;103(7):1057–1065. | |
Chang Q, Jurisica I, Do T, Hedley DW. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71(8):3110–3120. | |
Sun JD, Liu Q, Wang J, et al. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012;18(3):758–770. | |
Plentz R, Park JS, Rhim AD, et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009;136(5):1741–1749.e1746. | |
Cook N, Frese KK, Bapiro TE, et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209(3):437–444. | |
Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. | |
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. | |
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | |
Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. September 4, 2013. [Epub ahead of print.] | |
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. | |
Roberts EW, Deonarine A, Jones JO, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210(6):1137–1151. | |
Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148(1–2):362–375. | |
Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. | |
Tuveson DA, Neoptolemos JP. Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell. 2012; 148(1–2):21–23. | |
Brais RJ, Davies SE, O’Donovan M, et al. Direct histological processing of EUS biopsies enables rapid molecular biomarker analysis for interventional pancreatic cancer trials. Pancreatology. 2012;12(1):8–15. | |
Hirche TO, Ignee A, Barreiros AP, et al. Indications and limitations of endoscopic ultrasound elastography for evaluation of focal pancreatic lesions. Endoscopy. 2008;40(11):910–917. | |
Saftoiu A, Vilmann P, Gorunescu F, et al. Efficacy of an artificial neural network-based approach to endoscopic ultrasound elastography in diagnosis of focal pancreatic masses. Clin Gastroenterol Hepatol. 2012;10(1):84–90. e81. | |
Neesse A, Hahnenkamp A, Griesmann H, et al. Claudin-4-targeted optical imaging detects pancreatic cancer and its precursor lesions. Gut. 2013;62(7):1034–1043. | |
Eser S, Messer M, Eser P, et al. In vivo diagnosis of murine pancreatic intraepithelial neoplasia and early-stage pancreatic cancer by molecular imaging. Proc Natl Acad Sci U S A. 2011;108(24):9945–9950. | |
Bausch D, Thomas S, Mino-Kenudson M, et al. Plectin-1 as a novel biomarker for pancreatic cancer. Clin Cancer Res. 2011;17(2):302–309. | |
Neesse A, Griesmann H, Gress TM, Michl P. Claudin-4 as therapeutic target in cancer. Arch Biochem Biophys. 2012;524(1):64–70. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.